 Yu-Xing Liu
Yu-Xing Liu Man-Hua Ding3†
Man-Hua Ding3† Yang Zhang
Yang Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 January 2023
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.964990
This article is part of the Research Topic The Intersection of Gene Regulation and Metabolism in Cardiovascular Disease View all 9 articles
Background: Hyperhomocysteinemia (HHcy) is a risk factor for thromboembolic disease. Defects in one-carbon metabolism (1-CM)-related genes, such as methylenetetrahydrofolate reductase (MTHFR), methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1 (MTHFD1), can cause HHcy and may also affect the efficacy of folic acid therapy. The details of mechanisms are yet to be further investigated.
Method: We described a Chinese family with hereditary HHcy. The proband suffered from severe thromboembolic disease and experienced failure of folic acid therapy. Two sons of the proband were also diagnosed with HHcy but were sensitive to folic acid therapy. Whole-exome sequencing (WES) was conducted to evaluate the genetic lesion of this family.
Results: Compound heterozygous variants (a common polymorphism, p. A222V, and a novel variant, p. C631*fs*1) of the MTHFR gene and a homozygous missense variant (p. K134R) of the MTHFD1 gene were identified in the proband. The two sons, with successful intervention, only harbored the homozygous p. A222V variant of the MTHFR gene.
Conclusion: The clinical manifestations and genetic research synergistically confirmed the diagnosis of HHcy and clarified the failure of folic acid therapy in the proband caused by doubly bi-allelic variants of the MTHFR and MTHFD1 genes. Our study increased our understanding of the molecular basis of 1-CM-related gene defects on folic acid therapy in HHcy.
Hyperhomocysteinemia (HHcy) refers to abnormally high levels of plasma/serum homocysteine (Hcy) and is a risk factor for thromboembolic disease with highly variable symptoms including atherosclerosis, cerebral infarction, and pulmonary embolism (Son and Lewis, 2022). Some brain-related disorders, including epilepsy, have also been reported to be associated with HHcy (Hrncic et al., 2018). At present, folate supplementation is a common treatment for HHcy, while the efficacy may be affected by many factors (Du et al., 2018; Li et al., 2020). The invalid response to folate supplementation treatment remains a clinical challenge (Li et al., 2020).
The plasma Hcy level is closely associated with one-carbon metabolism (1-CM) which comprises methionine and Hcy metabolism. Genetic deficiencies in 1-CM can cause inherited HHcy, such as the methionine synthase deficiency, folic acid synthesis deficiency, methylenetetrahydrofolate reductase (MTHFR) deficiency, and methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1 (MTHFD1) deficiency (Burda et al., 2015a; Lyon et al., 2020; Steele et al., 2020; Biesalski et al., 2022; Sudheer et al., 2022). MTHFR deficiency results in homocystinuria and HHcy in an autosomal recessive pattern, and p. A222V of the MTHFR gene in the homozygous state is the most common genetic cause of elevated plasma Hcy. Compound heterozygous variants combined by the p. A222V mutation and other mono allelic MTHFR variants have also been reported in HHcy cases (Rummel et al., 2007; Clement et al., 2022; Fatima et al., 2022). Similarly, patients with MTHFD1 deficiency may present with HHcy merged with other phenotypes, including megaloblastic anemia and hemolytic uremic syndrome (Burda et al., 2015a). Compared with the MTHFR variant, few MTHFD1 mutations are identified in HHcy cases. Genetic polymorphisms of key enzymes in 1-CM, including betaine–homocysteine methyltransferase (BHMT), MTHFR, MTHFD1, methionine synthase reductase (MTRR), and methionine synthase (MTR), have also been reported to affect the efficacy of folate therapy in HHcy (Du et al., 2018; Li et al., 2019; Partearroyo et al., 2019). The details of mechanisms are yet to be further investigated.
Here, we reported a Chinese family with hereditary HHcy. The proband suffered from severe thromboembolic disease, and folic acid therapy was ineffective. Two sons of the proband were also diagnosed with HHcy but benefitted from folic acid therapy. Whole-exome sequencing (WES) was conducted to evaluate the genetic lesion of this family and identified doubly bi-allelic variants in MTHFR and MTHFD1 genes. Among these variants, the variant (p.C631*fs*1) of the MTHFR gene has not been published previously, and therefore, it is considered to be a novel mutation.
This study was approved by the Review Board of Xiangya Hospital, Changsha, China. Written informed consent was obtained from all adult participants and legal guardians of minor participants.
All family members (three patients and one healthy member) were enrolled and diagnosed by ultrasound examination, coronary angiogram, and laboratory inspection. The clinical data, including folate acid, vitamin B12 (VB12), ferritin, and erythropoietin (EPO), of each family member were collected for diagnosis or response evaluation. A total of 200 healthy subjects, as described in our previous study, were enrolled in this study to exclude polymorphisms (Wang et al., 2021).
