 Carine Ayoub1
Carine Ayoub1 Yara Azar1,2
Yara Azar1,2 Dina Maddah3
Dina Maddah3 Youmna Ghaleb1,2Sandy Elbitar1,2Yara Abou-Khalil1,2Selim Jambart3
Youmna Ghaleb1,2Sandy Elbitar1,2Yara Abou-Khalil1,2Selim Jambart3 Mathilde Varret2Catherine Boileau2,4Petra El Khoury1,2
Mathilde Varret2Catherine Boileau2,4Petra El Khoury1,2 Marianne Abifadel1,2*
Marianne Abifadel1,2*- 1Laboratory of Biochemistry and Molecular Therapeutics (LBTM), Faculty of Pharmacy, Pôle Technologie- Santé, Saint Joseph University of Beirut, Beirut, Lebanon
- 2Laboratory for Vascular Translational Science (LVTS), INSERM, Paris Cité University and Sorbonne Paris Nord University, Paris, France
- 3Faculty of Medicine, Saint Joseph University of Beirut, Beirut, Lebanon
- 4Genetic Department, AP-HP, Hôpital Bichat, Paris, France
Familial chylomicronemia syndrome is a rare autosomal recessive disorder of lipoprotein metabolism characterized by the presence of chylomicrons in fasting plasma and an important increase in plasma triglycerides (TG) levels that can exceed 22.58 mmol/l. The disease is associated with recurrent episodes of abdominal pain and pancreatitis, eruptive cutaneous xanthomatosis, lipemia retinalis, and hepatosplenomegaly. A consanguineous Syrian family who migrated to Lebanon was referred to our laboratory after perceiving familial chylomicronemia syndrome in two children. The LPL and PCSK9 genes were sequenced and plasma PCSK9 levels were measured. Sanger sequencing of the LPL gene revealed the presence of the p.(Val227Phe) pathogenic variant in exon 5 at the homozygous state in the two affected children, and at the heterozygous state in the other recruited family members. Interestingly, PCSK9 levels in homozygous carriers of the p.(Val227Phe) were ≈50% lower than those in heterozygous carriers of the variant (p-value = 0.13) and ranged between the 5th and the 7.5th percentile of PCSK9 levels in a sample of Lebanese children of approximately the same age group. Moreover, this is the first reported case of individuals carrying simultaneously an LPL pathogenic variant and PCSK9 variants, the L10 and L11 leucine insertion, which can lower and raise low-density lipoprotein cholesterol (LDL-C) levels respectively. TG levels fluctuated concomitantly between the two children, were especially high following the migration from a country to another, and were reduced under a low-fat diet. This case is crucial to raise public awareness on the risks of consanguineous marriages to decrease the emergence of inherited autosomal recessive diseases. It also highlights the importance of the early diagnosis and management of these diseases to prevent serious complications, such as recurrent pancreatitis in the case of familial hyperchylomicronemia.
Introduction
Genetic rare diseases usually require special care that could be challenging and hardly achievable, especially in war crises or migration situations. This is the case of type I hyperlipoproteinemia (T1HLP).
T1HLP (OMIM 238600), also known as familial hyperchylomicronemia, familial lipoprotein lipase deficiency (Pingitore et al., 2016), or familial chylomicronemia syndrome (FCS) (Caddeo et al., 2018), is a rare autosomal recessive disorder of lipoprotein metabolism characterized by the presence of chylomicrons in fasting plasma and an important increase in plasma triglycerides (TG) levels that can exceed 2,000 mg/dl (22.58 mmol/l) (Burnett et al., 1993). This condition is associated with recurrent episodes of abdominal pain and pancreatitis, eruptive cutaneous xanthomatosis, lipemia retinalis, and hepatosplenomegaly (Brunzell and Deeb, 2019). Recurrent episodes of pancreatitis in these patients can affect the functions of the pancreas and can, in severe cases, lead to a multi-organ failure and an increase in morbidity and mortality (Regmi and Rehman, 2021). The prevalence of the disease in the general population is estimated to be one to two per million (Abifadel et al., 2004; Rodrigues et al., 2016), however, it is higher in some isolated ethnic groups (i.e., French Canadians, Afrikaner) (Gagné et al., 1989; Henderson et al., 1992; Foubert et al., 1996; Pingitore et al., 2016), and in populations with a high incidence of consanguinity. T1HLP is caused in most cases by loss-of-function variants in the lipoprotein lipase (LPL) gene (Langlois et al., 1989; Caddeo et al., 2018). The latter encodes a secreted protein that plays a crucial role in lipid metabolism and homeostasis through the hydrolysis of TG transported by TG-rich lipoproteins [very low-density lipoproteins (VLDL) and chylomicrons] to decrease plasma TG and generate free fatty acids that are either stored in the adipose tissue or oxidized by the muscles (Wion et al., 1987; Pingitore et al., 2016; Rodrigues et al., 2016). In rarer cases, T1HLP is caused by variants in the apolipoprotein C2 (APOC2) (Catapano and Capurso, 1986; Cox et al., 1988; Zanelli et al., 1994) and apolipoprotein A5 (APOA5) (Pennacchio and Rubin, 2003; Priore Oliva et al., 2005) genes encoding respectively ApoC2 and ApoA5 which are LPL activators, the glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) gene which encodes a protein that plays a role in the transport and binding of LPL to the endothelial cell wall and its entry into the capillaries (Beigneux et al., 2009; Davies et al., 2010; Song et al., 2022), and in the lipase maturation factor 1 (LMF1) gene encoding an endoplasmic reticulum membrane protein, that plays a role in the posttranslational folding, assembly and stabilization of active homodimerized LPL (Péterfy et al., 2007; Péterfy, 2012).
An early diagnosis of the disease is important in order to instore as early as possible a very low-fat diet consisting of reducing the dietary fat to ≤20 g/day or 15% of the total daily energy intake to prevent abdominal pain and recurrent pancreatitis. The goal is to maintain plasma TG levels below 1,000 mg/dl (11.29 mmol/l). It is noted that recurrent abdominal pain is prevented when TG levels are maintained below 2,000 mg/dl (22.58 mmol/l) (Burnett et al., 1993). However, the compliance to the diet is usually poor (Caddeo et al., 2018), and its control is difficult, especially among migrants or low-income populations.
