 Tianyun Tao1,2Qianfeng Huang1,2Zhihao Zuo1,2
Tianyun Tao1,2Qianfeng Huang1,2Zhihao Zuo1,2 Yue Lu1,2Xiaomin Su1,2Yang Xu1,2
Yue Lu1,2Xiaomin Su1,2Yang Xu1,2 Pengcheng Li1,2
Pengcheng Li1,2 Chenwu Xu1,2*
Chenwu Xu1,2* Zefeng Yang1,2*
Zefeng Yang1,2*- 1Jiangsu Key Laboratory of Crop Genetics and Physiology/Key Laboratory of Plant Functional Genomics of the Ministry of Education/Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou, China
- 2Jiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou, China
Plant fw2.2-like (FWL) genes, encoding proteins harboring a placenta-specific eight domain, have been suggested to control fruit and grain size through regulating cell division, differentiation, and expansion. Here, we re-sequenced the nucleotide sequences of the maize ZmFWL7 gene, a member of the FWL family, in 256 elite maize inbred lines, and the associations of nucleotide polymorphisms in this locus with 11 ear-related traits were further detected. A total of 175 variants, including 159 SNPs and 16 InDels, were identified in the ZmFWL7 locus. Although the promoter and downstream regions showed higher nucleotide polymorphism, the coding region also possessed 61 SNPs and 6 InDels. Eleven polymorphic sites in the ZmFWL7 locus were found to be significantly associated with eight ear-related traits. Among them, two nonsynonymous SNPs (SNP2370 and SNP2898) showed significant association with hundred kernel weight (HKW), and contributed to 7.11% and 8.62% of the phenotypic variations, respectively. In addition, the SNP2898 was associated with kernel width (KW), and contributed to 7.57% of the phenotypic variations. Notably, the elite allele T of SNP2370 was absent in teosintes and landraces, while its frequency in inbred lines was increased to 12.89%. By contrast, the frequency of the elite allele A of SNP2898 was 3.12% in teosintes, and it was raised to 12.68% and 19.92% in landraces and inbred lines, respectively. Neutral tests show that this locus wasn’t artificially chosen during the process of domestication and genetic improvement. Our results revealed that the elite allelic variants in ZmFWL7 might possess potential for the genetic improvement of maize ear-related traits.
Introduction
Maize (Zea mays L.) is a major feed, food, and fuel (ethanol) crop in many parts of the world. Although global maize production has exceeded 1.2 billion tonnes in 2021/2022 (Statista 2021), the supply is still insufficient to meet the escalating demand. Breeders’ main goal has always been to increase yield. Grain yield is primarily determined by the morphological traits of the kernel and ear. Kernel yield is a complex quantitative trait that is influenced by various ear-related traits, including ear length (EL), ear weight (EW), ear grain weight (EGW), ear row number (ERN), kernel number per row (KNR), kernel size, and kernel weight (Liu et al., 2012; Zhang et al., 2017; Zhou et al., 2020). It is known that the yield components have a higher heritability compared with the kernel yield, and selecting certain yield components may be more advantageous than directly selecting the kernel yield alone (Gupta et al., 2006; Messmer et al., 2009). Among the ear-related traits, kernel size is a major factor that determines kernel weight, which is generally influenced by kernel length, width, and thickness (Li et al., 2013). Kernel weight has become a key factor affecting yield during breeding because of the fact that kernel weight reduction cannot be compensated for by other yield factors. In addition, ear-related traits, such as ER, ERN, and KNR, also have a significant impact on maize yield (Jia et al., 2020; Wang et al., 2021). Therefore, identifying the genes associated with ear-related traits and exploiting superior allelic variation may contribute to improvements in maize high-yield breeding.
Fruit-weight 2.2 (fw2.2), initially isolated from the tomato (Solanum lycopersicum L.), is a major gene that regulates fruit size and yield (Alpert et al., 1995). This gene represents a major quantitative trait locus that is responsible for around 30% of the difference in fruit size between wild and cultivated tomatoes and functions by negatively controlling cell division during fruit development (Frary et al., 2000; Nesbitt and Tanksley, 2001; Beauchet et al., 2021). Homologs of FWL genes encode proteins that contain a conserved cysteine-rich PLAC8 domain (CCXXXXCPC or CLXXXXCP), and are associated with various phenotypic traits, including organ size, plant architecture, and fruit weight (Guo and Simmons, 2011; Monforte et al., 2014). Recent studies have demonstrated that fw2.2 is transcribed specifically in the epidermis and sub-epidermis of the tomato fruit and produces new cells by regulating the division of transverse and longitudinal anticlinal cells, which promotes fruit growth (Renaudin et al., 2017; Shinozaki et al., 2018). Notably, fw2.2 homologs are also connected with organ size or cell division in rice (TGW2/OsCNR1; Ruan et al., 2020), cherry (CNR12 and CNR20; De Franceschi et al., 2013) and husk tomato (CNR1; Li and He, 2015); thus, they are also known as Cell Number Regulator (CNR) genes. Fw2.2 and its FWL/CNR homologs can not only regulate organ size and regulate cell division, but also endow plants with a response to rhizobia inoculation (Qiao et al., 2017), cadmium (Cd) resistance (Xiong et al., 2018; Wang et al., 2019), zinc (Zn), and manganese (Mn) tolerance (Qiao et al., 2019), and Ca2+ signal transduction (Rosa et al., 2017).
Fw2.2 and its FWL/CNR homologs belong to a complex multigene family. Thirteen FWL/CNR gene families have been identified in the 10 chromosomes of the maize genome. Of these, CNR1, is involved in a plant-specific cell proliferation mechanism and thus affects plant and fruit weight (Guo et al., 2010), whereas CNR2 expression has been shown to be adversely associated with tissue growth activity and hybrid seedling vigor (Guo et al., 2010; Guo and Simmons, 2011). Although FWL/CNR plays an important role in many biological processes, studies focusing on the genetic variants of FWL/CNR and their association with plant phenotypic traits in maize are scarce. In the present study, we identified the ZmFWL7 gene locus (GRMZM2G173742) from 256 inbred lines, 71 landraces, and 32 teosintes. We examined the relationship between this natural variation in this gene and several phenotypic features, including kernel yield and size. This study was designed to: 1) identify natural variations in ZmFWL7 connected to ear-related traits; 2) determine favourable alleles which are beneficial for HKW within ZmFWL7; and 3) assess the significance of ZmFWL7 in maize domestication and yield improvement.