Genomic DNA was prepared from peripheral blood lymphocytes of all the participants using the DNeasy Blood & Tissue Kit (QIAGEN, Valencia, CA, United States). The primary sequence for WES was provided by the Novogene Bioinformatics Institute (Beijing, China). Exomes were captured using the SureSelect Human All Exon V6 kit (Agilent, Santa Clara, CA, United States), and next-generation sequencing was conducted using a HiSeq X Ten system (Illumina, San Diego, CA, United States). The strategies of data filtering and the necessary bioinformatics analyses can be referred to in the previous studies published by our laboratory group (Fan et al., 2019; Wang et al., 2021).
Sanger sequencing was used to validate the candidate variants identified in WES. Segregation analysis was performed in the family members of this study. Sequences of the polymerase chain reaction (PCR) products were determined using the ABI 3100 Genetic Analyzer (ABI, Foster City, CA, United States).
SWISS-MODEL software (https://swissmodel.expasy.org/interactive) was used to identify the function of the mutation. The conservation analysis was performed by ConSurf Server software (http://consurf.tau.ac.il/).
The proband is a 51-year-old farmer from Jiangsu Province in China. He was referred to our hospital due to the progression of severe thrombotic disease. A medical history investigation showed that he suffered from cerebral infarction combined with secondary epilepsy, old myocardial infarction (OMI), and lower extremity arterial thrombosis. He was a non-smoker and had no previous history of diabetes, hypertension, or traumatic head injury. The head magnetic resonance angiography (MRA) showed severe stenosis of the right internal carotid artery and middle cerebral artery, and large softening lesions appeared in the right cerebral hemisphere (Figures 1A–C). A left ventricular mural thrombus was found in the proband (Figure 1D), as well as thrombosis in the left lower extremity artery and bilateral popliteal artery (Figure 1E). Laboratory examination showed a much higher level of total plasma Hcy (52.14 μmol/L), while the results for folate acid (5.55 ng/ml), VB12 (281 pg/ml), ferritin (169.3 ng/ml), and EPO (5.91 mIU/ml) were normal. Financial reasons prevented the patient from further hospitalization for testing and treatment after his condition improved slightly in the previous treatment. The major events in the medical records during 2016–2019 are shown in Figure 1F with several available data of Hcy. The proband was diagnosed with HHcy, and his thrombotic disease was related to a high level of plasma Hcy. A family history survey showed that two sons of the proband, II-1 and II-2, also had high levels of plasma Hcy. The eldest son (II-1) had developed pulmonary embolism and deep venous thrombosis of the right lower limb since he was 30 years old. The proband’s wife (I-1) had a normal plasma–Hcy level (Figure 1G). The levels of folate and VB12 detected in three family members (I-1, II-2, and II-3) were normal, denying the relationship between the abnormal Hcy level and a lack of folic acid or VB12 in this family and putting forward the possibility of genetic factors. No other malformations were detected in this family (Table 1). Under the diagnosis of inherited HHcy, all patients in this family were managed with folate (5 mg/day), thiamine (75 mg/day), and mecobalamin (1.5 mg/day), which decreased the two sons’ plasma–Hcy level to normal for 1 year after the diagnosis. Confusingly, the proband’s plasma–Hcy level remained elevated (49.29–52.14 μmol/L) despite 1 year of medications. Changes of various indexes, including Hcy, folic acid, VB12, ferritin, and EPO, during treatment are shown in Figures 1H–L.