Interestingly, patients with chylomicronemia syndrome generally present low low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) levels besides high TG levels (Fojo and Brewer, 1992; Hegele, 2013; Blom et al., 2018; O’Dea et al., 2019). More recently, PCSK9 has been identified as a major protagonist in lipid metabolism and familial hypercholesterolemia (FH) (Abifadel et al., 2003). Many studies have reported a potential correlation between plasma PCSK9 levels and LDL-C, but also TG, VLDL-C, and intermediate-density lipoprotein cholesterol (IDL-C). Investigations linking these actors of the lipid pathway are being conducted (Lakoski et al., 2009; Druce et al., 2015; Norata et al., 2016; Baragetti et al., 2018; Warden et al., 2020).
In this article, we report the case of a consanguineous family of Syrian refugees in Lebanon with two children suffering from familial hyperchylomicronemia. To our knowledge, this is the first study to measure circulating PCSK9 levels in familial hyperchylomicronemia and to identify individuals carrying simultaneously variants in the LPL and the PCSK9 genes.
Materials and methods
Study participants
A Syrian family who migrated to Lebanon was referred to our laboratory upon perceiving familial hyperchylomicronemia in two children. The parents and four of their children were recruited. We collected the clinical history and anthropometric data for the recruited members. The parents signed the informed consent to participate with their children in our study. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Hôtel Dieu de France Hospital and the Saint Joseph University of Beirut.
Laboratory and biochemical tests
Blood samples were obtained after overnight fasting, plasma, and serum were prepared and stocked at −80°C. Lipid measurements were determined on a COBAS INTEGRA® analyzer (Roche Diagnostics, Basel, Switzerland). Non-HDL-C was calculated by subtracting HDL-C from total cholesterol (TC).
DNA analysis and variant detection
Genomic DNAs of all the participants were extracted from peripheral blood leukocytes using the illustra™ blood genomicPrep Mini Spin Kit according to the manufacturer’s instructions. The exons and the flanking exon-intron boundaries of the LPL and PCSK9 genes were amplified by polymerase chain reaction (PCR) and sequenced using the Sanger method. PCR conditions and primers’ sequences are available upon request. For DNA sequence assembly and variant detection, the CodonCode Aligner® Software was used.
In silico analysis of the variant
The Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) was used for frequency determination of the variant. The Polymorphism Phenotyping version 2 (PolyPhen-2; http://genetics. bwh.harvard.edu/pph2/), Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org/index.php), Mutation Taster (http://www.mutationtaster.org/), and the Combined Annotation Dependent Depletion score (CADD score; https://cadd.gs.washington.edu/snv) were used to predict the pathogenicity of the variant.
PCSK9 measurements
We measured PCSK9 levels in the plasma of the recruited members using a commercial ELISA kit (Human Proprotein Convertase 9/PCSK9 Duoset catalog no. DY3888; R&D Systems, Minneapolis, MN, United States) and the Bio-Plex Pro assay technology (Luminex Corporation, Austin, TX, United States) as previously described (El Khoury et al., 2018).
Statistical analysis
The variables were analyzed using the GraphPad Prism version 9. Results for quantitative variables were expressed as median with its interquartile ranges (first quartile and third quartile). Spearman correlation was performed to measure the strength and direction of the linear relationship between PCSK9 and other quantitative variables. The Mann–Whitney U test was used to compare PCSK9 values between homozygous and heterozygous carriers of the p.(Val227Phe) variant in the LPL gene.
Results
Clinical characteristics and biochemical analysis
In the recruited family, the parents were first-degree cousins and two of their six children suffered from familial hyperchylomicronemia confirmed by lipoprotein electrophoresis and later by genetic sequencing (Figure 1).
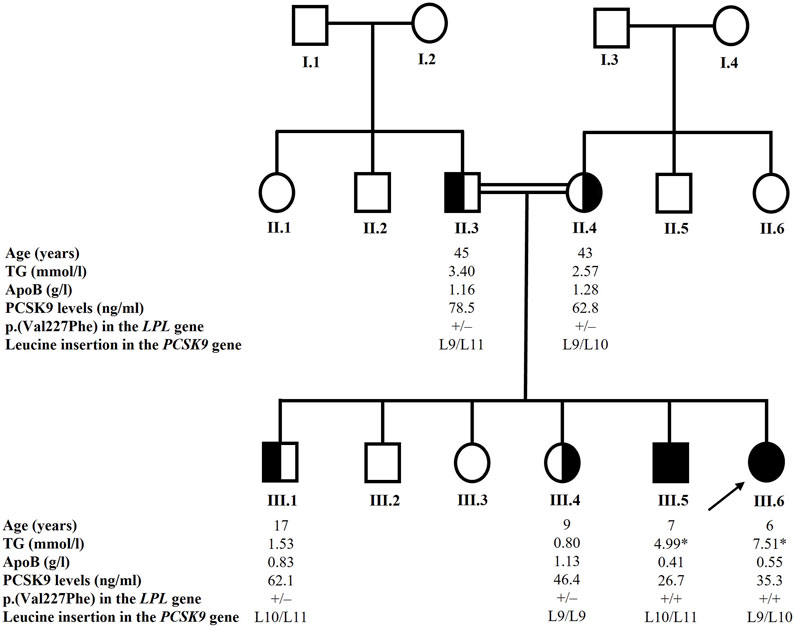
FIGURE 1. Pedigree of the family. The arrow indicates the proband. Blackened symbols indicate affected homozygous carriers of the p.(Val227Phe) variant in the LPL gene. Half-blackened symbols indicate heterozygous carriers of the p.(Val227Phe) variant. Only individuals with available and reported data were included in the study. The * sign indicates TG value while the patient was under a very strict low-fat diet. The +/+ sign indicates that the individual is homozygous for the p.(Val227Phe) variant in the LPL gene and the +/– sign indicates that the individual is heterozygous for the variant. The L9 designates the normal allele in exon 1 of the PCSK9 gene, the L10 designates the p.Leu21dup or p.L15_L16insL and the L11 variant designates the p.Leu21tri or p.L15_L16ins2L. Age at recruitment is given in years. TG levels are given in mmol/l, ApoB levels are given in g/l and PCSK9 levels are given in ng/ml. ApoB: apolipoprotein B; LPL: lipoprotein lipase; PCSK9: proprotein convertase subtilisin/kexin type 9; TG: triglycerides.
The proband (III.6) was a girl aged 6 years at the time of recruitment. She presented high TG levels (7.51 mmol/l) and low LDL-C and HDL-C levels (0.38 and 0.33 mmol/l respectively). At the age of 9 years, she was underweight with a BMI of 16.5 kg/m2. She suffered from recurrent abdominal pain, vomiting, and diarrhea almost every week, and the echography showed that she presented splenomegaly. Infectious gastrointestinal causes were ruled out. She suffered as well from chronic anemia. Thalassemia was excluded by the normal results of hemoglobin electrophoresis. Mediterranean fever was suspected and the MEFV gene (NM_000243), responsible for the disease was studied by Sanger sequencing. Analysis of the exons and the flanking intronic regions did not reveal the presence of any pathogenic variant. However, this study does not preclude large deletions. Recently, she had an outbreak of the disease with TG levels reaching 54.33 mmol/l and they were rapidly reduced to 4.29 mmol/l following a very strict low-fat diet.
