 Congfen Zhang*
Congfen Zhang* Xiaoqiang Guo
Xiaoqiang Guo- School of Pharmacy, Chengdu University, Chengdu, China
Recently, the species of the insect order Phasmatodea, have attracted the interest of more and more enthusiasts. Here, we obtained the complete mitochondrial genome of Ramulus irregulatiter dentatus (R. irregulatiter dentatus), which belongs to the subfamily of Phasmatidae, detected by Illumina next-generation sequencing. The entire mitochondrial genome is 16,060 bp in length and contains a standard set of 13 protein-coding genes, 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (rRNAs), and a putative A + T-rich region. The base composition and codon usage were typical of Phasmatodea species. The mitochondrial gene organization (37 genes) was consistent with that of other Phasmatidae. A phylogenetic tree was built from the sequence information of the 13 protein-coding genes by Bayesian analyses. The newly sequenced R. irregulatiter dentatus was most closely related to the family Phasmatidae. The complete mitochondrial genome of R. irregulatiter dentatus also provides valuable molecular information for future studies on Phasmatidae insect taxonomy and a framework to unveil more of their cryptic and unknown diversity, so that it can be used to control forest pests and protect crops.
Introduction
The walking stick insects Ramulus irregulatiter dentatus (R. irregulatiter dentatus), which are noted for crypsis by phenotypically and behaviorally mimicking twigs, bark, lichen, moss, and leaves, are part of the subfamily Clitumninae (Phasmatidae: Phasmatidae) (Lee et al., 2018; Robertson et al., 2018). Most of them are characterized by long, dark brown or green colored bodies and feed on leaves of shrubs or trees, which provide them with the most efficient natural camouflage on Earth (Xu et al., 2021a). Stick insect species, often called walking sticks, are a mesodiverse group of large terrestrial insects inhabiting various habitats worldwide. Therefore, it is difficult to identify the phylogenetic relationships of Phasmatodea from the characterization of morphology. The walking stick insects, with more than 3,200 species in approximately 570 genera, are predominantly distributed in tropical regions and temperate areas, including Yunnan, Hunan, Guizhou and Sichuan Provinces in China.
Except for those few species of stick insects beneficial to humans (E.g., Phyllium celebium, P. siccifolium and P. sinensis), most of the walking stick insects are typical phytophagous pests, which are very harmful to their hosts (Xu et al., 2021b). An outbreak of these insects will erode a whole mountain forest and can even wreak havoc on surrounding crops. However, most studies of Phasmatodea have focused on taxonomy (Yang et al., 2021), biology (Gao et al., 2020), morphology (Everts, 2016), bionics (Pan et al., 2021; Kobayashi et al., 2022), prevention and control (Xu et al., 2021b; Yano et al., 2021), but the genera of Phasmatodea within the Polyneoptera family, and the evolutionary relationship between them remain unclear, particularly for highly diverse arthropods.
With the dramatic improvement of DNA sequencing innovation, the mitochondrial genome (mitogenome) groupings have been broadly utilized for recognizing the hereditary variety, species beginning and development, and molecular scientific categorization of insects because of their simple and stable gene composition, absence of genetic recombination, and effective evolution rate (Liu et al., 2016). Insect mitochondrial genomes ordinarily share similar elements with a 14–20 kb double-stranded circular molecule that consists of 37 genes (thirteen protein-coding genes, two ribosomal RNAs, and 22 transfer RNA genes) and a control region (CR). Mitochondrial genomes have been used for determining phylogenetic relationships among insect orders, such as Corydalidae (Zhang et al., 2020), Lepidopteran (Dai et al., 2015), Decapoda (Xin et al., 2018), and Phasmatodea (Xu et al., 2021a). Additionally, mitogenomes have been shown to be valuable in the investigation of phasmatodean molecular evolution, phylogenetics, phylogeography and population genetics (Plazzi et al., 2011; Bradler et al., 2014).
In this Phasmatidae research, we obtained the complete mitogenome sequence of R. irregulatiter dentatus and compared the structure and composition of the mitogenome with those of other available Phasmatidae mitogenomes. Additionally, to investigate the phylogenetic relationship from the perspective of mitogenomes and explain the phylogenetic location of R. irregulatiter dentatus, we constructed phylogenetic positions according to 13 PCGs of 20 Phasmatidae by Bayesian analysis theory and methods. This new investigation of the pool of Phasmatidae mitogenomes provides useful sequence information that can be helpful for distinguishing the species and understanding the evolutionary relationship of R. irregulatiter dentatus from a biological perspective, so that it can be used to control forest pests and protect crops.
Materials and methods
Sample collection
All samples used for sequencing in this study were collected from the Southern Sichuan Bamboo Sea in Sichun Province. The owner of the land gave approval to study on this site, and the work did not contain endangered or protected species. Insects were rinsed with saline, snap frozen in liquid nitrogen for capture and stored at −80°C until DNA extraction.
DNA extraction, sequencing and gene annotation
Total genomic DNA was extracted from the whole body using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Subsequent high-throughput sequencing was performed by the Illumina NextSeq 500 Sequencing System (Illumina, CA, United States). The mitogenome was assembled using the SeqMan II program from the Lasergene software package (DNAStar Inc., Madison, United States). The beginning and stop codons of the protein-coding genes (PCGs) were determined by ORF Finder using invertebrate mitochondrial genetic codes, which was carried out by the NCBI website (https://www.ncbi.nlm.nih.gov/orffinder/). To acquire the base organization of nucleotide sequences, we determined composition skewness as depicted by Junqueira AT skew = [A−T]/[A + T], GC skew = [G−C]/[G + C]. The relative synonymous codon usage (RSCU) values were determined utilizing Codon W. The overlapping regions and intergenic spacers between genes were counted by manual means.
Phylogenetic analysis
The nucleotide sequences of 20 insect whole mitochondrial genomes, or nearly complete mitochondrial genomes (nearly complete refers to complete coding genes), were obtained from NCBI, and the whole mitochondrial genome sequence was obtained by ClustalX version 2.0. The mitochondrial genomes were acquired from GenBank, and the GenBank accession numbers are presented in Table 1. The mitogenomes of Drosophila incompta (NC_025936) and Trigoniophthalmus alternatus (EU016193), Bombyx mori (MK295811), and Atelura formicaria (EU084035) were downloaded and used as outgroups. BI analyses were performed in MrBayes v3.2 [(Ronquist et al., 2012)] using the GTR+Γ substitution model. The runs were set for 50 million generations with sampling every 5,000 generations. Twenty-five percent of the aging samples were omitted, and the average standard deviation of split frequencies was below 0.01.