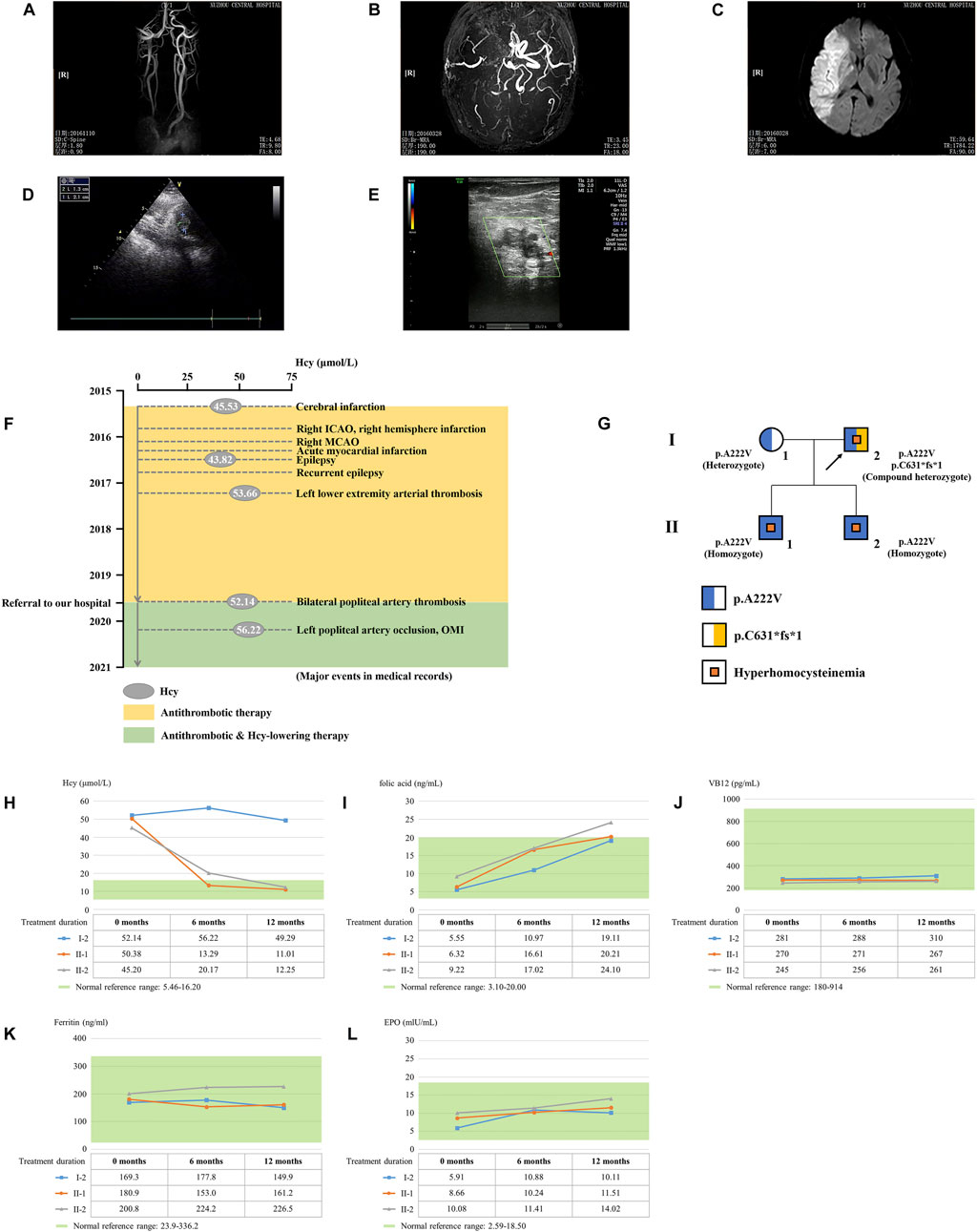
FIGURE 1. Clinical data on this HHcy family. The head MRA showed the (A) right internal carotid artery, (B) middle cerebral artery, and (C) right cerebral hemisphere of the proband (I-2). (D) Cardiac color ultrasound showed the left ventricular mural thrombosis of the proband. (E) Ultrasound examination showed lower-limb artery thrombosis of the proband. (F) Major events in the medical records of the proband. ICAO, internal carotid artery occlusion; MCAO, middle cerebral artery occlusion; OMI, old myocardial infarction. (G) Genealogy of this HHcy family. Squares indicate male family members; circles, female members; and arrow, proband. (H) Time course of Hcy-lowering therapy. The changes of Hcy, (I) folic acid, (J) VB12, (K) ferritin, and (L) EPO during treatment.
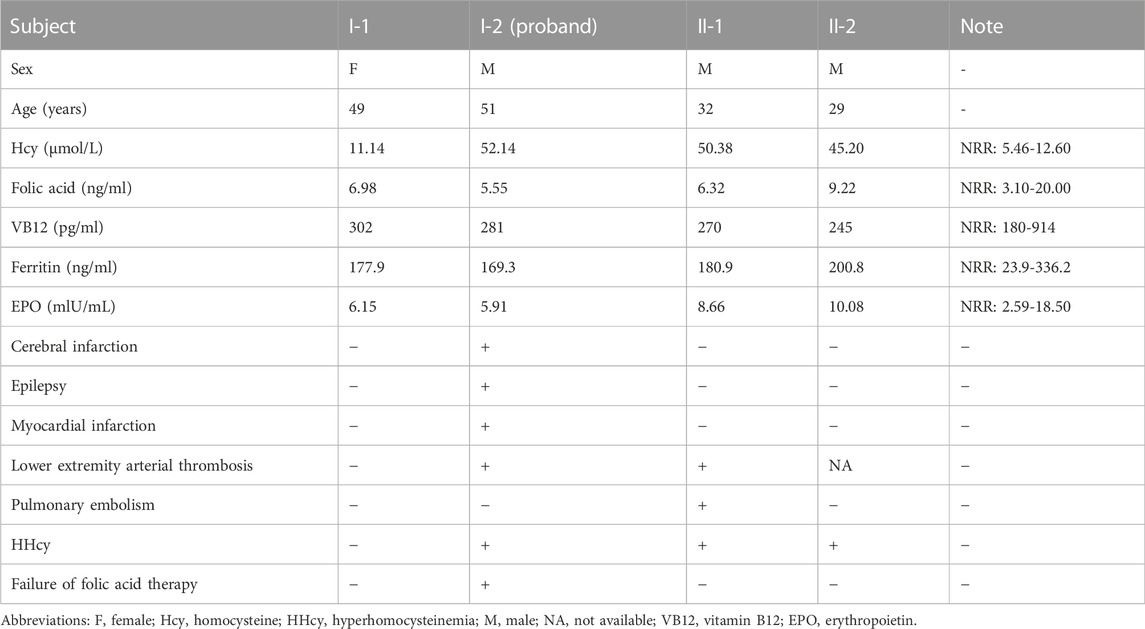
TABLE 1. Clinical data on the members in this family.