Her brother (III.5), aged 7 years at the time of recruitment, had TG levels fluctuating between 33.22 mmol/l for the highest value and 3.175 mmol/l for the lowest. At recruitment, he presented high TG levels (4.99 mmol/l) and low LDL-C and HDL-C levels (0.51 and 0.36 mmol/l respectively). At the age of 10 years, he was also underweight with a BMI of 14.6 kg/m2. Clinically, he presented cutaneous xanthomatosis on his elbows and asthenia. He was diagnosed with splenomegaly consecutive to echography. He presented hemoglobin levels that fluctuated between normal and low. The normal results of hemoglobin electrophoresis allowed to rule out thalassemia. Recently, he presented moderately high levels of TG.
Curiously, TG levels fluctuated concomitantly between the two children and were especially high following the migration from a country to another. For example, the TG levels were the highest upon the arrival of the family to Lebanon, and then they were gradually reduced.
Both children were under fenofibrate but in a discontinuous way, omega 3, vitamin D, folic acid, iron, and a low-fat diet.
Two of their siblings (III.1 and III.4) presented normal TG and TC levels (Table 1). Their parents (II.3 and II.4) presented moderate hypertriglyceridemia according to the 2018 ACC/AHA classification (Table 1) (Grundy et al., 2019). The father aged 45 years was obese.
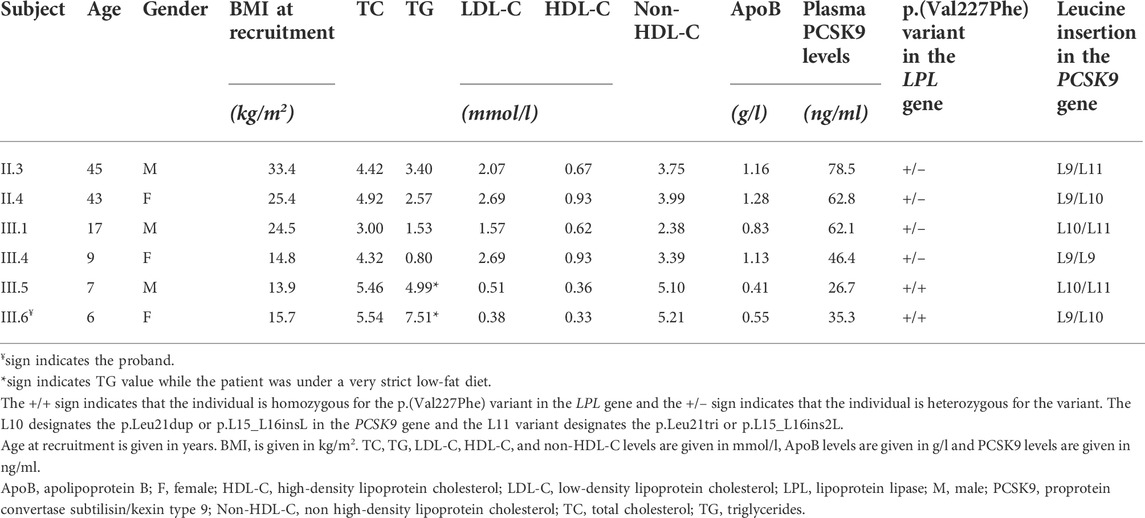
TABLE 1. Lipid measurements and characteristics of the recruited members.
PCSK9 measurements
Patients with T1HLP generally present low LDL-C levels and HDL-C levels besides high TG levels (Fojo and Brewer, 1992; Hegele, 2013; Blom et al., 2018; O’Dea et al., 2019). Moreover, many studies have reported a potential correlation between plasma PCSK9 levels and LDL-C, but also triglycerides-rich lipoproteins and investigations linking these actors are being conducted (Lakoski et al., 2009; Druce et al., 2015; Norata et al., 2016; Baragetti et al., 2018; Warden et al., 2020). For these reasons, we measured plasma PCSK9 levels in the recruited members of the family. The results are presented in Table 1 and Figure 2. Homozygous carriers of the p.(Val227Phe) variant (III.5 and III.6) presented a ≈50% decrease in PCSK9 levels compared to heterozygous carriers (II.3, II.4, III.1, and III.4) [median with its interquartile ranges (first quartile–third quartile) of 31.00 (26.70–35.30) ng/ml versus 62.45 (50.33–74.58) ng/ml, respectively] without being significant (p-value = 0.13). Moreover, using Spearman correlation PCSK9 levels were positively correlated with age, ApoB levels and with BMI in all family members (r = 0.94, p-value = 0.017; r = 0.89, p-value = 0.033 and r = 0.94, p-value = 0.017, respectively).
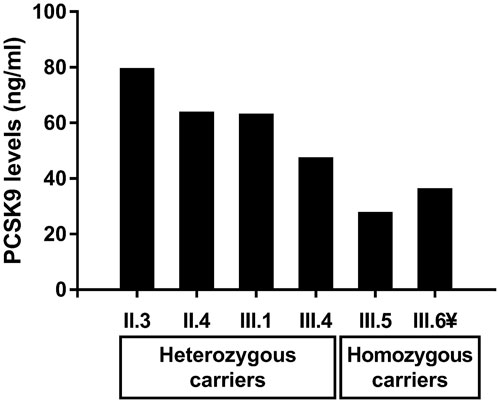
FIGURE 2. Levels of circulating PCSK9 in heterozygous and homozygous carriers of the p.(Val227Phe) variant in the LPL gene in the recruited family. The ¥ sign indicates the proband.
Genetic analysis
We sequenced all the exons and the flanking exon-intron boundaries of the LPL gene knowing that most cases of T1HLP are caused by loss-of-function variations in it. Sequencing of this gene revealed the presence of the c.679G > T variation in exon 5 (NM_000237.3) at the homozygous state in both affected children (III.5 and III.6) (Figure 1). This nucleotide change causes a valine to phenylalanine substitution at position 227 of the amino acid chain [p.(Val227Phe)]. The parents (II.3 and II.4) and the two recruited siblings (III.1 and III.4) were heterozygous for the p.(Val227Phe) variation (Figure 1).