Materials and methods
Plant materials, experimental design, and analysis of phenotypic data
In total, 256 elite inbred lines, 71 landraces, and 32 teosintes were collected in our study (Supplementary Table S1). All these inbred lines were planted in field in a randomized block design across three environments: the city of Sanya in Hainan province in 2015 and 2016, and the city of Yangzhou in Jiangsu province in 2017. Each line was planted with 13 plants in a single row, which made a plot 3 m long and 0.5 m wide. After harvesting and drying, three well-developed ears were used for measuring ear-related traits, including ear length (EL), ear diameter (ED), ear row number (ERN), kernel number per row (KNR), core diameter (CD), ear weight (EW), kernel length (KL), kernel width (KW), kernel thickness (KT), hundred kernel weight (HKW), and ear grain weight (EGW). Descriptive statistical analysis and ANOVA analysis of these phenotypic observations were performed using the “aov” function in R software. The best linear unbiased prediction (BLUP) values for each trait was calculated using the mixed linear model in the R package “lme4” (Bates et al., 2015). The parameters of the correlation coefficient were calculated using the R package psych.
DNA extraction and ZmFWL7 resequencing
Fresh leaves of 256 elite inbred lines were used to extract their genomic DNA using the modified cetyl trimethyl ammonium bromide (CTAB) method. Subsequently, the genomic sequences of the maize ZmFWL7 locus were re-sequenced using targeted sequence capture technology on the NimbleGen platform by BGI (Beijing Genomics Institute) Life Tech Co., China. The genomic sequences and positions of the ZmFWL7 (GRMZM2G173742) gene in the inbred line B73 (Schnable et al., 2009) were used as the reference for target capture sequencing. In order to estimate the neutral evolution of this locus, the sequences of 71 landraces and 32 teosintes were also captured using this method (Choi et al., 2009). The original re-sequencing data can be found in Supplementary Material S1.
Analysis of genotypic data
Multiple sequence alignment of the maize ZmFWL7 locus was performed using ClustalX software. The alignments were further manually edited to modify the putatively false alignment pairs (Larkin et al., 2007). The single nucleotide polymorphisms (SNPs), genetic and haplotype diversities of all tested lines were detected by DNASP5.0 software (Librado and Rozas, 2009). The nucleotide diversity (π) of the ZmFWL7 locus was estimated as the mean number of nucleotide differences per site between sequences using the R package PopGenome. A sliding window method was used with a window size of 200 base pairs (bp) and a step length of 50 bp (Pfeifer et al., 2014).
Marker-trait association analysis in inbred lines
TASSEL5.0 software was used to perform association analysis between the polymorphic sites of ZmFWL7 and the BLUP values of yield-related traits collected from the 256 inbred lines (Bradbury et al., 2007). The model of mixed linear was employed for association mapping. The kinship and population structure of the population, estimated using the principal component analysis (PCA) method, were used to decrease false-positive associations. Principal component analysis (PCA) and kinship were calculated using TASSEL5.0. A significant association was defined as polymorphic with p < 0.01. Linkage disequilibrium (LD) was estimated between any pairs of polymorphic sites in the sequenced region of ZmFWL7. The LD heatmap and r2 were generated using the R packages LDheatmap and pegas, respectively (Shin et al., 2006).
Results
Sequence polymorphisms in ZmFWL7
To detect nucleotide polymorphisms in ZmFWL7, the full-length sequence of this locus was re-sequenced in 256 inbred lines. A total of 3,450 bp sequences were recovered using a multiple sequence alignment, comprising the 1,572 bp upstream region, the 1,734 bp coding region and 144 bp of the downstream region. Nucleotide substitution, insertion, and deletion (Indel) variations within the ZmFWL7 locus are summarised in Table 1. In the genomic area surrounding ZmFWL7, a total of 175 variants, including 159 SNPs and 16 InDels, were discovered. SNPs and InDels were found every 21.7 and 251.6 kb on average. The overall nucleotide diversity (π) of the ZmFWL7 locus was 0.0087 for all 256 inbred lines; however, the coding region exhibited a much lower frequency of nucleotide polymorphisms compared with the downstream region. In addition, a sliding window of 200 bp with a step length of 50 bp was used to calculate π and θ (Figure 1). The 59–144 bp area in downstream region showed the highest nucleotide diversity, with π = 0.0285. The 746–845 bp area in the coding region exhibited the most nucleotide diversity, with π = 0.0242.
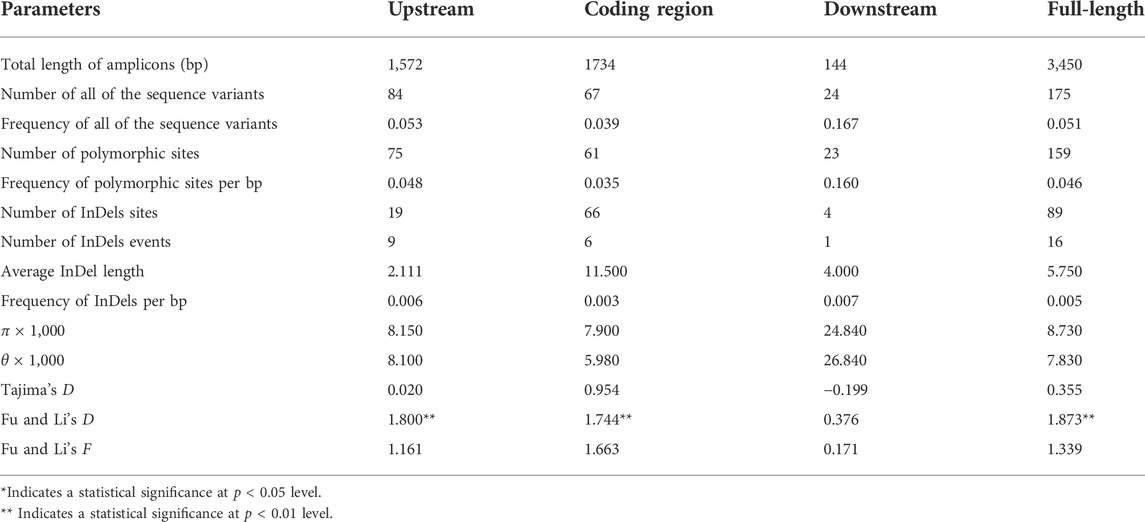
TABLE 1. Summary of parameters for the analysis of nucleotide polymorphisms of ZmFWL7 in inbred lines.