WES yielded 10.82 GB of data with 97.14% coverage of the target region, and 95.75% of the target covered over 10×. Totally, about 79,924 variants were detected in the proband. After data filtering, a previously not described heterozygous mutation (c.1888_1891dup; p.C631*fs*1) was identified in exon 12 of the MTHFR gene. In addition, another known heterozygous variant (c.665C>T; p. A222V, known as c. 677C>T; p. A222V) was also identified in exon 5 of the MTHFR gene (Fatima et al., 2022). Sanger sequencing was performed on all the family members. Co-segregation analysis showed that two sons of the proband (II-1 and II-2) harbored the homozygous p. A222V variant of the MTHFR gene. The proband’s wife (I-1) was healthy and harbored the heterozygous p. A222V variant of the MTHFR gene (Figures 1G, 2A). Thus, the patients of HHcy in this family were further diagnosed with congenital MTHFR deficiency. Alignment of MTHFR amino acid sequences revealed that the affected amino acid C631 is conserved in different species (Figure 2B). At the same time, ConSurf Server software (http://consurf.tau.ac.il/) predicted that two affected amino acids were evolutionarily conserved (Figure 2C). In addition, SWISS-MODEL software (https://swissmodel.expasy.org/) showed that the mutation at C631 may lead to a loss of C-terminal in the mutated MTHFR protein (Figure 2D). Furthermore, Metadome software (https://stuart.radboudumc.nl/metadome/dashboard) indicated that two affected residues are located in the intolerant region of MTHFR (Figure 2E).
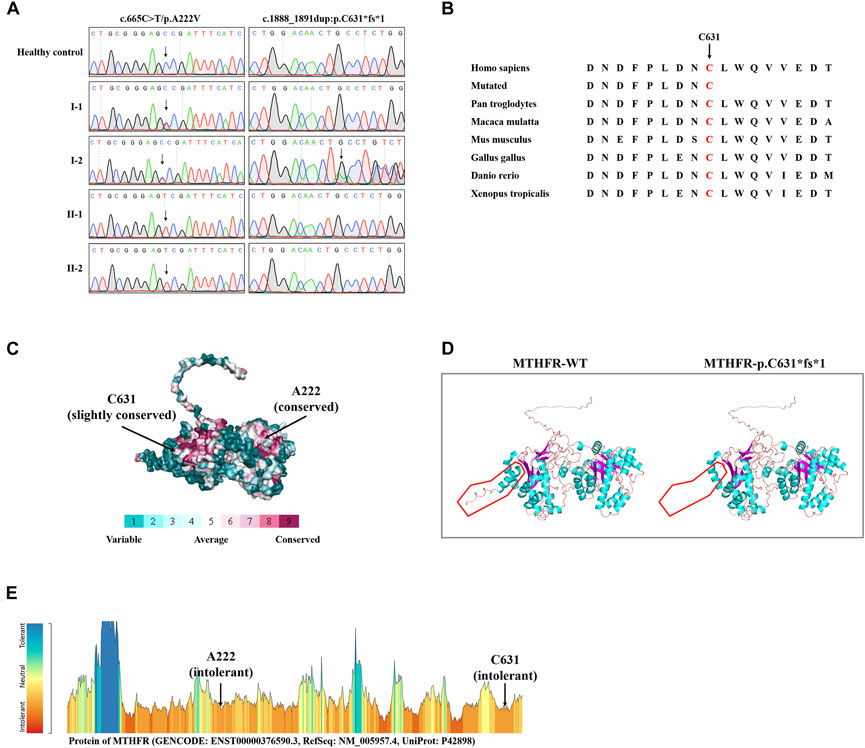
FIGURE 2. Sequencing data and bioinformatics analysis of MTHFR variants. (A) Sanger sequencing results of the MTHFR variants in this family. (B) Alignment of multiple MTHFR protein sequences across species. Letters in red show the C631 sites that are evolutionarily conserved. (C) Conservation analysis of the D612 amino acid was predicted by ConSurf Server software. (D) Structural prediction of the mutant protein. The wild-type MTHFR (MTHFR-WT) protein structure and the p. C631*fs*1 mutant MTHFR (MTHFR-p.C631*fs*1) protein structure were predicted by SWISS-MODEL online software. (E) MetaDome server was used to identify the intolerant regions in MTHFR. As depicted, the affected nucleotides/residues are located in the intolerant region.