In silico analysis revealed that the amino acid valine at position 227 is well conserved among species. The variation is not present in the gnomAD database and was predicted to be disease-causing on Mutation Taster (Grantham Matrix score of 50), deleterious on PROVEAN (with a score of -4.549), with probably damaging consequences on the functionality of the protein according to Polyphen-2 (score of 0.960, sensitivity: 0.63; specificity: 0.92), and presented a CADD score of 25.8 suggesting that this variant is predicted to be among the top 1% of the most deleterious variants in the human genome.
Interestingly, another LPL variant in the same codon (p.Val227Ala) was also reported to be responsible for T1HLP (Maruyama et al., 2004; Caddeo et al., 2018).
We also sequenced the PCSK9 gene to investigate the presence of an eventual variant that might explain the low levels of PCSK9 observed in some individuals of the family. The sequencing revealed the presence of two types of variations in exon 1 of the PCSK9 gene: the leucine insertion L10 also designated p.Leu21dup or p.L15_L16insL and the leucine insertion L11 also designated p.Leu21tri or p.L15_L16ins2L (Abifadel et al., 2008). The father (II.3) was heterozygous for the L11 variation while the mother (II.4) and one of the affected children (III.6) were heterozygous for the L10 variation. One child (III.4) carried the normal L9 alleles, and two children (III.1 and III.5) were compound heterozygotes for the L10 and L11 alleles. The results are presented in Figure 1.
Discussion
The p.(Val227Phe) pathogenic variant has been recently reported by Caddeo et al. (2018) in a University Hospital in Gothenburg (Sweden) at the homozygous state in a proband aged 29 years who presented a mean TG level of 18.7 ± 3.2 mmol/l and recurrent pancreatitis episodes. His brother also carried the pathogenic variant. They have likely migrated from the Middle East to Sweden where the center received migrants from Syria and Iran (Caddeo et al., 2018). Functional studies demonstrated the pathogenicity of the p.(Val227Phe) loss-of-function variant. These studies suggested that this variant affects protein production and secretion, but not its degradation. Results showed that the transfection of the HEK 293T/17 cells with the p.(Val227Phe) variant caused a reduction in protein synthesis by 35%–40% and a decrease by at least 80% in the secretion of the LPL in the media of these cells compared to the wild type LPL. Moreover, LPL enzymatic assay showed that LPL activity was absent in the media of the cells transfected with the variant. It is noteworthy that there was no difference in the intracellular degradation rate between the wild type and the mutant LPL transfected cells (Caddeo et al., 2018).
We identified the p.(Val227Phe) variant in the LPL gene causing T1HLP in a consanguineous Syrian family who migrated to Lebanon. Indeed, this is the first study to measure PCSK9 levels in T1HLP. In this family, homozygous carriers of the p.(Val227Phe) variant presented lower levels of PCSK9 than heterozygous carriers [median (first quartile–third quartile) of 31.00 (26.70–35.30) ng/ml versus 62.45 (50.33–74.58) ng/ml, respectively]. PCSK9 levels were positively correlated with age, ApoB levels and with BMI in all family members (p-values = 0.017, 0.033, and 0.017 respectively).Compared to a cohort of 279 Lebanese children of approximately the same age group (8–11 years) in whom PCSK9 levels were measured using the same method [median (first-third quartile) of 64.30 (46.95–93.38) ng/ml] (Azar et al., 2022), the two homozygous children (III.5 and III.6) presented PCSK9 levels that were approximately between the fifth (30.40 ng/ml) and the 7.5th percentile (33.25 ng/ml) of PCSK9 levels in the group, while their heterozygous sibling (III.4) presented PCSK9 levels (46.40 ng/ml) that were approximately within the 25th percentile (46.95 ng/ml). The sibling (III.1) presented PCSK9 levels of 62.1 ng/ml, which is normal compared to a group of 159 Lebanese adolescents aged 15–18 years [median (first-third quartile) of 60.55 (44.51–93.96) ng/ml] (Azar et al., 2022). The presence of loss-of-function variants in the PCSK9 gene has been studied by sequencing the whole gene. Interestingly, this is the first report of a family with an LPL pathogenic variant also carrying variants in PCSK9, more precisely two types of leucine insertion variants in the exon 1 of the PCSK9 gene (Table 1). These polymorphisms occur in the signal peptide region of the PCSK9 protein and are characterized by the insertion of one or two leucines into a stretch of nine leucines (Chen et al., 2005; Yue et al., 2006; Abifadel et al., 2008). They might lead to a structural change in the signal peptide causing an impairment in its cleavage and processing in the endoplasmic reticulum (Yue et al., 2006; Pisciotta et al., 2012; Benito-Vicente et al., 2022). In a study conducted on 1,745 apparently healthy individuals, plasma PCSK9 levels were significantly lower in individuals carrying a leucine insertion in exon 1 of the PCSK9 gene (Awan et al., 2013). Moreover, the insertion of two leucines in the signal peptide has been reported in a family with familial combined hyperlipidemia and two patients with FH (Abifadel et al., 2008) and in vitro studies have shown that it causes a reduction in the secretion of the mature form of PCSK9 compared to the wild type PCSK9 (Benito-Vicente et al., 2022). However, these variations might not alone explain the very low levels of PCSK9 observed in homozygous carriers of the LPL variant in our study.
Moreover, homozygous carriers of the p.(Val227Phe) variant in this family presented low levels of LDL-C and HDL-C. This was also observed in the study conducted by Caddeo et al. (2018), as well as in patients suffering from T1HLP described in the literature (O’Dea et al., 2019; Susheela et al., 2021). The low levels of LDL-C in our studied family cannot be explained by the presence of the leucine insertions in the signal peptide of PCSK9. On one hand, the leucine insertion L10 also designated p.Leu21dup or p.L15_L16insL is a common variation associated with lower levels of LDL-C in populations with normal to low LDL-C levels, but also in patients suffering from FH carrying the same p.(Cys681X) mutation in the LDL receptor (LDLr) gene (Abifadel et al., 2009). On the other hand, the leucine insertion L11 also designated p.Leu21tri or p.L15_L16ins2L is a rare variant that has been associated with familial combined hyperlipidemia and was found at a low frequency in subjects presenting LDL-C levels of 2.96–4.90 mmol/L and coronary lesions in the American population (Chen et al., 2005; Abifadel et al., 2008; Benito-Vicente et al., 2022). In fact, low levels of LDL-C and HDL-C in T1HLP may be explained by the disruption in the activity of the LPL (Hegele et al., 2018). Variations in LPL activity in humans result in changes in lipoproteins metabolism (Goldberg et al., 1990). Indeed, low or absent activity of LPL results in an impairment in the conversion of TG-rich particles to their remnant lipoproteins, including chylomicron remnants, VLDL, VLDL remnants, intermediate density lipoprotein and LDL. The subsequent decrease in the available cholesterol from VLDL, LDL, and peripheral tissues causes a decrease in HDL-C levels (Hegele et al., 2018). Other studies attribute the low levels of LDL-C and HDL-C to an increase in their catabolism, besides the decrease in their synthesis (Fojo and Brewer, 1992). Interestingly, the low levels of LDL-C might explain the observed low levels of plasma PCSK9. In fact, many studies have shown a positive correlation between circulating plasma PCSK9 and LDL-C (Shapiro et al., 2019). It is suggested that this correlation is due to the fact that PCSK9 acts as an important regulator of LDL metabolism through targeting the LDLR for lysosomal degradation, but also due to a direct interaction between PCSK9 and LDL (Abifadel et al., 2010; Tavori et al., 2015). Further studies of PCSK9 levels in patients with hyperchylomicronemia would be interesting in order to verify if all homozygous patients with T1HLP have low PCSK9 levels, or if it is specific to the patients in the studied family, and to decipher the causes of this decrease, as well as the mechanism, the role of PCSK9, and its correlation with ApoB levels (Ayoub et al., 2021).