TABLE 1. Taxon sampling with accession numbers from NCBI.
Results
Mitochondrial genome organization and composition
The length of the complete mitochondrial genome of the walking stick insect R. irregulatiter dentatus was 16,060 bp. (Table 2), which is similar to a previous study (GenBank: AB477463.1). According to recent studies on the total mitochondrial genomes of stick and leaf insects, we found that the varying lengths of the Phasmatodea mitogenomes (15,590–18,248 bp) were caused by the size of the A + T-rich district, gene overlaps, and different intergenic nucleotides (IGNs). The mitogenome of R. irregulatiter dentatus (16,060 bp) contains a A + T-rich region of 1,465 bp. The mitochondrial genomes of the species had similar genes and gene organization as those of other reported stick insects, which have 37 genes, including 13 PCGs, 22 tRNA genes, and two rRNA genes. The gene organization pattern of the R. irregulatiter dentatus mitochondrial genome is similar to the assumed normal insect precursors (Xu et al., 2021c).
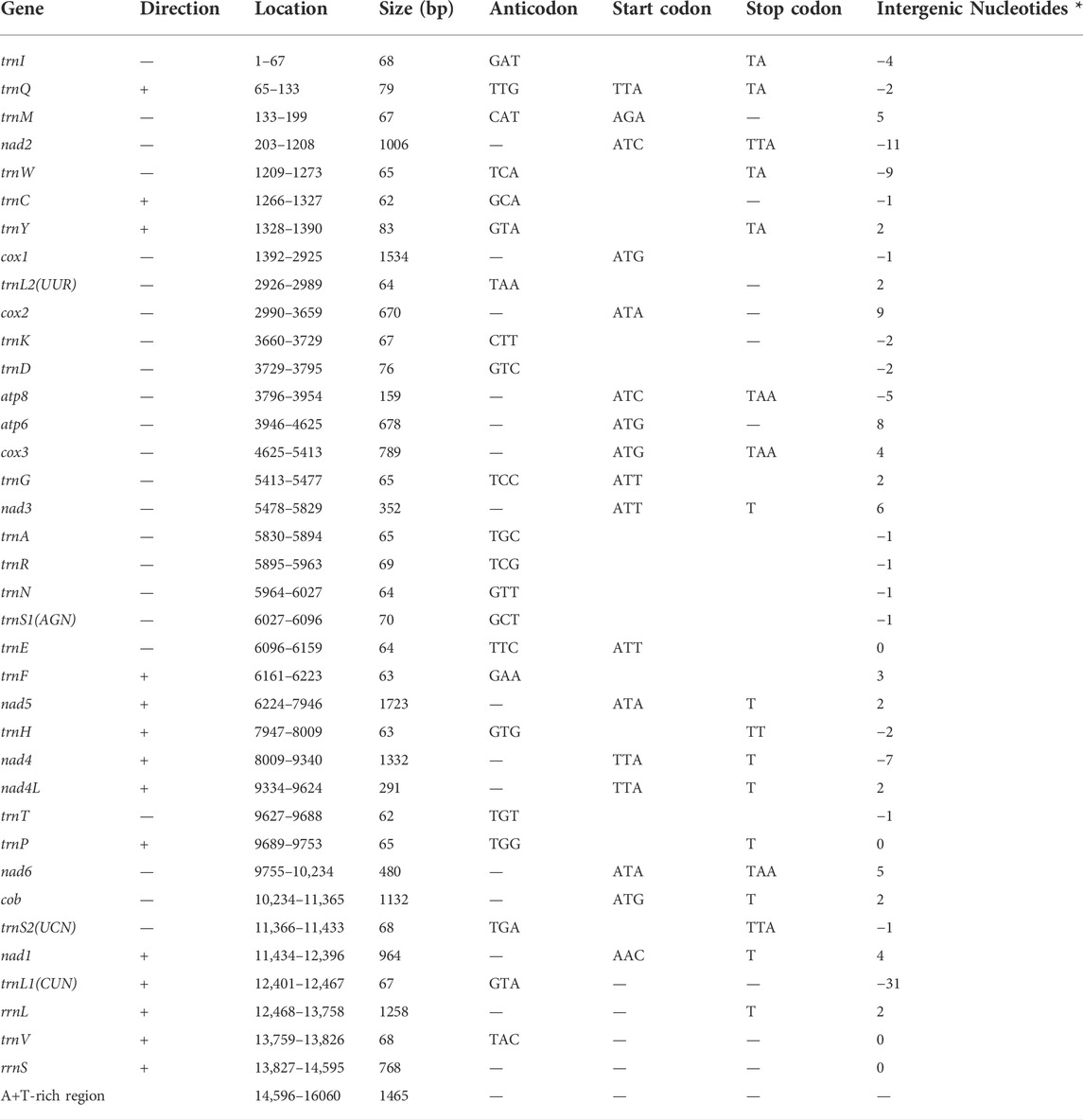
TABLE 2. Annotation and gene arrangement of the Ramulus irregulatiter dentatus mitogenome.
We found that the 13 PCGs of R. irregulatiter dentatus were 11,095 bp in length and accounted for 69.08% of the mitogenome length. Nine of these PCGs (nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, and cob) were coded by the H-strand, while the other four PCGs (nad5, nad4, nad4L, and nad1) were coded by the L-strand. All PCGs started with the typical putative start codons (ATN), except for the nad4 and nad4L genes with TTA. Five genes shared the complete termination codon TAA, while six genes adopted partial stop codons of a single T. A single T as a stop codon for nad4 and nad5 has been described in most of the sequenced Phasmatodea mitogenomes and even in some mammalian mitogenomes (Song et al., 2020). (Table 2).
Eleven overlapping sequences with lengths ranging from 1 to 31 bp were recognized in the R. irregulatiter dentatus mitogenome. The regions of 31 bp overlap were trnL1–rrnL 11 bp nad2-trnW and 9 bp trnW–trnC (Table 2). Other overlapping regions were shorter than 5 bp. The intergenic spacers of R. irregulatiter dentatus mitogenomes were distributed in 15 regions and varied in size from 1 to 9 bp with a total length of 58 bp. The longest spacer (9 bp) occurred between cox2 and trnK. The 7 bp conserved sequence containing “ATGATAA” between atp8 and atp6 (Figure 4A) has also been documented in several Phasmatodea sequenced to date and the motif commonly exists in other insects, such as lepidopterans and megalopterans (Yang et al., 2020; Zhang et al., 2020). The intergenic spacers in R. irregulatiter dentatus were similar to those of the complete mitogenomes of other Phasmatodea insects (Song et al., 2020).