The plasma–Hcy levels of two sons were successfully intervened after folic acid therapy, while the proband’s plasma–Hcy level remained elevated despite 1 year of medications. Since genetic polymorphisms of key enzymes in 1-CM may affect the efficacy of folate therapy in patients with HHcy (Du et al., 2018), we further investigated other possibilities underlying his Hcy level besides compound heterozygous MTHFR variants. The results indicated that the patient carried an additional homozygous variant (c.401A>G; p.K134R) of the MTHFD1 gene. Sanger sequencing confirmed that two sons of the proband (II-1 and II-2) harbored this heterozygous variant; this variant was not detected in the proband’s wife (I-1) (Figure 3A).
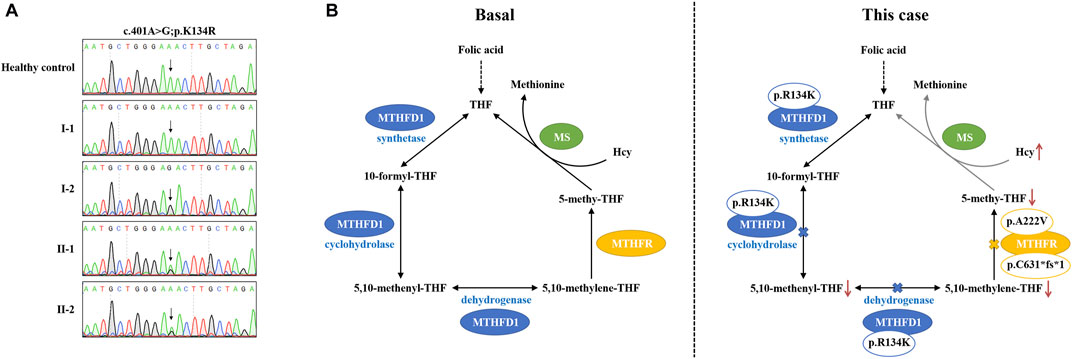
FIGURE 3. Pathogenesis hypothesis of the proband. (A) Sequencing data on MTHFD1 variants in this family. (B) Both MTHFD1 and MTHFR participated in the cytoplasmic folate cycle. Three reactions are catalyzed by MTHFD1: the conversion of formate and THF to 10-formyl-THF (synthetase); the interconversion of 10-formyl-THF and 5,10-methenyl-THF (cyclohydrolase); and the reduction of 5,10-methenyl-THF to 5,10-methylene-THF (dehydrogenase). MTHFR catalyzes the reduction of 5,10-methylene-THF to 5,10-methenyl-THF. 5,10-Methenyl-THF is utilized by methionine synthase to convert homocysteine to methionine. In our case, the p. R134K variant is located within the dehydrogenase/cyclohydrolase domain of the MTHFD1 protein which may lead to reduction of 5,10-methenyl-THF and 5,10-methylene-THF syntheses. On the other hand, the compound heterozygous variants, p. A222V and p. C631*fs*1, may result in dysfunction of the MTHFR enzyme, causing reduction of the 5,10-methenyl-THF synthesis. As a result, the conversion of homocysteine to methionine is inhibited by the low level of 5,10-methenyl-THF, which may contribute to the abnormal accumulation of plasma–Hcy and finally lead to HHcy and failure of folic acid therapy, as observed in our patient.
In this study, we reported a Chinese family with hereditary HHcy. The proband suffered from severe thromboembolic disease and failed to accept folic acid therapy. Two sons of the proband were diagnosed with HHcy but were sensitive to folic acid therapy. Exome sequencing was conducted to evaluate the genetic cause in this family. Simultaneously, compound heterozygous variants and a homozygous variant of the MTHFR gene were identified in the HHcy individuals (I-2, II-1, and II-2) of this family. Furthermore, an additional homozygous variant of the MTHFD1 gene was identified in the proband (I-2) within whom folic acid therapy was invalid. Our study suggests that doubly bi-allelic variants of the MTHFR and MTHFD1 genes are associated with HHcy and caused a failure of folic acid therapy.