It is noteworthy that anemia has been described as a clinical sign of T1HLP that occurs in some cases (Rahalkar and Hegele, 2008; Brahm and Hegele, 2013). In a case series of infants with T1HLP, 7 out of 16 infants presented normocytic anemia which cause was unknown, and both males and females were affected (Feoli-Fonseca et al., 1998). In another study conducted to determine the phenotype-genotype relationships between different subgroups of T1HLP, all the female participants (n = 7) suffered from anemia (Chokshi et al., 2014).
Although the parents (II.3 and II.4) were heterozygous carriers of the p.(Val227Phe) variant, they presented moderately high levels of TG. The phenotypic expression of heterozygous LPL deficiency is not clinically and biochemically well described yet. It has been reported that heterozygous carriers of one defective allele in the LPL gene do not present chylomicronemia nor other manifestations of the disease. They may present normal or moderately increased fasting TG levels, especially when fed a high-calorie, high-fat diet (Miesenböck et al., 1993; Hölzl et al., 2000; Abifadel et al., 2004). The presence of precipitating factors such as age, obesity, pregnancy, hyperinsulinemia, and lipid-raising drugs would contribute to the phenotypic expression of heterozygous LPL deficiency (Wilson et al., 1990; Abifadel et al., 2004).
The identification of heterozygous carriers of an LPL mutation is of major importance, especially in countries with a high frequency of consanguineous marriages (Abifadel et al., 2004). It is also necessary for the prevention of precipitating factors knowing that the lipoprotein phenotype in heterozygous carriers of a defective LPL allele has been considered atherogenic (Reymer et al., 1995; Bijvoet et al., 1996; Wittrup et al., 1997; Hölzl et al., 2000).
The high prevalence of consanguineous marriages in Middle Eastern countries increases the risk of autosomal recessive genetic diseases (Al-Herz and Al-Mousa, 2013) such as hyperchylomicronemia.
A study conducted to determine the prevalence of consanguineous marriages in Syria showed that the overall frequency was 30.3% in urban areas and 39.8% in rural ones, with an overall rate of 35.4%. In some provinces, the frequency could reach 67.5%. Among this type of marriage, first cousins’ marriages were the most common with a rate of 20.9% (Othman and Saadat, 2009). It is noteworthy that the natality rate increases in refugees’ camps or migrant populations, as well as the risk of consanguineous marriages.
To date, more than 200 pathogenic variants in the LPL gene have been reported to cause T1HLP (Caddeo et al., 2018). However, rare cases have been described in the Middle East and the Mediterranean regions. A summary of the described mutations in this region is presented in Table 2.

TABLE 2. Summary of the described variations in the LPL gene in the Middle East and the Mediterranean regions.
Conclusion
This is the first study to measure plasma PCSK9 levels in T1HLP. We found that homozygous affected patients presented low levels of PCSK9 compared to heterozygous non-affected members of the family and to children from the same age group. Further studies of PCSK9 levels in patients with hyperchylomicronemia would be interesting in order to verify if all homozygous patients with T1HLP have low PCSK9 levels. This would help decipher the causes of this decrease, as well as the mechanism and the role of PCSK9 in this disease. It might also be interesting to elucidate pathways linking PCSK9 to apolipoprotein B or eventually to triglycerides-rich lipoproteins metabolism.
The identification of the same mutation in the LPL gene in two distinct families with T1HLP originated most probably from the same region in Syria and that migrated to either Lebanon or Sweden, should lead us to search for this mutation as the first cause of hyperchylomicronemia in patients originated from Syria. A founder effect could be hypothesized but needs further investigation and cases to be confirmed.
Public awareness and education concerning the medical risks of consanguineous marriages are important and should be included in the international effort and politics that provide care, birth control, and genetic counseling when needed. They might help in decreasing the emergence of inherited autosomal recessive diseases.
Finally, handling a metabolic disease that needs specific care and compliance to a drastic regimen or treatment to prevent some fatal complications is not easy, especially in a crisis. The main solution is to prevent the emergence of these diseases by raising awareness of the risks of consanguineous marriages.
It is more and more urgent to address genetic lipid disorders and more generally genetic diseases that need special treatment, diagnosis, management, and prevention especially in refugee populations or in war or economic crisis countries.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committee of the Hôtel Dieu de France Hospital and the Saint Joseph University of Beirut. Written informed consent to participate in this study was provided by the participants or by the participants’ legal guardian/next of kin for children.
Author contributions
Conceptualization: MA, PE, and SJ; methodology: CA, YA, YA-K, YG, SE, PE, and MA; investigation: DM; writing—original draft preparation: CA, YA, YA-K, YG, SE, PE, and MA; writing—review and editing: CA, PE, SE, YG, YA, YA-K, MV, CB, and MA; supervision: MA and PE; funding acquisition: MA, MV, and CB. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Conseil de la Recherche de l’Université Saint-Joseph, Beirut, Lebanon; Lebanese National Council for Scientific Research (CNRS-L) and Program franco-libanais CEDRE. This work was also supported by Institut National de la Santé et de la Recherche Médicale (INSERM). Carine Ayoub and Yara Azar are supported by grants from Lebanese National Council for Scientific Research (CNRS-L) and Council of Research of Saint-Joseph University of Beirut, Lebanon; Carine Ayoub is granted by “Bourses de Séjour Scientifiques de Haut Niveau (SSHN-2022)” from l’Ambassade de France/Institut français du Liban. Youmna Ghaleb was supported by a grant by the International Atherosclerosis Society (IAS).