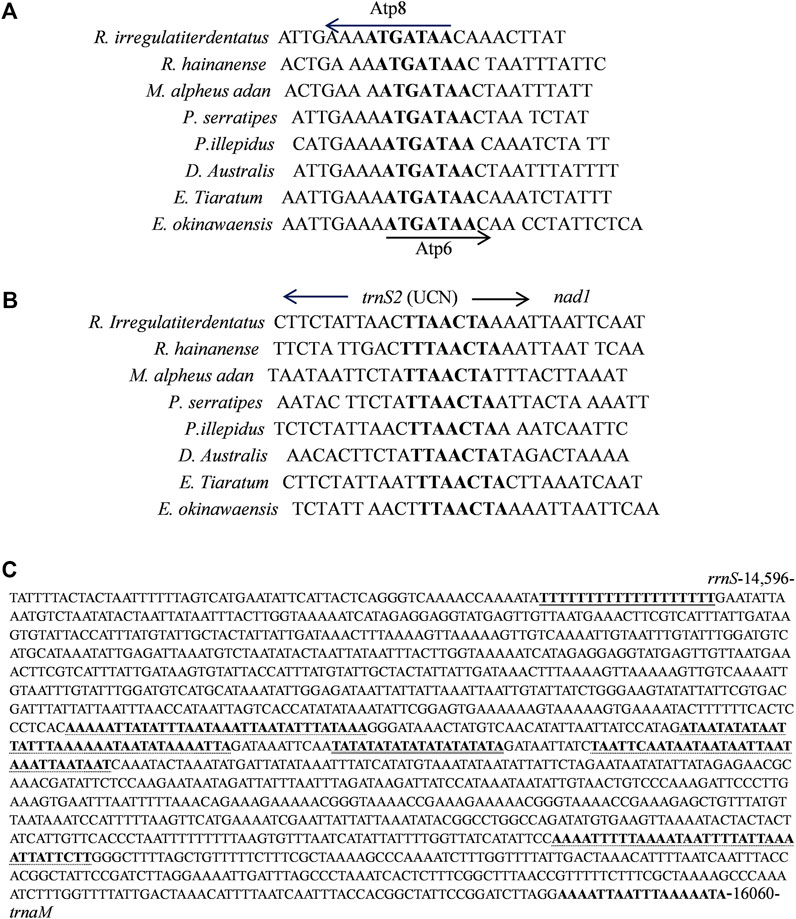
FIGURE 4. (A) Sequence analysis of the partial Atp6 and Atp8 regions of 8 Phasmatidae insects. The boxed nucleotides indicate the “ATAGATAA” conserved motif. (B) Sequence alignment of the partial trnS2 and nad1 regions of 8 Phasmatidae insects. The boxed nucleotides indicate the “TTACTA” conserved motif. (C) Features present in the A + T-rich region of R. irregulariter dentatus. The poly-T stretch is underlined, while the poly-A stretch is underlined in bold black letters. The single microsatellite T/A repeat sequence is indicated by double underlined, and long T/A repeats are indicated by dotted underlined.
On the other hand, we identified the nucleotide composition of the R. irregulatiter dentatus mitochondrial genome. The frequencies of adenine, cytosine, guanine, and thymine were 37.5%, 9.34%, 15.07%, and 38.09%, respectively. A + T% shared 75% of the whole mitogenome length, which was similar to those of other previously reported phasmatodean mtDNAs (Kômoto et al., 2011). The A + T% values of 13 PCG, 22 tRNA and 2 rRNA genes accounted for 73.56%, 76.82%, and 81.66%, respectively. The 87.06% percent of A + T% in the A + T region was highest (Table 3). Thirteen PCGs and 2 rRNAs obtained negative scores, and 22 tRNA obtained positive scores. The estimated G + C skews and obtained negative scores were, similar to other species. All of the mtDNA sequenced in this study shared fairly common gene composition patterns among Hexapoda (Kômoto et al., 2011).
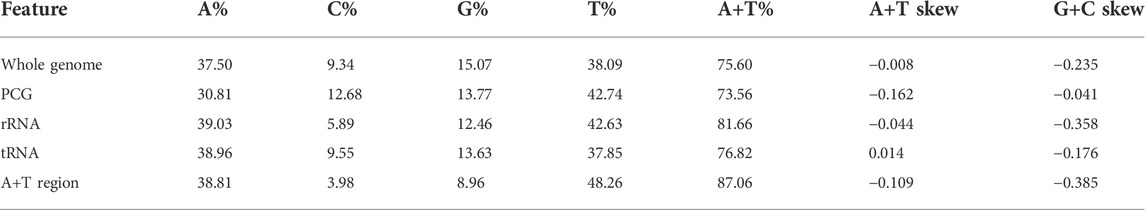
TABLE 3. Nucleotide composition of the Ramulus irregulatiter dentatus mitochondrial genome.
Protein-coding genes and codon usage
The behavior of codon usage of the protein-coding genes in the R. irregulatiter dentatus mitochondrial genome in the PCGs was investigated. We found that all codons were present, and AUA (M), AAA (K), AAU (N), UUA (L), UAU (Y), AUU (I), and UUU (F) were the most abundant codons in the 13 PCGs (Table 4). Here, we compared the RSCU values among 8 PCGs calculated from the Phasmatidae mitogenomes (Figure 1). AGN codons (coding for Arg2), TAN (coding for Ter1), and TCN (coding for Ser2) were more frequently used than CGN codons (coding for Arg1), TGA (coding for Ter2), and AGN (coding for Ser1), respectively. Interestingly, TTN (coding for Leu2) was higher than CTN (coding for Leu1) in the mitogenomes of the 8 Phasmatidae insect species except Ramulus hainanense. From the codon distribution in Phasmatidae CDspT, we found that Asn and Ter1 were the richest. The CDspTs of Ile(47.0) and Lys(18.3) of the R. irregulatiter dentatus mitogenome are significantly lower than those of other Phasmatidae insects, but Leu2(68.1) and Phe(60) are the highest among them (Figure 2). This phenomenon is similar to that found in the mitogenomes of ditrysian (Wei et al., 2013), neuropterid (Enrico Negrisolo and Patarnello, 2011) and lepidopteran (Dai et al., 2016; Qiu-Ning Liu et al., 2016; Zou et al., 2017) insects.