MTHFR is considered as the key enzyme in controlling the plasma–Hcy levels (Rummel et al., 2007). It catalyzes an unusual two-step reaction. The first step is reducing the equivalents that are transferred from nicotinamide adenine dinucleotide phosphate (NADPH) to the cofactor flavin adenine dinucleotide (FAD). The second step is reducing equivalents that were further passed on to 5,10-methylenetetrahydrofolate (5,10-methylene-THF), forming 5-methyltetrahydrofolate (5-methyl-THF) (Guenther et al., 1999). 5-Methyl-THF is the predominant circulatory form of folate and is also the major carbon donor in the remethylation of homocysteine to methionine. Thus, the MTHFR enzyme is essential for maintaining the pool of circulating folate and methionine and reducing the levels of homocysteine (Zaric et al., 2019; Raghubeer and Matsha, 2021). The MTHFR protein is composed of an N-terminal catalytic domain and a C-terminal regulatory domain. The binding sites for the substrates NADPH, 5,10-methylene-THF, and the co-factor FAD are located in the N-terminal catalytic domain (Guenther et al., 1999). A previous study has indicated that missense mutations in the N-terminal catalytic domain may lead to a loss of MTHFR activity, whereas in the C-terminal regulatory domain, only truncating mutations have such a severe effect (Burda et al., 2015b). The novel frameshift variant p. C631*fs*1 identified in this study is located in the C-terminal regulatory domain, leading to a truncated MTHFR protein. Although the change of the MTHFR enzyme activity has not been assayed, we presumed this variant is pathogenic, according to previous studies. Homozygosity for the p. A222V variant is the most common genetic cause of HHcy (Guenther et al., 1999), and A222V substitution is a known cause for reduced levels of specific activity and increased thermolability. Furthermore, compound heterozygous p. A222V variant and another MTHFR gene variant have also been reported in intermediate HHcy subjects (Rummel et al., 2007). Consistent with previous reports, we reported an HHcy patient with the compound heterozygous state for the known variant p. A222V and a novel variant of p. C631*fs*1 in the MTHFR gene. A previous study also indicated that the p. A222V variant combined with severe MTHFR mutations in the compound heterozygous state may require further therapy in addition to folate substitution to normalize homocysteine levels (Rummel et al., 2007), which is similar to the features in this proband. Notably, the mutation (p. C631*fs*1) detected in this research has not been published, and therefore, it is considered novel.
MTHFD1 is a tri-functional protein comprising 5,10-methenyltetrahydrofolate (5,10-methenyl-THF), cyclohydrolase, 5,10-methylene-THF dehydrogenase, and 10-formyltetrahydrofolate (10-formyl-THF) synthetase. As a 1-CM-related enzyme, MTHFD1 catalyzes three reactions involved in folate metabolism and plays an important role in Hcy metabolism (Christensen et al., 2009; Khaki et al., 2019; Bidla et al., 2020). MTHFD1 is encoded by the gene MTHFD1. Homozygous mutations at MTHFD1 may lead to disorders of the folate-mediated Hcy pathway and could affect folate treatment response (Bidla et al., 2020; Li et al., 2020). The p. R134K variant (rs1950902) is a common non-synonymous SNP in the MTHFD1 gene. Epidemiological studies have reported a connection between the p. R134K variant, and diseases relate to folate metabolism or Hcy levels (Sutherland et al., 2014). A recent study illustrated that rs1950902 significantly affected changes in the Hcy level and further indicated the relevance of rs1950902 in the folate treatment response (Li et al., 2020), although in the absence of functional consequences (Sutherland et al., 2014). In our study, the HHcy proband failed to reach the normal Hcy range after 1 year of folate supplementation. Aside from compound heterozygous variants in the MTHFR gene, an additional homozygous variant (p.K134R) of the MTHFD1 gene was detected. The p. R134K variant, which is located within the dehydrogenase/cyclohydrolase domain of the MTHFD1 protein, may affect the biosynthesis of 5,10-methenyl-THF and 5,10-methylene-THF (Burda et al., 2015a). In this scenario, we hypothesize the pathogenesis that doubly bi-allelic variants of MTHFR and MTHFD1 lead to a reduction of 5,10-methenyl-THF/5,10-methylene-THF synthesis when combined with dysfunctional MTHFR, which eventually results in HHcy and the failure of folic acid therapy presented in our patient (Figure 3B). Although this suspicion will only be proven by further study, additional functional analysis of the doubly bi-allelic variants is recommended and may result in additional information about the pathogenetic mechanism of HHcy. Based on the genetic findings, the proband was further managed with betaine (6 g/day), thiamine (75 mg/day), mecobalamin (1.5 mg/day), and 5-methyl-THF (600 mg/day) (Guenther et al., 1999; Servy et al., 2018). A follow-up visit has also been scheduled to ensure the patients’ benefit from personalized treatment.
