
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 September 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.951829
This article is part of the Research Topic Biomedical Application of DNA Modifications View all 5 articles
 Fang Fu1,2*
Fang Fu1,2* Ru Li1Xiao Dang1
Ru Li1Xiao Dang1 Qiuxia Yu1
Qiuxia Yu1 Ke Xu3Weiyue Gu3Dan Wang1
Ke Xu3Weiyue Gu3Dan Wang1 Xin Yang1
Xin Yang1 Min Pan1Li Zhen1Yongling Zhang1Fatao Li1Xiangyi Jing2Fucheng Li2Dongzhi Li2
Min Pan1Li Zhen1Yongling Zhang1Fatao Li1Xiangyi Jing2Fucheng Li2Dongzhi Li2 Can Liao2*
Can Liao2*Balanced chromosomal abnormalities (BCAs) are the most common chromosomal abnormalities and the frequency of congenital abnormalities is approximately twice as high in newborns with a de novo BCA, but a prenatal diagnosis based on BCAs is subject to evaluation. To detect translocation breakpoints and conduct a prenatal diagnosis, we performed whole-genome sequencing (WGS) in 21 subjects who were found BCAs, 19 balanced chromosome translocations and two inversions, in prenatal screening. In 16 BCAs on non-N-masked regions (non-NMRs), WGS detected 13 (81.2%, 13/16) BCAs, including all the inversions. All the breakpoints of 12 (12/14) cases of sufficient DNA were confirmed by Sanger sequencing. In 13 interrupted genes, CACNA1E (in case 12) and STARD7 (in case 17) are known causative and PDCL was found in subject (case 11) with situs inversus for the first time. Case 12 with abnormal ultrasound reached a definitive genetic diagnosis of CACNA1E-disease, while STARD7 exon deletion has never been found causative in patients. WGS provides the possibility of prenatal diagnosis in fetuses with BCAs, and its clinical significance also lies in providing data for postnatal diagnosis.
Balanced chromosomal abnormalities (BCAs) can be categorized into reciprocal translocations, changes in either localization or orientation between the two non-homologous chromosomes, inversions in the same chromosome, and Robertsonian translocations, a result of the fusion of two acrocentric chromosomes 13, 14, 15, 21 or 22. BCAs are the most common chromosomal abnormalities in both healthy and diseased individuals and the estimated frequency of all balanced structural rearrangements in the general population is 0.2%–0.4% (Jacobs et al., 1992; Blake et al., 2014). The frequency of congenital abnormalities, e.g., multiple congenital anomalies or intellectual disability (ID), is approximately twice as high in newborns with a de novo BCA (6.1% for reciprocal translocation, 9.4% for inversions, and 3.7% for Robertsonian translocations) compared with the risk in the general population. (Warburton, 1991). This suggests a causative link between BCAs and clinical phenotype.
The consequences of de novo BCAs prenatally diagnosed are unpredictable, which introduce uncertainty into pathogenicity reports and genetic counseling decisions. The clinical phenotype in newborns with BCAs can be caused by disruption of specific gene(s) at a translocation or inversion breakpoint(s), therefore, characterization of the breakpoints in BCAs has been a key to elucidate the potential Mendelian disorders. (Gianfrancesco et al., 2008; Moller et al., 2008; Kalscheuer et al., 2009). Therefore, prenatal diagnosis of BCAs relies on a high-resolution method of genomic structural variations (SVs) detection, while chromosomal microarray analysis (CMA), the method commonly used in copy number variants (CNVs) testing, cannot detect balanced translocations.
Recently, short-read sequencing-based whole-genome sequencing (WGS) has emerged as a comprehensive diagnostic tool with a continuously improving cost-effectiveness. Redin et al. (2017) performed WGS in 273 subjects with BCAs and congenital abnormalities and revealed that WGS could revise 93% of karyotyping results by at least one sub-band, who also determined that in BCAs, 33.9% resulted in gene disruptions, 5.2% were associated with pathogenic genomic imbalances, and 7.3% disrupted topologically associated domains (TADs). (Redin et al., 2017). Using WGS, Yu et al. (2020) identified BCA breakpoints in 3 cases and reached definitive genetic diagnosis in 4 cases, out of 10 cases.