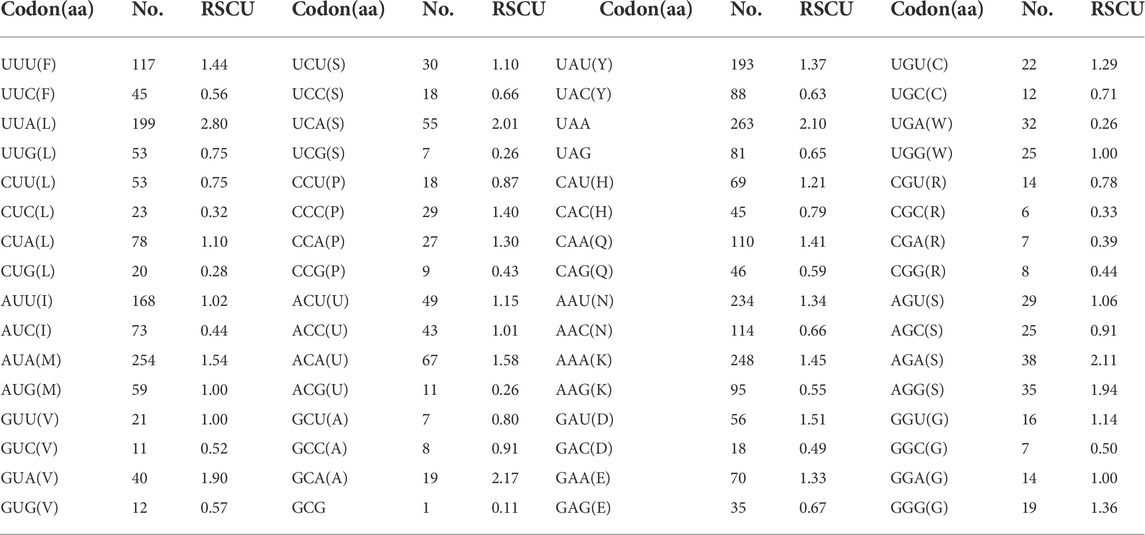
TABLE 4. Codon usage of the protein-coding genes in the Ramulus irregulatiter dentatus mitochondrial genome.
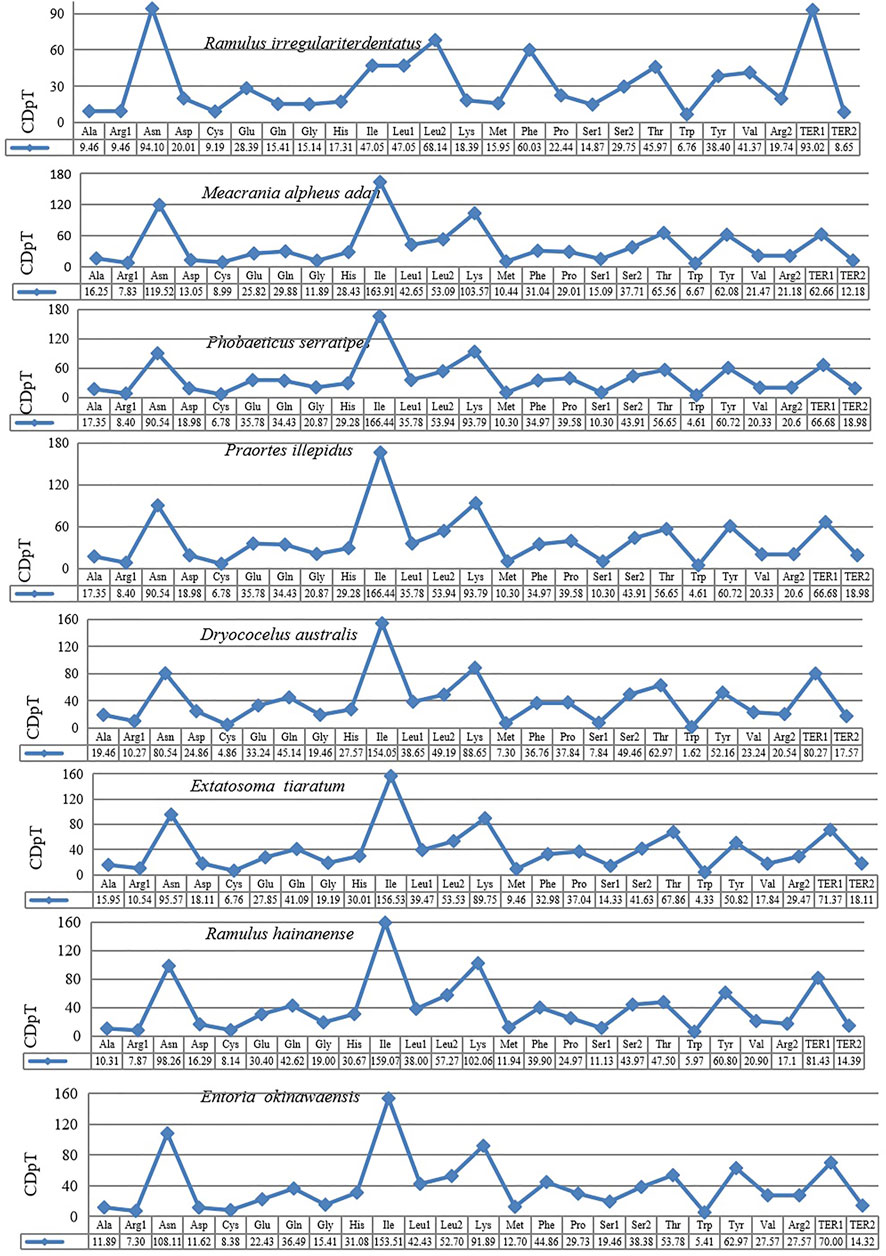
FIGURE 1. Codon distribution in Phasmatidae CDspT, codons per thousand codons. The protein-coding genes are from the following species: Ramulus irregulariter dentatus, Megacrania alpheus adan (NC_014688.1), Phobaeticus serratipes (NC_014678.1), Praortes illepidus (NC_014695.1), Dryococelus australis (AP018522), Extatosoma tiaratum (AB642680.1), Ramulus hainanense (FJ156750.1), and Entoria okinawaensis (NC_014694.1).