We used WES to evaluate the genetic cause in a Chinese family with hereditary HHcy and revealed that doubly bi-allelic variants of MTHFR and MTHFD1 genes can result in an HHcy phenotype with failure of folic acid therapy. Our study provided data for genetic counseling and personalized treatment to this family, which might contribute to a more comprehensive genetic counseling in HHcy patients, and further demonstrated the important role for 1-CM in HHcy control.
The data presented in the study are deposited in the GSEHuman repository, accession number HRA003374 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA003374).
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Xiangya Hospital of Central South University, China. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YZ and M–FS enrolled the family members. YS performed DNA isolation and Sanger sequencing. Y–XL and M–HD performed genetic analysis. Y–XL wrote the manuscript. YZ and LL supported the project. All authors reviewed the manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
This study was supported by National Natural Science Foundation of China (82000427 and 82000079), National Natural Science Foundation of Hunan province (2020JJ5785 and 2021JJ40849), the Fundamental Research Funds for the Central Universities of Central South University (2021zzts0079), Natural Science Project of Changsha City (kq2014233), Research Project of Hunan Provincial Health Commission (202203023480), the Open Sharing Fund for the Large-scale Instruments and Equipments of Central South University (CSUZC202249) and Development Fund of Affiliated Hospital of Xuzhou Medical University (XYFY2020030).
The authors thank all the subjects for participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bidla, G., Watkins, D., Chery, C., Froese, D. S., Ells, C., Kerachian, M., et al. (2020). Biochemical analysis of patients with mutations in MTHFD1 and a diagnosis of methylenetetrahydrofolate dehydrogenase 1 deficiency. Mol. Genet. Metab. 130, 179–182. doi:10.1016/j.ymgme.2020.04.008
Biesalski, A. S., Hoffjan, S., Schneider, R., Nguyen, H. P., Dekomien, G., Lucke, T., et al. (2022). Phoenix from the ashes: Dramatic improvement in severe late-onset methylenetetrahydrofolate reductase (MTHFR) deficiency with a complete loss of vision. J. Neurol. 269, 2206–2209. doi:10.1007/s00415-021-10841-x
Burda, P., Kuster, A., Hjalmarson, O., Suormala, T., Burer, C., Lutz, S., et al. (2015). Characterization and review of MTHFD1 deficiency: Four new patients, cellular delineation and response to folic and folinic acid treatment. J. Inherit. Metab. Dis. 38, 863–872. doi:10.1007/s10545-015-9810-3
Burda, P., Schafer, A., Suormala, T., Rummel, T., Burer, C., Heuberger, D., et al. (2015). Insights into severe 5, 10-methylenetetrahydrofolate reductase deficiency: Molecular genetic and enzymatic characterization of 76 patients. Hum. Mutat. 36, 611–621. doi:10.1002/humu.22779
Christensen, K. E., Rohlicek, C. V., Andelfinger, G. U., Michaud, J., Bigras, J. L., Richter, A., et al. (2009). The MTHFD1 p.Arg653Gln variant alters enzyme function and increases risk for congenital heart defects. Hum. Mutat. 30, 212–220. doi:10.1002/humu.20830
Clement, P., Alvarez, S., Jacquesson-Fournols, L., Cornet, D., Clement, A., Brack, M., et al. (2022). T677T methylenetetrahydrofolate reductase single nucleotide polymorphisms increased prevalence in a subgroup of infertile patients with endometriosis. J. Womens Health 31, 1501–1506. doi:10.1089/jwh.2022.0019
Du, B., Tian, H., Tian, D., Zhang, C., Wang, W., Wang, L., et al. (2018). Genetic polymorphisms of key enzymes in folate metabolism affect the efficacy of folate therapy in patients with hyperhomocysteinaemia. Br. J. Nutr. 119, 887–895. doi:10.1017/S0007114518000508
Fan, L. L., Liu, J. S., Huang, H., Du, R., and Xiang, R. (2019). Whole exome sequencing identified a novel mutation (p.Ala1884Pro) of beta-spectrin in a Chinese family with hereditary spherocytosis. J. Gene Med. 21, e3073. doi:10.1002/jgm.3073
Fatima, T., Arif, U., Shaharyar, S., Khan, S., Ashraf, M., Rafaqat, W., et al. (2022). MTHFR-c 677C>T polymorphism and male infertility: An analysis in a cohort of Pakistani men. Rev. Int. Androl. 20, 274–280. doi:10.1016/j.androl.2021.05.001