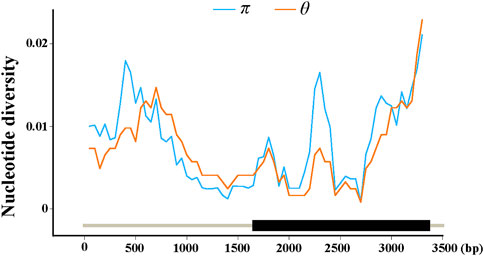
FIGURE 1. Nucleotide diversity (π and θ) estimated along the sequences of maizeZmFWL7. π and θ were calculated using the method of sliding windows of 200 bp with a step of 50 bp. The gene structure was indexed at the bottom of the coordinates.
Nucleotide diversity and selection of ZmFWL7 in inbred lines, landraces, and teosintes
To see whether artificial selection played a role in the domestication of ZmFWL7, we compared the genetic diversity of this gene and its flanking areas in inbred lines, landraces, and teosintes (Table 2). Compared with teosintes, inbred lines and landraces exhibited lower genetic diversity in the ZmFWL7 coding region, implying that selection occurred along the entire length of the gene sequence. In addition, we observed that the highest divergence between inbred lines and teosintes occurred in the upstream and downstream regions, whereas the coding regions were associated with a low divergence (Figure 2). These results indicate that this uneven distribution of polymorphisms may result from a lower frequency of variants in the coding region of ZmFWL7.
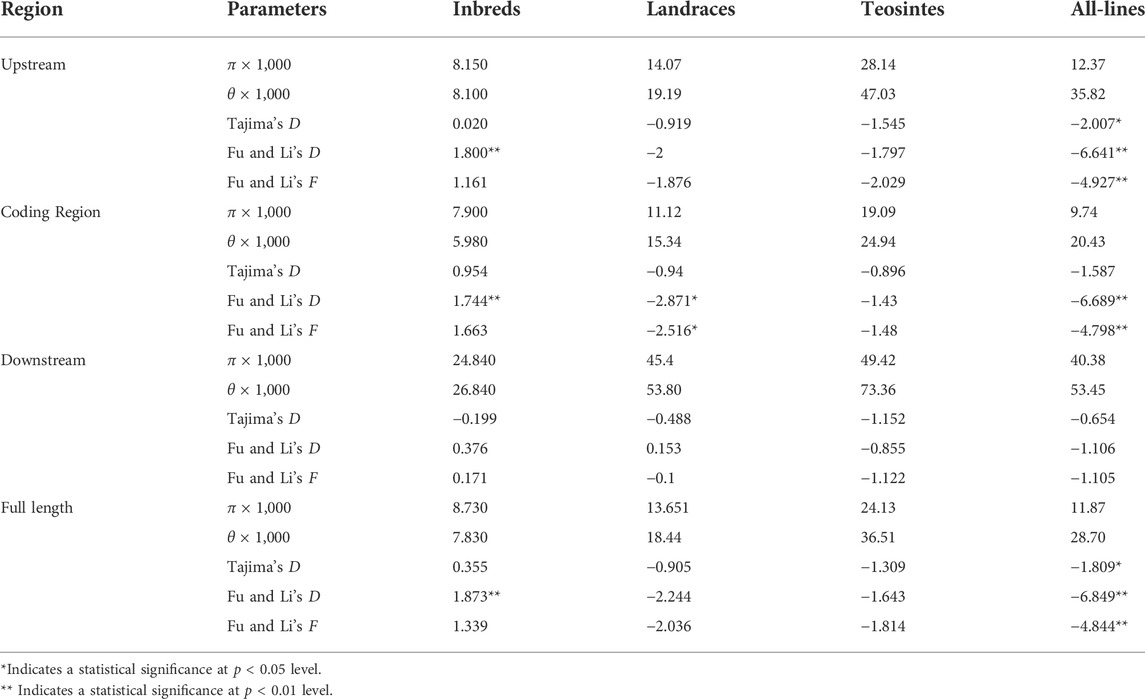
TABLE 2. The estimated parameters of nucleotide diversity, Tajima’D, Fu and Li’s D, and Fu and Li’s F of ZmFWL7.
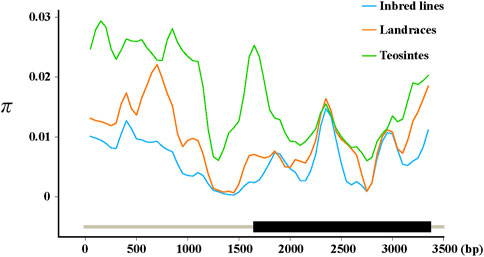
FIGURE 2. Nucleotide diversity in inbred lines, landraces, and teosintes. π was calculated using the sliding window method with a window size of 200 bp and a step length of 50 bp. Three lines were fitted by locally weighted regression (Loess) in R software. The parameter controlling the degree of smoothing was set as 0.1.
To further investigate the role of ZmFWL7 in the domestication and refinement of maize, ZmFWL7 sequences from three populations were examined by a neutral test, including Tajima’s D, and Fu and Li’s D* and F*. All of the estimation values from Tajima’s D in the teosintes and landraces were negative, suggesting an excess of low-frequency alleles in the landraces and teosintes. For the three populations, the Tajima’s D values did not reach a statistically significant level. Additionally, we found that the values for Tajima’s D in the inbred lines were positive, except for the downstream region, while Fu and Li’s D* estimations for this gene were significantly higher than zero, suggesting that moderate frequency alleles are present in this population.