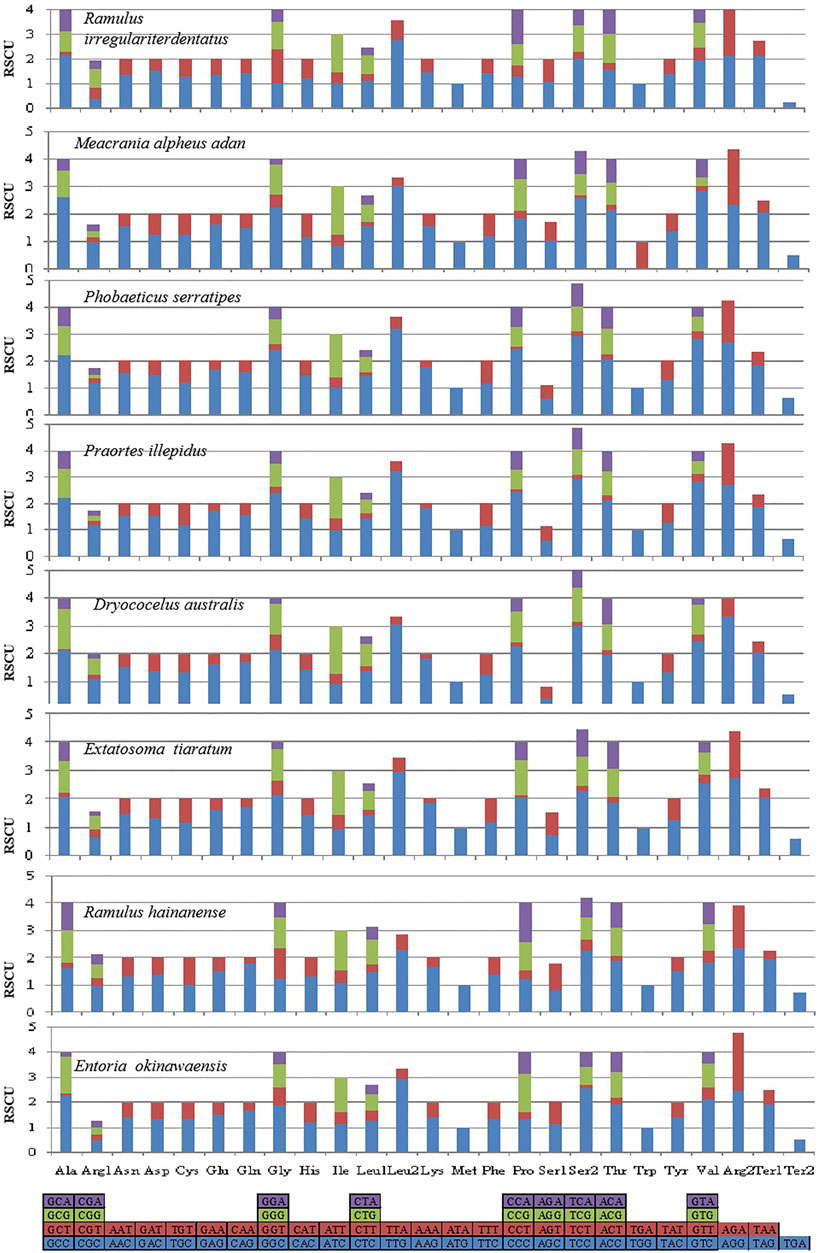
FIGURE 2. The mitochondrial genome (mitogenome) relative synonymous codon usage (RSCU) in Phasmatidae. Codon families are provided on the X axis. Codons indicated above the bar are not present in the mitogenome. The protein-coding genes are from the following species: Ramulus irregulariter dentatus, Megacrania alpheus adan (NC_014688.1), Phobaeticus serratipes (NC_014678.1), Praortes illepidus (NC_014695.1), Dryococelus australis (AP018522), Extatosoma tiaratum (AB642680.1), Ramulus hainanense (FJ156750.1), and Entoria okinawaensis (NC_014694.1).
Ribosomal RNA and transfer RNA genes
The mitochondrial genome of R. irregulatiter dentatus had 22 tRNA genes, ranging from 62 bp (trnC, trnT) to 83 bp (trnY), as in other Phasmatodea mitogenomes. Fourteen genes were encoded on the H-strand, and the others were encoded on the L-strand (Table 2). The total tRNA size of R. irregulatiter dentatus was 1,484 bp, with a high A + T bias of 76.8% (Table 3).
The AT skew was positive 0.014 compared with GC being negative 0.176 (Table 3). All tRNAs could be folded into normal secondary cloverleaf structures except for the trnH, trnM and trnF genes, which lack TΨC loops (Figure 3). This phenomenon was also observed in the mitochondrial genomes of the other three Phasmatodea insects, including trnN (Orestes guangxiensis and Peruphasma schultei) and trnP (O. guangxiensis) losing the TΨC loops, and trnS1 (O. guangxiensis) lacking the dihydrouridine (DHC) arm (Xu et al., 2021a). Generally, in the absence of DHC arms or TΨC loops in other stick insects and other species of insects, there is lower translational activity compared with that of the typical structures (Wang et al., 2019; Zhang et al., 2020; Xu et al., 2021a). We also found some incorrect pairs, such as unmatched U-U base pairs in trnV, A-G base pairs in trnW, A-A base pairs in trnS1, A-G base pairs in trnW, C-A base pairs in trnG, and U-U base pairs in trnV, trnA, trnY, trnS2, and trnL1, in three of the other Phasmatodea insects (O. guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis) (Xu et al., 2021a). There were 2 or 3 G-U pairs in trn A, trn C, trn F, trn G, and trn H, and one mismatched G-U base pair included trnD, trnL2, trnK, trnM, trnS1, trnP, trnR, trnY, and trnT (Figure 3). Most mismatched pairs were located at the amino acid acceptor stem and DHU stem of the transfer RNA gene secondary structures. Mismatched pairs may affect aminoacylation and the function of binding to ribosomes.