Acknowledgments
We thank all the family members for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abifadel, M., Bernier, L., Dubuc, G., Nuel, G., Rabès, J.-P., Bonneau, J., et al. (2008). A PCSK9 variant and familial combined hyperlipidaemia. J. Med. Genet. 45, 780–786. doi:10.1136/jmg.2008.059980
Abifadel, M., Jambart, S., Allard, D., Rabès, J.-P., Varret, M., Derré, A., et al. (2004). Identification of the first Lebanese mutation in the LPL gene and description of a rapid detection method. Clin. Genet. 65, 158–161. doi:10.1111/j.0009-9163.2004.00205.x
Abifadel, M., Pakradouni, J., Collin, M., Samson-Bouma, M.-E., Varret, M., Rabès, J.-P., et al. (2010). Strategies for proprotein convertase subtilisin kexin 9 modulation: a perspective on recent patents. Expert Opin. Ther. Pat. 20, 1547–1571. doi:10.1517/13543776.2010.518615
Abifadel, M., Rabès, J.-P., Jambart, S., Halaby, G., Gannagé-Yared, M.-H., Sarkis, A., et al. (2009). The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum. Mutat. 30, E682–E691. doi:10.1002/humu.21002
Abifadel, M., Varret, M., Rabès, J.-P., Allard, D., Ouguerram, K., Devillers, M., et al. (2003). Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156. doi:10.1038/ng1161
Al-Herz, W., and Al-Mousa, H. (2013). Combined immunodeficiency: the Middle East experience. J. Allergy Clin. Immunol. 131, 658–660. doi:10.1016/j.jaci.2012.11.033
Awan, Z., Delvin, E. E., Levy, E., Genest, J., Davignon, J., Seidah, N. G., et al. (2013). Regional distribution and metabolic effect of PCSK9 insLEU and R46L gene mutations and apoE genotype. Can. J. Cardiol. 29, 927–933. doi:10.1016/j.cjca.2013.03.004
Ayoub, C., Azar, Y., Abou-Khalil, Y., Ghaleb, Y., Elbitar, S., Halaby, G., et al. (2021). Identification of a variant in APOB gene as a major cause of hypobetalipoproteinemia in Lebanese families. Metabolites 11, 564. doi:10.3390/metabo11090564
Azar, Y., Gannagé-Yared, M.-H., Naous, E., Ayoub, C., Abou Khalil, Y., Chahine, E., et al. (2022). Circulating PCSK9 linked to dyslipidemia in Lebanese schoolchildren. Metabolites 12, 504. doi:10.3390/metabo12060504
Baragetti, A., Grejtakova, D., Casula, M., Olmastroni, E., Jotti, G. S., Norata, G. D., et al. (2018). Proprotein Convertase Subtilisin-Kexin type-9 (PCSK9) and triglyceride-rich lipoprotein metabolism: facts and gaps. Pharmacol. Res. 130, 1–11. doi:10.1016/j.phrs.2018.01.025
Beigneux, A. P., Franssen, R., Bensadoun, A., Gin, P., Melford, K., Peter, J., et al. (2009). Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 29, 956–962. doi:10.1161/ATVBAHA.109.186577
Benito-Vicente, A., Uribe, K. B., Larrea-Sebal, A., Palacios, L., Cenarro, A., Calle, X., et al. (2022). Leu22_Leu23 duplication at the signal peptide of PCSK9 promotes intracellular degradation of LDLr and autosomal dominant hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 42, e203–e216. doi:10.1161/ATVBAHA.122.315499
Bijvoet, S., Gagné, S. E., Moorjani, S., Gagné, C., Henderson, H. E., Fruchart, J. C., et al. (1996). Alterations in plasma lipoproteins and apolipoproteins before the age of 40 in heterozygotes for lipoprotein lipase deficiency. J. Lipid Res. 37, 640–650. doi:10.1016/S0022-2275(20)37605-7
Blom, D. J., O’Dea, L., Digenio, A., Alexander, V. J., Karwatowska-Prokopczuk, E., Williams, K. R., et al. (2018). Characterizing familial chylomicronemia syndrome: baseline data of the APPROACH study. J. Clin. Lipidol. 12, 1234–1243. e5. doi:10.1016/j.jacl.2018.05.013
Bouabdellah, M., Iraqi, H., Benlian, P., Berqia, I., Benchekroun, L., Chraïbi, A., et al. (2015). Familial hypertriglyceridemia: biochemical, clinical and molecular study in a Moroccan family. Ann. Biol. Clin. 73, 474–484. doi:10.1684/abc.2015.1058
Brahm, A., and Hegele, R. A. (2013). Hypertriglyceridemia. Nutrients 5, 981–1001. doi:10.3390/nu5030981
Brunzell, J. D., and Deeb, S. S. (2019). “Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency,” in The online metabolic and molecular bases of inherited disease. Editors D. L. Valle, S. Antonarakis, A. Ballabio, A. L. Beaudet, and G. A. Mitchell (New York, NY: McGraw-Hill Education). Available at: https://www.ommbid.mhmedical.com/content.aspx?aid=1181449576 (Accessed June 3, 2022).
Burnett, J. R., Hooper, A. J., and Hegele, R. A. (1993). “Familial lipoprotein lipase deficiency,” in GeneReviews®. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, and K. Stephens. Editors (Seattle (WA): University of Washington, Seattle). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1308/(Accessed September 18, 2019).