Phenotypic variations and association analysis
Eleven ear-related traits were determined, and the descriptive statistics are presented in Supplementary Table S2. All of these ear-related features exhibited significant variations across inbred lines in ANOVA analysis, indicating that this population has genetic characteristics for association analyses. In addition, a significant phenotypic variation in all of these traits was observed in different environments and genotype-environment interactions. It is worth noting that the block effect significantly affected all traits except CD and KT (Supplementary Table S2). Therefore, to reduce the influence of environmental effects and block effects on genetic assessment and to obtain individual stability in genetic phenotypes, the BLUP model was used for analyzing the phenotypic data. A pairwise correlation analysis was performed to evaluate the association between these phenotypic traits, and Pearson correlation coefficients (r) between any two phenotypic traits were determined. Interestingly, most traits were statistically significantly correlated, with EW/EGW exhibiting the greatest correlation (r = 0.98). Only 8 of the 55 pairwise correlation tests for ear-related traits did not reach a significant level (EL/ED, EL/ERN, EL/CD, EL/KT, ED/KNR, ERN/HKW, KNR/HKW, and KL/KT) (Figure 3). Notably, HKW showed a significant positive correlation with KL, KW, and KT, of which KW exhibited the highest correlation with HKW (r = 0.98, p < 0.001). Therefore, the study of kernel length, width, and thickness should be conducted in the context of kernel weight.
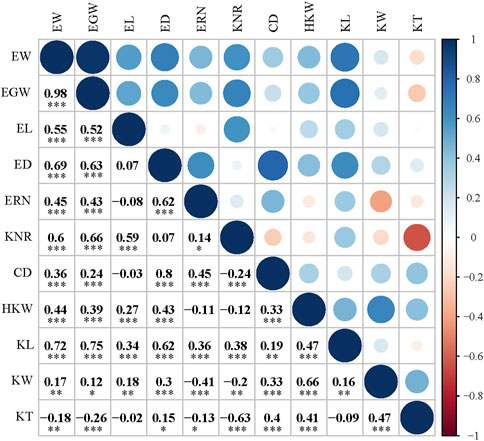
FIGURE 3. Pearson correlation coefficients for the BLUP value of 11 ear traits in 256 maize inbred lines. Abbreviations for traits are as follows: CD, core diameter; ED, ear diameter; EGW, ear grain weight; EL, ear length; ERN, ear row number; EW, ear weight; HKW, hundred kernel weight; KNR, kernel number per row; KW, kernel width. * indicates a statistical significance at p < 0.05 level, ** indicates a statistical significance at p < 0.01 level, *** indicates statistical significance at p < 0.001 level.
To find significant variants related with phenotypic traits, a statistical analysis of 128 variants was performed, comprising 101 SNPs and 27 InDels with a minor allele frequency (MAF) ≥ 0.01. A marker-trait association study was carried out using mixed linear models (PCA + Kinship) (Figure 4). The quantile quantile (QQ) plot indicated that the PCA and kinship were well controlled for association study in the mixed linear models (Supplementary Figure S1). And the results indicated that 11 pleiotropic sites were significantly associated, including EW, EGW, EL, ED, ERN, CD, HKW, and KW (p < 0.01, Figure 5A). We further analysed two pleiotropic sites (SNP2370 and SNP2898) in the coding region in which the p-value was less than 0.001. SNP2370 (serine to alanine) and SNP2898 (lysine to glutamine) were significantly associated with HKW, respectively, which explains the 7.11% and 8.62% phenotypic variation (Figure 5B). Moreover, SNP2898 was also significantly associated with KW, which could explain the phenotypic variation rate of 7.57%. These results demonstrate that ZmFWL7 may be associated with HKW and KW.
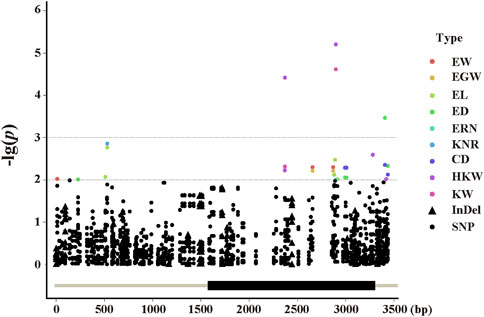
FIGURE 4. Manhattan plot by using the MLM (PCA + Kinship) model. Dots and triangles represent SNPs and Indels, respectively. Abbreviations for traits are as follows: CD, core diameter; ED, ear diameter; EGW, ear grain weight; EL, ear length; ERN, ear row number; EW, ear weight; HKW, hundred kernel weight; KNR, kernel number per row; KW, kernel width.
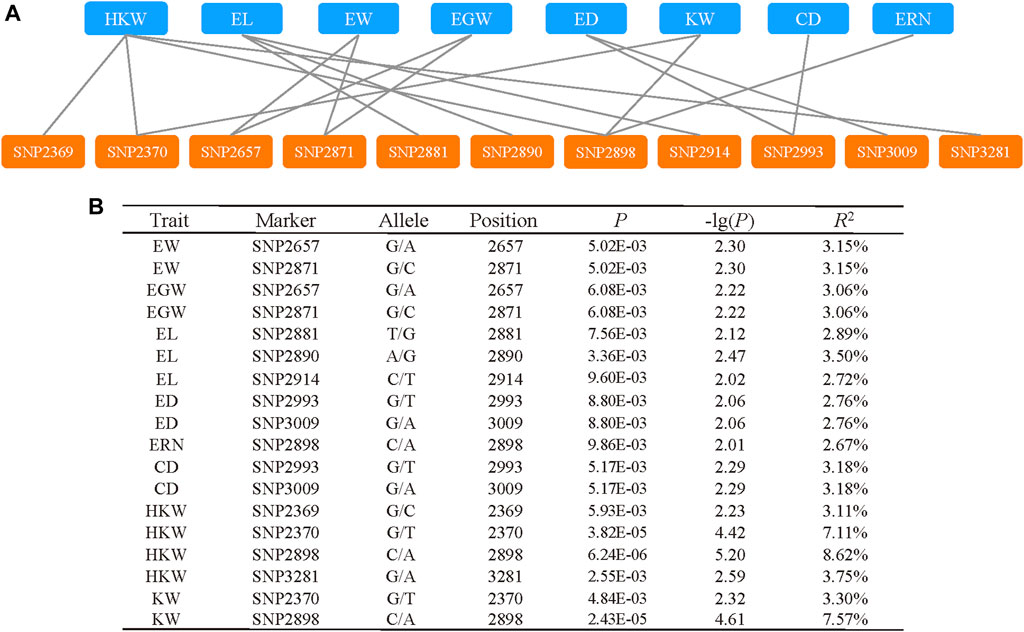
FIGURE 5. Significant markers associated with phenotypic traits. (A) The network between pleiotropic site and associated traits. (B) The information of significant markers which associated with phenotypic.