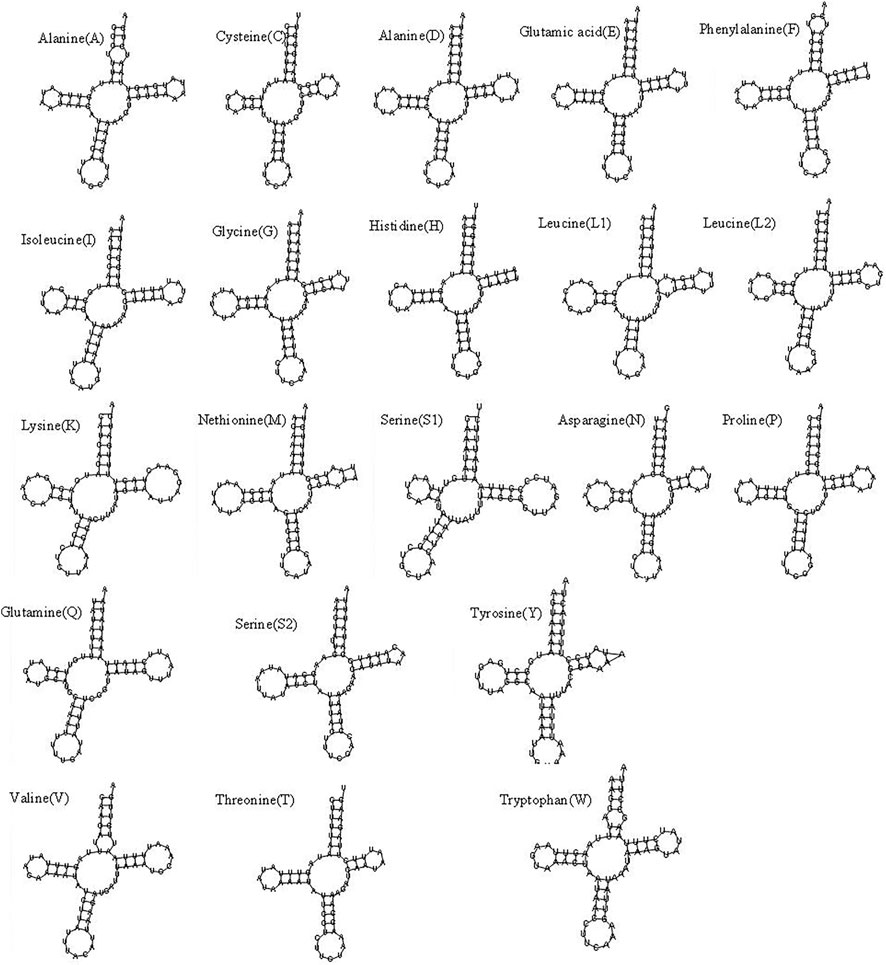
FIGURE 3. Putative secondary structures of the 22 tRNA genes of the Ramulus irregulariter dentatus mitogenome.
The 7 bp spacer sequence between trnS2 and nad1 contained the “TTAACTA” motif (Figure 4B), which is a common feature among Phasmatidae insects. Several similar motifs were found in other insects, including lepidopterans with “ATACTAA” (Yang et al., 2020) and pentanucleotide TCTAA conservative motifs existing in the overlap regions between COX1 and trnL2 of several other stick insects (Plazzi et al., 2011).
The A + T-rich region
The mitogenome of R. irregulatiter dentatus includes an A + T-rich region of 1,465 bp that shares the highest A + T content (87.06%), negative AT skew (−0.109) and GC skew (−0.385) (Table 3). These A + T content values were similar to those found in the presented phasmatodean mitogenomes, such as 78.0% for Pharnaciini spec. indet., 76.3% for M. brachptera, and 76.9% for Phraortes sp. (Song et al., 2020). The control region was characterized by a 19 bp poly-T stretch, a microsatellite-like (TA)10 and a terminal poly-A element (Figure 4C). Multiple tandem repeat elements, as a characteristic of the insect A + T-rich region, have been found in the mitogenome sequence of R. irregulatiter dentatus. The A + T-rich region having four sequences with more than 30 bp long repeats included “AAAAATTATATTTAATAAATTAATATTTATAAA,” “ATAATATATAATTATTTAAAAAATA ATATAAAATTA,” “TAATTCAATAATAATAATTAATAAATTAATAAT,” and “AAAATTTTTAAAATAATTTTATTAAAATTATTCTT.” Copies of tandem repeats regions in the AT-rich region were also detected among other Phasmatodea mitogenomes including Ramulus hainanense, Extatosoma tiaratum and Phraortes sp. 1 NS-2020 (Shuichiro Tomita and Kômoto, 2011).
Phylogenetic relationships
According to the phylogenetic tree of R. irregulatiter dentatus, it can be confirmed that Lonchodinae, Necrosciinae, Platycraninae, Eurycanthinae, Clitumninae, Heteropterygidae, Aschiphasmatoidea, Pseudophasmatoidea, etc., are all on the same large branch. It revealed that R. irregulatiter dentatus is most closely related to E. okinawaensis and R. hainanense in the Clitumninae subfamily. In addition, Phasmatidae and Lonchodidae are the families that are most closely related, followed by Bacilloidea (Figure 5). In the present research, the relationships at the superfamily level are consistent with previous investigations of Phasmatodea family-level groups by wing venation characters. Our data suggested that mitochondrial genome sequences are evolutionarily conserved among different Phasmatodea species. Molecular phylogenetic analysis based on mitochondrial genomes can provide profoundly helpful information on the scientific classification of Phasmatodea.
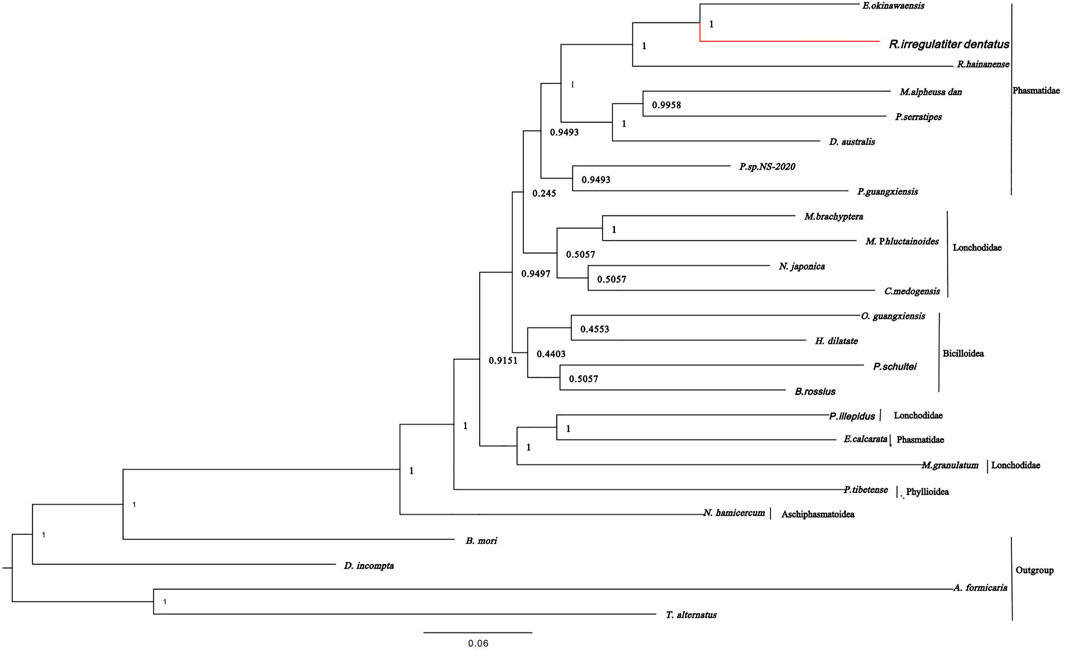
FIGURE 5. Phylogenetic relationships were built from the mitochondrial genome sequences of 20 insects by Bayesian inference methods.