Caddeo, A., Mancina, R. M., Pirazzi, C., Russo, C., Sasidharan, K., Sandstedt, J., et al. (2018). Molecular analysis of three known and one novel LPL variants in patients with type I hyperlipoproteinemia. Nutr. Metab. Cardiovasc. Dis. 28, 158–164. doi:10.1016/j.numecd.2017.11.003
Catapano, A. L., and Capurso, A. (1986). “Apolipoprotein C-II deficiency,” in Human apolipoprotein mutants: impact on atherosclerosis and longevity NATO ASI series. Editors C. R. Sirtori, A. V. Nichols, and G. Franceschini (Boston, MA: Springer US), 187–195. doi:10.1007/978-1-4615-9474-1_22
Chen, S. N., Ballantyne, C. M., Gotto, A. M., Tan, Y., Willerson, J. T., and Marian, A. J. (2005). A common PCSK9 haplotype, encompassing the E670G coding single nucleotide polymorphism, is a novel genetic marker for plasma low-density lipoprotein cholesterol levels and severity of coronary atherosclerosis. J. Am. Coll. Cardiol. 45, 1611–1619. doi:10.1016/j.jacc.2005.01.051
Chokshi, N., Blumenschein, S. D., Ahmad, Z., and Garg, A. (2014). Genotype-phenotype relationships in patients with type I hyperlipoproteinemia. J. Clin. Lipidol. 8, 287–295. doi:10.1016/j.jacl.2014.02.006
Cox, D. W., Wills, D. E., Quan, F., and Ray, P. N. (1988). A deletion of one nucleotide results in functional deficiency of apolipoprotein CII (apo CII Toronto). J. Med. Genet. 25, 649–652. doi:10.1136/jmg.25.10.649
Davies, B. S. J., Beigneux, A. P., Barnes, R. H., Tu, Y., Gin, P., Weinstein, M. M., et al. (2010). GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 12, 42–52. doi:10.1016/j.cmet.2010.04.016
Deeb, S. S., and Peng, R. L. (1989). Structure of the human lipoprotein lipase gene. Biochemistry 28, 4131–4135. doi:10.1021/bi00436a001
Druce, I., Abujrad, H., and Ooi, T. C. (2015). PCSK9 and triglyceride-rich lipoprotein metabolism. J. Biomed. Res. 29, 429–436. doi:10.7555/JBR.29.20150052
El Khoury, P., Roussel, R., Fumeron, F., Abou-Khalil, Y., Velho, G., Mohammedi, K., et al. (2018). Plasma proprotein-convertase-subtilisin/kexin type 9 (PCSK9) and cardiovascular events in type 2 diabetes. Diabetes Obes. Metab. 20, 943–953. doi:10.1111/dom.13181
Feoli-Fonseca, J. C., Lévy, E., Godard, M., and Lambert, M. (1998). Familial lipoprotein lipase deficiency in infancy: clinical, biochemical, and molecular study. J. Pediatr. 133, 417–423. doi:10.1016/S0022-3476(98)70280-X
Fojo, S. S., and Brewer, H. B. (1992). Hypertriglyceridaemia due to genetic defects in lipoprotein lipase and apolipoprotein C-II. J. Intern. Med. 231, 669–677. doi:10.1111/j.1365-2796.1992.tb01256.x
Foubert, L., Benlian, P., and Turpin, G. (1996). Lipoprotein lipase: a multifunctional enzyme in lipoprotein metabolism. Presse Med. 25, 207–210.
Gagné, C., Brun, L. D., Julien, P., Moorjani, S., and Lupien, P. J. (1989). Primary lipoprotein-lipase-activity deficiency: clinical investigation of a French Canadian population. CMAJ 140, 405–411.
Goldberg, I. J., Blaner, W. S., Vanni, T. M., Moukides, M., and Ramakrishnan, R. (1990). Role of lipoprotein lipase in the regulation of high density lipoprotein apolipoprotein metabolism. Studies in normal and lipoprotein lipase-inhibited monkeys. J. Clin. Invest. 86, 463–473. doi:10.1172/JCI114732
Grundy, S. M., Stone, N. J., Bailey, A. L., Beam, C., Birtcher, K. K., Blumenthal, R. S., et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation 139, e1082–e1143. doi:10.1161/CIR.0000000000000625
Hegele, R. A., Berberich, A. J., Ban, M. R., Wang, J., Digenio, A., Alexander, V. J., et al. (2018). Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J. Clin. Lipidol. 12, 920–927. e4. doi:10.1016/j.jacl.2018.03.093
Hegele, R. A. (2013). “Chapter 96 - lipoprotein and lipid metabolism,” in Emery and rimoin’s principles and practice of medical genetics. Editors D. Rimoin, R. Pyeritz, and B. Korf. Sixth Edition (Oxford: Academic Press), 1–33. doi:10.1016/B978-0-12-383834-6.00100-2
Henderson, H. E., Hassan, F., Berger, G. M., and Hayden, M. R. (1992). The lipoprotein lipase gly188----glu mutation in South Africans of Indian descent: evidence suggesting common origins and an increased frequency. J. Med. Genet. 29, 119–122. doi:10.1136/jmg.29.2.119
Hölzl, B., Kraft, H. G., Wiebusch, H., Sandhofer, A., Patsch, J., Sandhofer, F., et al. (2000). Two novel mutations in the lipoprotein lipase gene in a family with marked hypertriglyceridemia in heterozygous carriers. Potential interaction with the polymorphic marker D1S104 on chromosome 1q21-q23. J. Lipid Res. 41, 734–741. doi:10.1016/s0022-2275(20)32382-8
Kavazarakis, E., Stabouli, S., Gourgiotis, D., Roumeliotou, K., Traeger-Synodinos, J., Bossios, A., et al. (2004). Severe hypertriglyceridaemia in a Greek infant: a clinical, biochemical and genetic study. Eur. J. Pediatr. 163, 462–466. doi:10.1007/s00431-004-1474-1
Lakoski, S. G., Lagace, T. A., Cohen, J. C., Horton, J. D., and Hobbs, H. H. (2009). Genetic and metabolic determinants of plasma PCSK9 levels. J. Clin. Endocrinol. Metab. 94, 2537–2543. doi:10.1210/jc.2009-0141
Langlois, S., Deeb, S., Brunzell, J. D., Kastelein, J. J., and Hayden, M. R. (1989). A major insertion accounts for a significant proportion of mutations underlying human lipoprotein lipase deficiency. Proc. Natl. Acad. Sci. U. S. A. 86, 948–952. doi:10.1073/pnas.86.3.948
Maruyama, T., Yamashita, S., Matsuzawa, Y., Bujo, H., Takahashi, K., Saito, Y., et al. (2004). Mutations in Japanese subjects with primary hyperlipidemia--results from the research committee of the ministry of health and welfare of Japan since 1996--. J. Atheroscler. Thromb. 11, 131–145. doi:10.5551/jat.11.131
Miesenböck, G., Hölzl, B., Föger, B., Brandstätter, E., Paulweber, B., Sandhofer, F., et al. (1993). Heterozygous lipoprotein lipase deficiency due to a missense mutation as the cause of impaired triglyceride tolerance with multiple lipoprotein abnormalities. J. Clin. Invest. 91, 448–455. doi:10.1172/JCI116222