Linkage disequilibrium (LD) analysis indicated that SNP2369, SNP2370, SNP2657, and SNP2871 showed a relatively high linkage with SNP2898 (Figure 6A). Among them, SNP2370 and SNP2898 showed a relatively high LD, which was associated with HKW and KW. Three haplotypes were divided based on these two SNPs, hap2, and hap3 have a significantly higher HKW than hap1. While Hap3 has a significantly higher KW than hap1 (Figures 6B,C). For SNP2370, the allele T group had a significantly higher HKW than the allele G group (Figures 6D,E). In addition, we noticed that the allele A group also contained significantly higher values for HKW and KW compared with allele C in SNP2898 (Figures 6F,G). These results indicate that allele A of SNP2898 increased maize KW and thus resulted in the HKW variant. Furthermore, we analysed SNP2370 and SNP2898 allele frequencies in three separate groups. According to the findings, the frequency of SNP2898A in teosintes was 3.12%, while the frequency increased to 12.68% and 19.92% in landraces and inbred lines, respectively. By contrast, the elite allele T of SNP2370 was absent in teosintes and landraces, while its frequency was increased to 12.89% in inbred lines (Figures 6H,I). Based on these results, we conclude that allele A of SNP2898 and allele T of SNP2370 have a positive effect on the increase of HKW in maize.
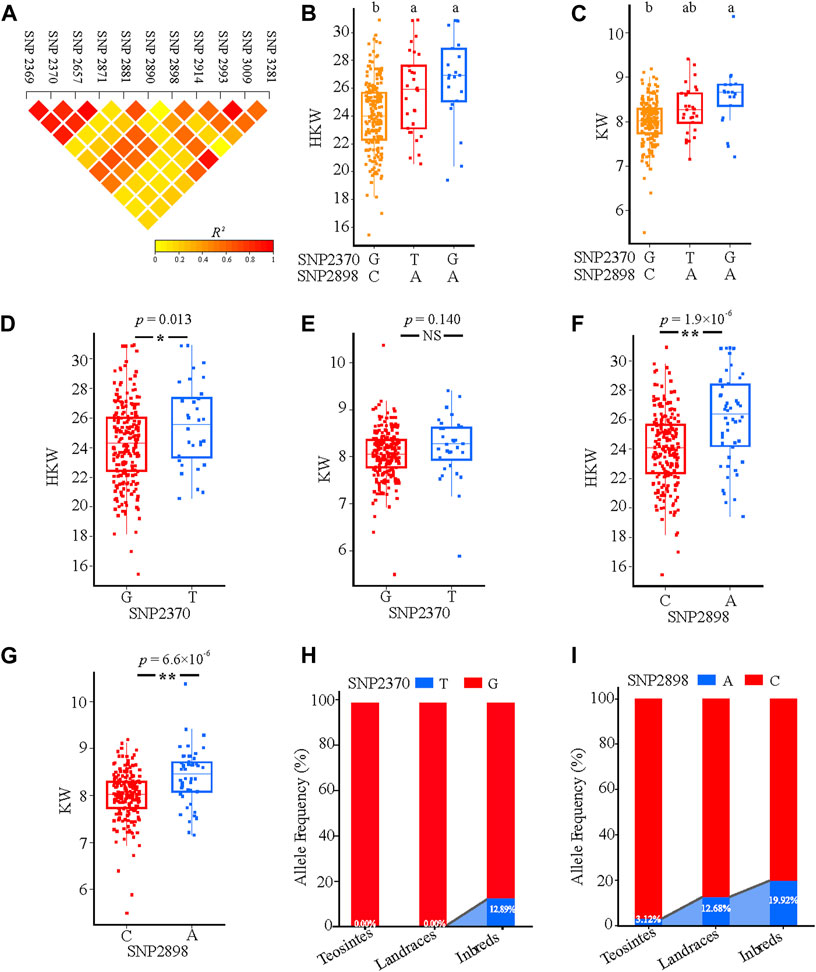
FIGURE 6. Natural variations in ZmFWL7 were significantly associated with hundred kernel weight (HKW) and kernel width (KW). (A) Linkage disequilibrium (LD) heatmap for eleven significant variants associated with ear traits. (B,C) Haplotypes of ZmFWL7 among natural variations based on two nonsynonymous mutation SNP2370 and SNP2898. Different letters indicate significant difference (Tukey’s HSD test at p < 0.05). (D–G) Comparison of hundred kernel weight (HKW) and kernel width (KW) between different alleles of SNP2370 and SNP2898. (H,I) The allele frequencies of SNP2370 and SNP2898 in inbred lines, landraces, and teosintes. p value for the t test comparing two groups carrying different alleles were indexed on the top (**p < 0.01; *p < 0.05).