Discussion
The mitogenome of R. irregulatiter dentatus is a typical circular DNA molecule 16,060 bp long. The mitogenome sequence had the same genes and gene organization as those formerly investigated for other stick insect species, with 37 genes involving 13 protein-coding genes (PCGs), 22 tRNA genes, and two rRNA genes (Xu et al., 2021c). All protein-coding genes (PCGs) start with standard ATN initiation codons except for nad4 and nad4L. Twenty-two tRNAs are predicted to fold into a characteristic secondary cloverleaf structure, except for the trnH, trnM, and trnF genes, which lack TΨC loops. The sizes of the large and small ribosomal RNA genes are 1,258 bp and 768 bp, respectively. The A + T control region contained a 19 bp poly-T stretch, a microsatellite-like (TA)10 and a terminal poly-A element. In addition, the phylogenetic analyses confirmed that R. irregulatiter dentatus belongs to Phasmatidae.
Phasmatodea, as a large group of mostly nocturnal herbivore insects, is regarded as a potential forest pest not only in China but also in Korea, Japan and North America (Lee et al., 2018; Liu, 2021; Yano et al., 2021). In China, stick insects have defoliated trees and damaged more than 60 plant species in the Provinces of Jiangxi, Guangdong, Hubei and Guizhou since the first record in 1985. However, the existence of unique morphological features in some species within the genus Phasmatidae led to the emergence of a new genus, which included new taxa and new nomenclature of Medaurini (Phasmatidae: Clitumninae) from China (Ho, 2021), Cretophasmomima traceyae sp. nov. (Phasmatodea: Susumanioidea) in southern England (Hennemann, 2020), Diapheromera arena n.sp. t (Phasmatodea: Diapheromeridae: Diapheromera) from western Texas and New Mexico (Stidham and Stidham, 2018), the genus Parapachymorpha Brunner von Wattenwyl, 1893 (Phasmatidae, Phasmatidae, Clitumninae) from Laos, etc. At present, the control methods and technologies used for Phasmatidae pests mainly include physical methods, chemical methods and biological methods. Although chemicals are an effective control method, they will lead to drug resistance and pesticide residues by repeated use. Therefore, biological control measures are more popular to control these pests. In this paper, the sequence analysis of the mitochondrial genome of R. irregulatiter dentatus also provides a reference for the use of biotechnology to control forest pests. On the other hand, despite Phasmatidae as a phytophagous and predatory species with important agricultural value, their evolutionary relationships remained ineffectively explored. The mitogenome data of R. irregulatiter dentatus would be useful for further genetic studies, phylogenetic analysis, taxonomic resolution and pest control of Phasmatidae insects.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
CZ designed the research, collected and identified the sample. XG revised the version to be published. All authors agree to be accountable for all aspects of the work.
Funding
This work was supported by Foundation for Scientific Research Startup Project of Chengdu University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bradler, S., Robertson, J. A., and Whiting, M. F. (2014). A molecular phylogeny of Phasmatodea with emphasis on Necrosciinae, the most species-rich subfamily of stick insects. Syst. Entomol. 39 (2), 205–222. doi:10.1111/syen.12055
Dai, L.-S., Zhu, B. J., Zhao, Y., Zhang, C. F., and Liu, C. L. (2016). Comparative of Eligma narcissus and other Lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. Sci. Rep. 6, 26387. doi:10.1038/srep26387
Dai, L. S., Qian, C., Zhang, C., Wang, L., Wei, G., Li, J., et al. (2015). Characterization of the complete mitochondrial genome of Cerura menciana and comparison with other Lepidopteran insects. Plos One 10 (8), e0132951. doi:10.1371/journal.pone.0132951
Enrico Negrisolo, M. B., and Patarnello, Tomaso (2011). The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics 12, 221. doi:10.1186/1471-2164-12-221
Everts, S. (2016). Stick insects stole enzyme genes from their microbiome. C&EN Glob. Enterp. 94 (23), 9–10. doi:10.1021/cen-09423-scicon002
Gao, K., Horng, J. H., Liu, Y., and Chang, C. C. (2020). A reversible secret image sharing scheme based on stick insect matrix. Ieee Access 8, 130405–130416. doi:10.1109/access.2020.3009410
Hennemann, F. H. (2020). Megacraniinae: The palm stick insects: A new subfamily of old world Phasmatodea and a redefinition of Platycraninae Brunner v. Wattenwyl, 1893 (Phasmatodea: "Anareolatae"). Zootaxa 4896 (2), 151–179. doi:10.11646/zootaxa.4896.2.1
Ho, G. W. C. (2021). Contribution to the knowledge of Chinese Phasmatodea X: Eight new species of cnipsomorpha from China (Phasmatidae: Clitumninae: Medaurini). Zootaxa 5026 (1), 102–126. doi:10.11646/zootaxa.5026.1.4
Kobayashi, S., Takaoka, C., Tanimoto, H., Arimitsu, S., and Izawa, M. (2022). Effect of spraying behavior and body size on predators of the big head stick insect Megacrania tsudai (Phasmatodea: Phasmatidae). Entomological Sci. 25 (2), e12508. doi:10.1111/ens.12508
Kômoto, N., Yukuhiro, K., Ueda, K., and Tomita, S. (2011). Exploring the molecular phylogeny of phasmids with whole mitochondrial genome sequences. Mol. Phylogenet. Evol. 58 (1), 43–52. doi:10.1016/j.ympev.2010.10.013
Lee, J., Baek, S., Kang, C., Lee, Y. S., and Lee, Y. (2018). Temperature-dependent development and oviposition models of Ramulus irregulariterdentatus (Phasmida: Phasmatidae). J. Asia-Pacific Entomology 21 (3), 903–913. doi:10.1016/j.aspen.2018.07.003