Norata, G. D., Tavori, H., Pirillo, A., Fazio, S., and Catapano, A. L. (2016). Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc. Res. 112, 429–442. doi:10.1093/cvr/cvw194
O’Dea, L. S. L., MacDougall, J., Alexander, V. J., Digenio, A., Hubbard, B., Arca, M., et al. (2019). Differentiating familial chylomicronemia syndrome from multifactorial severe hypertriglyceridemia by clinical profiles. J. Endocr. Soc. 3, 2397–2410. doi:10.1210/js.2019-00214
Othman, H., and Saadat, M. (2009). Prevalence of consanguineous marriages in Syria. J. Biosoc. Sci. 41, 685–692. doi:10.1017/S0021932009003411
Pennacchio, L. A., and Rubin, E. M. (2003). Apolipoprotein A5, a newly identified gene that affects plasma triglyceride levels in humans and mice. Arterioscler. Thromb. Vasc. Biol. 23, 529–534. doi:10.1161/01.ATV.0000054194.78240.45
Péterfy, M., Ben-Zeev, O., Mao, H. Z., Weissglas-Volkov, D., Aouizerat, B. E., Pullinger, C. R., et al. (2007). Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat. Genet. 39, 1483–1487. doi:10.1038/ng.2007.24
Péterfy, M. (2012). Lipase maturation factor 1: A lipase chaperone involved in lipid metabolism. Biochim. Biophys. Acta 1821, 790–794. doi:10.1016/j.bbalip.2011.10.006
Pingitore, P., Lepore, S. M., Pirazzi, C., Mancina, R. M., Motta, B. M., Valenti, L., et al. (2016). Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia. J. Clin. Lipidol. 10, 816–823. doi:10.1016/j.jacl.2016.02.015
Pisciotta, L., Sallo, R., Rabacchi, C., Wunsch, A., Calandra, S., and Bertolini, S. (2012). Leucine 10 allelic variant in signal peptide of PCSK9 increases the LDL cholesterol-lowering effect of statins in patients with familial hypercholesterolaemia. Nutr. Metab. Cardiovasc. Dis. 22, 831–835. doi:10.1016/j.numecd.2011.04.003
Priore Oliva, C., Pisciotta, L., Li Volti, G., Sambataro, M. P., Cantafora, A., Bellocchio, A., et al. (2005). Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 25, 411–417. doi:10.1161/01.ATV.0000153087.36428.dd
Rahalkar, A. R., and Hegele, R. A. (2008). Monogenic pediatric dyslipidemias: classification, genetics and clinical spectrum. Mol. Genet. Metab. 93, 282–294. doi:10.1016/j.ymgme.2007.10.007
Regmi, M., and Rehman, A. (2021). Familial hyperchylomicronemia syndrome. StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK551655/(Accessed June 1, 2022).
Reymer, P. W., Gagné, E., Groenemeyer, B. E., Zhang, H., Forsyth, I., Jansen, H., et al. (1995). A lipoprotein lipase mutation (Asn291Ser) is associated with reduced HDL cholesterol levels in premature atherosclerosis. Nat. Genet. 10, 28–34. doi:10.1038/ng0595-28
Rodrigues, R., Artieda, M., Tejedor, D., Martínez, A., Konstantinova, P., Petry, H., et al. (2016). Pathogenic classification of LPL gene variants reported to be associated with LPL deficiency. J. Clin. Lipidol. 10, 394–409. doi:10.1016/j.jacl.2015.12.015
Shapiro, M. D., Minnier, J., Tavori, H., Kassahun, H., Flower, A., Somaratne, R., et al. (2019). Relationship between low-density lipoprotein cholesterol and lipoprotein(a) lowering in response to PCSK9 inhibition with evolocumab. J. Am. Heart Assoc. 8, e010932. doi:10.1161/JAHA.118.010932
Song, W., Beigneux, A. P., Winther, A.-M. L., Kristensen, K. K., Grønnemose, A. L., Yang, Y., et al. (2022). Electrostatic sheathing of lipoprotein lipase is essential for its movement across capillary endothelial cells. J. Clin. Invest. 132, e157500. doi:10.1172/JCI157500
Susheela, A. T., Vadakapet, P., Pillai, L., and Thampi, S. (2021). Familial chylomicronemia syndrome: a case report. J. Med. Case Rep. 15, 5. doi:10.1186/s13256-020-02609-0
Tavori, H., Rashid, S., and Fazio, S. (2015). On the function and homeostasis of PCSK9: reciprocal interaction with LDLR and additional lipid effects. Atherosclerosis 238, 264–270. doi:10.1016/j.atherosclerosis.2014.12.017
Warden, B. A., Fazio, S., and Shapiro, M. D. (2020). The PCSK9 revolution: current status, controversies, and future directions. Trends cardiovasc. Med. 30, 179–185. doi:10.1016/j.tcm.2019.05.007
Wilson, D. E., Emi, M., Iverius, P. H., Hata, A., Wu, L. L., Hillas, E., et al. (1990). Phenotypic expression of heterozygous lipoprotein lipase deficiency in the extended pedigree of a proband homozygous for a missense mutation. J. Clin. Invest. 86, 735–750. doi:10.1172/JCI114770
Wion, K. L., Kirchgessner, T. G., Lusis, A. J., Schotz, M. C., and Lawn, R. M. (1987). Human lipoprotein lipase complementary DNA sequence. Science 235, 1638–1641. doi:10.1126/science.3823907
Wittrup, H. H., Tybjaerg-Hansen, A., Abildgaard, S., Steffensen, R., Schnohr, P., and Nordestgaard, B. G. (1997). A common substitution (Asn291Ser) in lipoprotein lipase is associated with increased risk of ischemic heart disease. J. Clin. Invest. 99, 1606–1613. doi:10.1172/JCI119323
Yue, P., Averna, M., Lin, X., and Schonfeld, G. (2006). The c.43_44insCTG variation in PCSK9 is associated with low plasma LDL-cholesterol in a Caucasian population. Hum. Mutat. 27, 460–466. doi:10.1002/humu.20316
Keywords: chylomicronemia syndrome, type 1 hyperlipoproteinemia, LPL gene, triglycerides, Lebanon, Syria, PCSK9 levels
Citation: Ayoub C, Azar Y, Maddah D, Ghaleb Y, Elbitar S, Abou-Khalil Y, Jambart S, Varret M, Boileau C, El Khoury P and Abifadel M (2022) Low circulating PCSK9 levels in LPL homozygous children with chylomicronemia syndrome in a syrian refugee family in Lebanon. Front. Genet. 13:961028. doi: 10.3389/fgene.2022.961028
Received: 03 June 2022; Accepted: 26 July 2022;
Published: 19 August 2022.
Edited by:
Alberico Luigi Catapano, University of Milan, ItalyReviewed by:
Mariko Harada-Shiba, Osaka Medical and Pharmaceutical University, JapanWenxin Song, David Geffen School of Medicine, UCLA, United States
Copyright © 2022 Ayoub, Azar, Maddah, Ghaleb, Elbitar, Abou-Khalil, Jambart, Varret, Boileau, El Khoury and Abifadel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianne Abifadel, bWFyaWFubmUuYWJpZmFkZWxAdXNqLmVkdS5sYg==