Discussion
Candidate gene association mapping is widely employed to detect functional SNPs or alleles linked to gene-related agronomic parameters in maize, such as Dwarf8 (Thornsberry et al., 2001) for flowering period; ZmPGP1 (Li et al., 2019b), GS3 (Li et al., 2010b) and GW2 (Li et al., 2010a) for kernel shape and weight; Zmisa2 (Yang et al., 2014) and ZmBT1 (Xu et al., 2014) for starch properties; ZmHKT1 (Li et al., 2019a) and ZmMADS60 (Li et al., 2020) for root morphology; and ZmTD1 (Liu et al., 2019). Through the method of the candidate gene association analysis, we seek to uncover the link between natural sequence variation of the maize gene ZmFWL7 and variation of ear-related variables such as grain weight and grain width in this study. We found that a total of 11 variants in the maize ZmFWL7 gene show associations with multiple ear-related traits, including EW, EGW, EL, ED, ERN, CD, HKW, and KW. The ZmFWL7 gene was found to be significantly associated with kernel weight and KW, and the excellent haplotypes significantly increased HKW and KW. Endosperm filling, kernel weight and kernel size have become key parameters for maize yield, and studies have shown that kernel weight is closely related to KW, which is regulated by cell division and cell expansion (Li et al., 2018). Endosperm growth determines the majority of maize kernel size, and cell expansion runs through the whole process of endosperm development (Ji et al., 2022). Studies have shown that the FWL gene promotes cell division, produces new cells, and increases fruit size by regulating the division of transverse and longitudinal anticlinal cells (Renaudin et al., 2017; Shinozaki et al., 2018). In addition, the maize CNR1 gene, a member of FWL gene family, increases the grain width through a plant-specific cell proliferation function (Guo et al., 2010). In this analysis, two nonsynonymous SNPs (SNP2370 and SNP2898) showed a significant association with HKW and KW simultaneously. Further analyses suggested that allele A of the SNP2898 variants significantly increased KW and HKW. The results of a phenotypic correlation analysis also revealed that the most relevant trait to HKW was KW. Therefore, we speculated that allele A of SNP2898 may increase HKW through positive regulation of KW. In addition, allele T of the SNP2370 variants significantly increased HKW. Haplotype analysis based on these two nonsynonymous SNPs also showed that hap2 with SNP2370-T and SNP 2898-A had higher HKW than hap1 with SNP2370-G and SNP 2898-C. These results showed that both SNP2370 and SNP2898 can be used as the target sites to regulate kernel weight, and therefore has important application value.
Although the selection strategies for crop genes vary at different selection stages (domestication or improvement), it is undeniable that many genes that control important agronomic traits have been subject to crop domestication and subsequent selective breeding (Liu et al., 2021). The functions and underlying mechanisms of these genes have been extensively studied and utilised (Doebley et al., 2006). The domesticated or enhanced genes in maize, wheat, and rice include a variety of biological traits, such as grain filling (GIF1; Wang et al., 2008, ZmSWEET4c; Sosso et al., 2015), flowering time (ZmCOL3; Jin et al., 2018), grain quality (Wx; Huang et al., 2020), and crop yield (KNR2; Chen et al., 2022). The main objectives include easy cultivation, high yield, and rich nutrition. FWL/CNR gene family plays a crucial function in plant development regulation (Thibivilliers et al., 2020). In this study, we detected sequence polymorphisms in ZmFWL7 from 256 inbred lines, which yielded approximately one SNP per 35 bp in the coding region, indicating that this population has rich genetic diversity. The sequences of ZmFWL7 in three populations were tested by the neutral evolution test. The results indicated that all of the Tajima’s D values for the three separate groups did not reach a statistically significant level, indicating that the ZmFWL7 locus did not escape from neutral evolution. Of note, the allele A of SNP2898 was rare in the teosintes (3.12%), but its frequency trend increased and was 4 times higher in landraces and more than 6 times higher in inbred lines. In addition, the allele T of SNP2370 was completely absent in teosintes and landraces, but the frequency of this allele was increased to 12.89% in inbred lines. These findings indicated that these excellent allele variations have the potential values in maize breeding.
In summary, it is possible to increase kernel yield by using this gene in the maize population. Although the functional characteristics of ZmFWL7 need to be further investigated, our findings indicate that SNP2370 and SNP2898 may be used for marker-assisted selection to improve maize breeding.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
TT, CX, and ZY conceived and designed the research. TT, QH, ZZ, YL, and XS conducted the experiments. TT, QH, ZZ, YX, and PL analyzed the data. TT, CX, and ZY wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the grant from the National Natural Science Foundation of China (32070558, 32061143030, 32100448, 32170636, and 31972487), Natural Science Foundation of Jiangsu Province (BK20210799), China Postdoctoral Science Foundation (2019T120470), Open Project Program of State Key Laboratory of Rice Biology (20190102), A project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Seed Industry Revitalization Project of Jiangsu Province (JBGS[2021]009).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.960529/full#supplementary-material
References
Alpert, K. B., Grandillo, S., and Tanksley, S. D. (1995). Fw 2.2:a major QTL controlling fruit weight is common to both red- and green-fruited tomato species. Theor. Appl. Genet. 91 (6-7), 994–1000. doi:10.1007/BF00223911