Liu, H. (2021). Biology and ecology of the northern walkingstick, Diapheromera femorata (say) (Phasmatodea: Diapheromerinae): A review. J. Appl. Entomol. 145 (7), 635–647. doi:10.1111/jen.12902
Liu, Q. N., Chai, X. Y., Bian, D. D., Zhou, C. L., and Tang, B. P. (2016). The complete mitochondrial genome of Plodia interpunctella (Lepidoptera: Pyralidae) and comparison with other pyraloidea insects. Genome 59 (1), 37–49. doi:10.1139/gen-2015-0079
Pan, J. S., Jeng-Shyang Pan, P. C. S., Pei-Cheng Song, C. A. P., and Chun-An Pan, A. A. (2021). The Phasmatodea population evolution algorithm and its application in 5G heterogeneous network downlink power allocation problem. J. Internet Technol. 22 (6), 1199–1213. doi:10.53106/160792642021112206001
Plazzi, F., Ricci, A., and Passamonti, M. (2011). The mitochondrial genome of Bacillus stick insects (Phasmatodea) and the phylogeny of orthopteroid insects. Mol. Phylogenet. Evol. 58 (2), 304–316. doi:10.1016/j.ympev.2010.12.005
Qiu-Ning Liu, Z.-Z. X., Bian, D-D., Chai, X-Y., Zhou, C-L., Tang, B-P., and Tang, B. P. (2016). The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci. Rep. 6, 35878. doi:10.1038/srep35878
Robertson, J. A., Bradler, S., and Whiting, M. F. (2018). Evolution of oviposition techniques in stick and leaf insects (Phasmatodea). Front. Ecol. Evol. 6, 1–15. doi:10.3389/fevo.2018.00216
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi:10.1093/sysbio/sys029
Shuichiro Tomita, K. Y., and Kômoto, N. (2011). The mitochondrial genome of a stick insect Extatosoma tiaratum (Phasmatodea) and the phylogeny of polyneopteran insects. J. Insect Biotechnol. Sericology 80, 79–88. doi:10.11416/jibs.80.3_079
Song, N., Li, X., and Na, R. (2020). Mitochondrial genomes of stick insects (Phasmatodea) and phylogenetic considerations. PLoS One 15 (10), e0240186. doi:10.1371/journal.pone.0240186
Stidham, J. A., and Stidham, T. A. (2018). A new species of stick insect (Phasmatodea: Diapheromeridae: Diapheromera) from holocene sandy areas in western Texas and new Mexico (USA). Entomol. News 128 (1), 1–10. doi:10.3157/021.128.0102
Wang, J., Dai, X. Y., Xu, X. D., Zhang, Z. Y., Yu, D. N., Storey, K. B., et al. (2019). The complete mitochondrial genomes of five longicorn beetles (Coleoptera: Cerambycidae) and phylogenetic relationships within Cerambycidae. PeerJ 7, e7633. doi:10.7717/peerj.7633
Wei, S. J., Shi, B.-C., Gong, Y-J., Li, Q., and Chen, X. X. (2013). Characterization of the mitochondrial genome of the Diamondback Moth Plutella xylostella (Lepidoptera: Plutellidae) and phylogenetic analysis of advanced moths and butterflie. DNA Cell Biol. 32 (4), 173–187. doi:10.1089/dna.2012.1942
Xin, Z. Z., Liu, Y., Tang, B., Zhou, C., and Liu, Q. (2018). A comprehensive phylogenetic analysis of grapsoidea crabs (Decapoda: Brachyura) based on mitochondrial cytochrome oxidase subunit 1 (CO1) genes. Turk. J. Zool. 42 (1), 46–52. doi:10.3906/zoo-1703-46
Xu, F. L., Jiang, Y. J., Yang, M. F., Da, W., Yang, X. W., and Shi, T. Y. (2021). Three first records of stick insects attacking plants (Inseect: Phasmida) in Tibet. Braz J. Biol. 83, e245862. doi:10.1590/1519-6984.245862
Xu, K. K., Chen, Q. P., Ayivi, S. P. G., Guan, J. Y., Storey, K. B., Yu, D. N., et al. (2021). Three complete mitochondrial genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and their phylogeny. Insects 12 (9), 779–818. doi:10.3390/insects12090779
Xu, K. K., Chen, Q. P., Guan, J. Y., Zhang, Z. Y., Storey, K. B., Yu, D. N., et al. (2021). The mitochondrial genome of Eurycantha calcarata Lucas, 1869 (Phasmatodea: Lonchodinae) and its phylogeny. Mitochondrial DNA. B Resour. 6 (11), 3109–3111. doi:10.1080/23802359.2021.1964403
Yang, H. R., Shi, C., Engel, M. S., Zhao, Z., Ren, D., and Gao, T. (2021). Early specializations for mimicry and defense in a Jurassic stick insect. Natl. Sci. Rev. 8 (1), 146–155. doi:10.1093/nsr/nwaa056
Yang, L., Dai, J., Gao, Q., Yuan, G., Liu, J., Sun, Y., et al. (2020). Characterization of the complete mitochondrial genome of Orthaga olivacea warre (Lepidoptera: Pyralidae) and comparison with other Lepidopteran insects. PLoS One 15 (3), e0227831. doi:10.1371/journal.pone.0227831
Yano, K., Ozaki, T., Suzuki, T., Yamazaki, H., Nasuno, M., Degawa, Y., et al. (2021). Outbreak of the stick insect, Ramulus mikado (Phasmatodea, Phasmatidae), in the akashina area of Japan (azumino city, nagano prefecture). Entomological Sci. 24 (2), 196–200. doi:10.1111/ens.12467
Zhang, C. F., Liu, X., Liu, C., and Luo, X. (2020). Characterization of the complete mitochondrial genome of Acanthacorydalis fruhstorferi van der Weele (Megaloptera: Corydalidae). J. Kans. Entomological Soc. 93 (4), 267–281. doi:10.2317/0022-8567-93.4.267
Zou, Z., Min, Q., Cheng, S., Xin, T., and Xia, B. (2017). The complete mitochondrial genome of Thitarodes sejilaensis (Lepidoptera: Hepialidae), a host insect of Ophiocordyceps sinensis and its implication in taxonomic revision of Hepialus adopted in China. Gene 601, 44–55. doi:10.1016/j.gene.2016.11.039
Keywords: phasmatodea, mitochondrial genome, Ramulus irregulatiter dentatus, gene organization, phylogenetic tree
Citation: Zhang C and Guo X (2022) Organization of the mitochondrial genome of Ramulus irregulatiter dentatus (Phasmatidae: Phasmatidae). Front. Genet. 13:967113. doi: 10.3389/fgene.2022.967113
Received: 12 June 2022; Accepted: 28 July 2022;
Published: 29 August 2022.
Edited by:
Qiu-Ning Liu, Yancheng Teachers University, ChinaReviewed by:
Li-Shang Dai, Wenzhou Medical University, ChinaBaojian Zhu, Anhui Agricultural University, China
Copyright © 2022 Zhang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Congfen Zhang, emhhbmdjb25nZmVuQGNkdS5lZHUuY24=