Guenther, B. D., Sheppard, C. A., Tran, P., Rozen, R., Matthews, R. G., and Ludwig, M. L. (1999). The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 6, 359–365. doi:10.1038/7594
Hrncic, D., Rasic-Markovic, A., Macut, D., Mladenovic, D., Susic, V., Djuric, D., et al. (2018). Sulfur - containing amino acids in seizures: Current state of the art. Curr. Med. Chem. 25, 378–390. doi:10.2174/0929867324666170609090613
Khaki, A. A., Tanoomand, A., Hajibemani, A., and Abouhamzeh, B. (2019). Detection of polymorphisms in MTHFD1 G1958A and its possible association with idiopathic male infertility. Urol. J. 16, 586–591. doi:10.22037/uj.v0i0.4647
Li, D., Yang, J., Zhao, Q., Zhang, C., Ren, B., Yue, L., et al. (2019). Genetic and epigenetic regulation of BHMT is associated with folate therapy efficacy in hyperhomocysteinaemia. Asia Pac. J. Clin. Nutr. 28, 879–887. doi:10.6133/apjcn.201912_28(4).0025
Li, D., Zhao, Q., Huang, X., Zhang, C., Godfrey, O., and Zhang, W. (2020). Association of genetic and epigenetic variants in one-carbon metabolism gene with folate treatment response in hyperhomocysteinaemia. Eur. J. Clin. Nutr. 74, 1073–1083. doi:10.1038/s41430-020-0611-x
Lyon, P., Strippoli, V., Fang, B., and Cimmino, L. (2020). B vitamins and One-Carbon metabolism: Implications in human health and disease. Nutrients 12, 2867. doi:10.3390/nu12092867
Partearroyo, T., Murillo-Cuesta, S., Vallecillo, N., Bermudez-Munoz, J. M., Rodriguez-de, L. R. L., Mandruzzato, G., et al. (2019). Betaine-homocysteine S-methyltransferase deficiency causes increased susceptibility to noise-induced hearing loss associated with plasma hyperhomocysteinemia. Faseb J. 33, 5942–5956. doi:10.1096/fj.201801533R
Raghubeer, S., and Matsha, T. E. (2021). Methylenetetrahydrofolate (MTHFR), the One-Carbon cycle, and cardiovascular risks. Nutrients 13, 4562. doi:10.3390/nu13124562
Rummel, T., Suormala, T., Haberle, J., Koch, H. G., Berning, C., Perrett, D., et al. (2007). Intermediate hyperhomocysteinaemia and compound heterozygosity for the common variant c.677C>T and a MTHFR gene mutation. J. Inherit. Metab. Dis. 30, 401. doi:10.1007/s10545-007-0445-x
Servy, E. J., Jacquesson-Fournols, L., Cohen, M., and Menezo, Y. (2018). MTHFR isoform carriers. 5-MTHF (5-methyl tetrahydrofolate) vs folic acid: A key to pregnancy outcome: A case series. J. Assist. Reprod. Genet. 35, 1431–1435. doi:10.1007/s10815-018-1225-2
Steele, J. W., Kim, S. E., and Finnell, R. H. (2020). One-carbon metabolism and folate transporter genes: Do they factor prominently in the genetic etiology of neural tube defects? Biochimie 173, 27–32. doi:10.1016/j.biochi.2020.02.005
Sudheer, P., Agarwal, A., Garg, A., Padma, S. M., and Vishnu, V. Y. (2022). MTHFR deficiency: A potentially treatable cause of adult-onset hereditary spastic paraparesis. Ann. Indian Acad. Neurol. 25, 300–301. doi:10.4103/aian.aian_340_21
Sutherland, H. G., Hermile, H., Sanche, R., Menon, S., Lea, R. A., Haupt, L. M., et al. (2014). Association study of MTHFD1 coding polymorphisms R134K and R653Q with migraine susceptibility. Headache 54, 1506–1514. doi:10.1111/head.12428
Wang, T., Liu, Y. X., Luo, F. M., Dong, Y., Li, Y. L., and Fan, L. L. (2021). A novel homozygous variant of TMEM231 in a case with hypoplasia of the cerebellar vermis and polydactyly. Front. Pediatr. 9, 774575. doi:10.3389/fped.2021.774575
Keywords: MTHFR, MTHFD1, hyperhomocysteinemia, folic acid, whole-exome sequencing
Citation: Liu Y-X, Ding M-H, Sheng Y, Sun M-F, Liu L and Zhang Y (2023) Doubly bi-allelic variants of MTHFR and MTHFD1 in a Chinese patient with hyperhomocysteinemia and failure of folic acid therapy. Front. Genet. 13:964990. doi: 10.3389/fgene.2022.964990
Received: 11 July 2022; Accepted: 30 November 2022;
Published: 04 January 2023.
Edited by:
Julie Pires Da Silva, Université Toulouse III Paul Sabatier, FranceReviewed by:
Rami M. Elshazli, Horus University, EgyptCopyright © 2023 Liu, Ding, Sheng, Sun, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lv Liu, ZG9jbGl1bHZAY3N1LmVkdS5jbg==; Yang Zhang, aHVheWluZ2xpZWh1b0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.