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2015). Fitting linear mixed-effects models Usinglme4. J. Stat. Softw. 67 (1), 1–48. doi:10.18637/jss.v067.i01
Beauchet, A., Gevaudant, F., Gonzalez, N., and Chevalier, C. (2021). In search of the still unknown function of FW2.2/CELL NUMBER REGULATOR, a major regulator of fruit size in tomato. J. Exp. Bot. 72 (15), 5300–5311. doi:10.1093/jxb/erab207
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). Tassel: Software for association mapping of complex traits in diverse samples. Bioinformatics 23 (19), 2633–2635. doi:10.1093/bioinformatics/btm308
Chen, W., Chen, L., Zhang, X., Yang, N., Guo, J., Wang, M., et al. (2022). Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science 375 (6587), eabg7985. doi:10.1126/science.abg7985
Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., et al. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 106 (45), 19096–19101. doi:10.1073/pnas.0910672106
De Franceschi, P., Stegmeir, T., Cabrera, A., van der Knaap, E., Rosyara, U. R., Sebolt, A. M., et al. (2013). Cell number regulator genes in Prunus provide candidate genes for the control of fruit size in sweet and sour cherry. Mol. Breed. 32, 311–326. doi:10.1007/s11032-013-9872-6
Doebley, J. F., Gaut, B. S., and Smith, B. D. (2006). The molecular genetics of crop domestication. Cell. 127 (7), 1309–1321. doi:10.1016/j.cell.2006.12.006
Frary, A., Nesbitt, T. C., Grandillo, S., Knaap, E., Cong, B., Liu, J., et al. (2000). fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289 (5476), 85–88. doi:10.1126/science.289.5476.85
Guo, M., Rupe, M. A., Dieter, J. A., Zou, J., Spielbauer, D., Duncan, K. E., et al. (2010). Cell number Regulator1 affects plant and organ size in maize: Implications for crop yield enhancement and heterosis. Plant Cell. 22 (4), 1057–1073. doi:10.1105/tpc.109.073676
Guo, M., and Simmons, C. R. (2011). Cell number counts--the fw2.2 and CNR genes and implications for controlling plant fruit and organ size. Plant Sci. 181 (1), 1–7. doi:10.1016/j.plantsci.2011.03.010
Gupta, P. K., Rustgi, S., and Kumar, N. (2006). Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 49 (6), 565–571. doi:10.1139/g06-063
Huang, L., Sreenivasulu, N., and Liu, Q. (2020). Waxy editing: Old meets new. Trends Plant Sci. 25 (10), 963–966. doi:10.1016/j.tplants.2020.07.009
Ji, C., Xu, L., Li, Y., Fu, Y., Li, S., Wang, Q., et al. (2022). The O2-ZmGRAS11 transcriptional regulatory network orchestrates the coordination of endosperm cell expansion and grain filling in maize. Mol. Plant 15 (3), 468–487. doi:10.1016/j.molp.2021.11.013
Jia, H., Li, M., Li, W., Liu, L., Jian, Y., Yang, Z., et al. (2020). A serine/threonine protein kinase encoding gene KERNEL NUMBER PER ROW6 regulates maize grain yield. Nat. Commun. 11 (1), 988. doi:10.1038/s41467-020-14746-7
Jin, M., Liu, X., Jia, W., Liu, H., Li, W., Peng, Y., et al. (2018). ZmCOL3, a CCT gene represses flowering in maize by interfering with the circadian clock and activating expression of ZmCCT. J. Integr. Plant Biol. 60 (6), 465–480. doi:10.1111/jipb.12632
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23 (21), 2947–2948. doi:10.1093/bioinformatics/btm404
Li, C., Li, Y., Sun, B., Peng, B., Liu, C., Liu, Z., et al. (2013). Quantitative trait loci mapping for yield components and kernel-related traits in multiple connected RIL populations in maize. Euphytica 193 (3), 303–316. doi:10.1007/s10681-013-0901-7
Li, P., Ge, Z., Wang, H., Wei, J., Wang, Y., Xu, Y., et al. (2020). Nucleotide diversity and association analysis of ZmMADS60 with root length in the maize seedling stage. Agronomy 10 (3), 342. doi:10.3390/agronomy10030342
Li, P., Pan, T., Wang, H., Wei, J., Chen, M., Hu, X., et al. (2019a). Natural variation of ZmHKT1 affects root morphology in maize at the seedling stage. Planta 249 (3), 879–889. doi:10.1007/s00425-018-3043-2
Li, P., Wei, J., Wang, H., Fang, Y., Yin, S., Xu, Y., et al. (2019b). Natural variation and domestication selection of ZmPGP1 affects plant architecture and yield-related traits in maize. Genes. (Basel) 10 (9), E664. doi:10.3390/genes10090664
Li, Q., Li, L., Yang, X., Warburton, M. L., Bai, G., Dai, J., et al. (2010a). Relationship, evolutionary fate and function of two maize co-orthologs of rice GW2 associated with kernel size and weight. BMC Plant Biol. 10, 143. doi:10.1186/1471-2229-10-143
Li, Q., Yang, X., Bai, G., Warburton, M. L., Mahuku, G., Gore, M., et al. (2010b). Cloning and characterization of a putative GS3 ortholog involved in maize kernel development. Theor. Appl. Genet. 120 (4), 753–763. doi:10.1007/s00122-009-1196-x
Li, W., Yang, Z., Yao, J., Li, J., Song, W., and Yang, X. (2018). Cellulose synthase-like D1 controls organ size in maize. BMC Plant Biol. 18 (1), 239. doi:10.1186/s12870-018-1453-8
Li, Z., and He, C. (2015). Physalis floridana Cell Number Regulator1 encodes a cell membrane-anchored modulator of cell cycle and negatively controls fruit size. J. Exp. Bot. 66 (1), 257–270. doi:10.1093/jxb/eru415
Librado, P., and Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11), 1451–1452. doi:10.1093/bioinformatics/btp187
Liu, C., Zhou, Y., Zhang, X., Zhang, J., Zhou, Z., Weng, J., et al. (2019). Natural variation in the THICK TASSEL DWARF1 (TD1) gene in the regulation of maize (Zea mays L.) ear-related traits. Breed. Sci. 69 (2), 323–331. doi:10.1270/jsbbs.18170
Liu, J., Fernie, A. R., and Yan, J. (2021). Crop breeding - from experience-based selection to precision design. J. Plant Physiol. 256, 153313. doi:10.1016/j.jplph.2020.153313
Liu, R., Jia, H., Cao, X., Huang, J., Li, F., Tao, Y., et al. (2012). Fine mapping and candidate gene prediction of a pleiotropic quantitative trait locus for yield-related trait in Zea mays. PLoS One 7 (11), e49836. doi:10.1371/journal.pone.0049836
Messmer, R., Fracheboud, Y., Banziger, M., Vargas, M., Stamp, P., and Ribaut, J. M. (2009). Drought stress and tropical maize: QTL-by-environment interactions and stability of QTLs across environments for yield components and secondary traits. Theor. Appl. Genet. 119 (5), 913–930. doi:10.1007/s00122-009-1099-x
Monforte, A. J., Diaz, A., Cano-Delgado, A., and van der Knaap, E. (2014). The genetic basis of fruit morphology in horticultural crops: Lessons from tomato and melon. J. Exp. Bot. 65 (16), 4625–4637. doi:10.1093/jxb/eru017
Nesbitt, T. C., and Tanksley, S. D. (2001). fw2.2 directly affects the size of developing tomato fruit, with secondary effects on fruit number and photosynthate distribution. Plant Physiol. 127 (2), 575–583. doi:10.1104/pp.010087
Pfeifer, B., Wittelsburger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31 (7), 1929–1936. doi:10.1093/molbev/msu136
Qiao, K., Wang, F., Liang, S., Wang, H., Hu, Z., and Chai, T. (2019). Improved Cd, Zn and Mn tolerance and reduced Cd accumulation in grains with wheat-based cell number regulator TaCNR2. Sci. Rep. 9 (1), 870. doi:10.1038/s41598-018-37352-6
Qiao, Z., Brechenmacher, L., Smith, B., Strout, G. W., Mangin, W., Taylor, C., et al. (2017). The GmFWL1 (FW2-2-like) nodulation gene encodes a plasma membrane microdomain-associated protein. Plant Cell. Environ. 40 (8), 1442–1455. doi:10.1111/pce.12941
Renaudin, J. P., Deluche, C., Cheniclet, C., Chevalier, C., and Frangne, N. (2017). Cell layer-specific patterns of cell division and cell expansion during fruit set and fruit growth in tomato pericarp. J. Exp. Bot. 68 (7), 1613–1623. doi:10.1093/jxb/erx058
Rosa, M., Abraham-Juarez, M. J., Lewis, M. W., Fonseca, J. P., Tian, W., Ramirez, V., et al. (2017). The maize MID-COMPLEMENTING ACTIVITY homolog CELL NUMBER REGULATOR13/NARROW ODD DWARF coordinates organ growth and tissue patterning. Plant Cell. 29 (3), 474–490. doi:10.1105/tpc.16.00878
Ruan, B., Shang, L., Zhang, B., Hu, J., Wang, Y., Lin, H., et al. (2020). Natural variation in the promoter of TGW2 determines grain width and weight in rice. New Phytol. 227 (2), 629–640. doi:10.1111/nph.16540
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: Complexity, diversity, and dynamics. Science 326 (5956), 1112–1115. doi:10.1126/science.1178534
Shin, J.-H., Blay, S., Graham, J., and McNeney, B. (2006). LDheatmap: AnRFunction for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 16, 1–9. doi:10.18637/jss.v016.c03
Shinozaki, Y., Nicolas, P., Fernandez-Pozo, N., Ma, Q., Evanich, D. J., Shi, Y., et al. (2018). High-resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening. Nat. Commun. 9 (1), 364. doi:10.1038/s41467-017-02782-9
Sosso, D., Luo, D., Li, Q. B., Sasse, J., Yang, J., Gendrot, G., et al. (2015). Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 47 (12), 1489–1493. doi:10.1038/ng.3422
Thibivilliers, S., Farmer, A., and Libault, M. (2020). Biological and cellular functions of the microdomain-associated FWL/CNR protein family in plants. Plants (Basel) 9 (3), E377. doi:10.3390/plants9030377
Thornsberry, J. M., Goodman, M. M., Doebley, J., Kresovich, S., Nielsen, D., and Buckler, E. S. T. (2001). Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28 (3), 286–289. doi:10.1038/90135
Wang, E., Wang, J., Zhu, X., Hao, W., Wang, L., Li, Q., et al. (2008). Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 40 (11), 1370–1374. doi:10.1038/ng.220
Wang, F., Tan, H., Han, J., Zhang, Y., He, X., Ding, Y., et al. (2019). A novel family of PLAC8 motif-containing/PCR genes mediates Cd tolerance and Cd accumulation in rice. Environ. Sci. Eur. 31 (1), 82. doi:10.1186/s12302-019-0259-0
Wang, X., Liao, C., Wang, X., Yang, R., Zhai, L., and Huang, J. (2021). Construction of maize–teosinte introgression line population and identification of major quantitative trait loci. Euphytica 217 (9), 179. doi:10.1007/s10681-021-02912-x
Xiong, W., Wang, P., Yan, T., Cao, B., Xu, J., Liu, D., et al. (2018). The rice "fruit-weight 2.2-like" gene family member OsFWL4 is involved in the translocation of cadmium from roots to shoots. Planta 247 (5), 1247–1260. doi:10.1007/s00425-018-2859-0
Xu, S., Yang, Z., Zhang, E., Jiang, Y., Pan, L., Chen, Q., et al. (2014). Nucleotide diversity of Maize ZmBT1 gene and association with starch physicochemical properties. PLoS One 9 (8), e103627. doi:10.1371/journal.pone.0103627
Yang, Z., Zhang, E., Jiang, Y., Xu, S., Pan, L., Chen, Q., et al. (2014). Sequence polymorphisms in Zmisa2 gene are significantly associated with starch pasting and gelatinization properties in maize (Zea mays L.). Mol. Breed. 34 (4), 1833–1842. doi:10.1007/s11032-014-0142-z
Zhang, C., Zhou, Z., Yong, H., Zhang, X., Hao, Z., Zhang, F., et al. (2017). Analysis of the genetic architecture of maize ear and grain morphological traits by combined linkage and association mapping. Theor. Appl. Genet. 130 (5), 1011–1029. doi:10.1007/s00122-017-2867-7
Keywords: maize, ear -related traits, nucleotide polymorphisms, the ZmFWL7 gene, association analysis
Citation: Tao T, Huang Q, Zuo Z, Lu Y, Su X, Xu Y, Li P, Xu C and Yang Z (2022) Nucleotide polymorphisms of the maize ZmFWL7 gene and their association with ear-related traits. Front. Genet. 13:960529. doi: 10.3389/fgene.2022.960529
Received: 03 June 2022; Accepted: 18 July 2022;
Published: 10 August 2022.
Edited by:
Himanshu Sharma, National Agri-Food Biotechnology Institute, IndiaReviewed by:
Guosheng Xie, Huazhong Agricultural University, ChinaBeata Alicja Myśków, West Pomeranian University of Technology, Poland
Copyright © 2022 Tao, Huang, Zuo, Lu, Su, Xu, Li, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenwu Xu, Y3d4dUB5enUuZWR1LmNu; Zefeng Yang, emZ5YW5nQHl6dS5lZHUuY24=