The aim of this study was to assess the clinical utility of WGS in the prenatal setting, and to explore the potential implications on the decision-making process during perinatal period.
This study was a single center pilot cohort study performed at the Department of Prenatal Diagnostic Centre, Guangzhou Medical University (China) between January 2019 and December 2021, including all cases of BCA(s), not Robertsonian translocation, prenatally suspected after a dedicated prenatal karyotyping. The study was approved by the ethics committee of the Guangzhou Women and Children’s Medical Center and adheres to the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. (2019356B-01). All individuals enrolled in this study from Guangzhou Women and Children’s Medical Center and informed consent forms were signed by the participants.
Genomic DNA was extracted from amniotic fluid according to standard protocols and purified using a QIAamp DNA Blood Maxi Kit (Qiagen, Hilden, Germany). Genomic DNA from each sample was sheared as input material for library construction, and the fragmented DNA was further processed with end repair, A-tailing, adapter ligation, and PCR amplification following the manufacturer’s recommended protocols. High-throughput sequencing was performed on NovaSeq6000 (Illumina, United States). A minimal read depth was 30-fold for WGS. Quality control for the paired-end reads was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and fastp(Chen et al., 2018) was used for adapter trimming and quality filtering.
Sequencing reads were aligned to the human reference genome GRCh37/UCSC hg19 using BWA-MEM (Li and Durbin, 2010), then SAMtools (Li et al., 2009) and Picard (https://broadinstitute.github.io/picard/) was performed to sort and mark duplicated reads, respectively. SVs were screened using MANTA (Chen et al., 2016) and LUMPY (Layer et al., 2014), and CNVnator (Abyzov et al., 2011) and AMYCNE (Eisfeldt et al., 2018) software packages were used to detect CNVs. Repeat expansions at known loci were called by ExpansionHunter (Dolzhenko et al., 2019).
SVs and CNVs were annotated and ranked by AnnotSV (Geoffroy et al., 2018), they were evaluated by comparison with literature values and databases, such as the Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home), the Database of genomic variation and Phenotype in Humans Using Ensembl Resources (DECIPHER, https://www.deciphergenomics.org/), ClinGen Dosage Sensitivity Map (ClinGen, https://www.clinicalgenome.org/) and PubMed (https://pubmed.ncbi.nlm.nih.gov/). According to the guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) (Richards et al., 2015; Abou Tayoun et al., 2018; Brandt et al., 2020; Riggs et al., 2020), the clinical significance of all identified variations was interpreted and classified into five categories: pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign, and benign. To complement the uncertainty in the interpretation of CNV breakpoints in the current ACMG guidelines(Brandt et al., 2020; Riggs et al., 2020), we performed extra analysis of the genes in which those breakpoints are located. The clinical phenotypes of patients were extracted to specific Human Phenotype Ontology (HPO, https://hpo.jax.org/) terms to facilitate identifying disease-causing variants. Screening and prioritizing clinically relevant variants on the basis of variants quality, allele frequency, functional impact, inheritance model or familial segregation, available information in published literature, and agreement to explain the patient clinical phenotype. The size of aberration, gene disruptions as well as functional elements like enhancers and regulatory elements in topologically associated domain (TAD) (Wang et al., 2018) regions were used for further interpretation for SVs.
PCR amplification and Sanger sequencing of the junction fragments were performed to validate SVs using standard protocols. In brief, PCR products were purified by exonuclease I and sequenced on an ABI 3730 DNA Analyzer using BigDye Terminator v3.1 (Applied Biosystems, United States). Sanger sequencing was applied across the WGS detected breakpoints to generate a unique junction fragment sequence of SVs. Except for insufficient DNA in case 1 and 20, the information of Sanger sequencing and the primers in other 11 subjects with gene interruption were shown in Supplementary Material S1.
In the 21 subjects, 19 were balanced chromosome translocations and two were inversions. All the subjects had no significant pathogenic genomic variation revealed by previous prenatal screening, including trio karyotyping with a band resolution of 400–650 bands per haploid chromosome set and/or cell-free fetal DNA testing in each subject. Two fetuses were found abnormality by ultrasonography screening (Table 1), in which significant macrocephaly and extremities anomaly were found in case 12 (Figure 1A).
TABLE 1. Prenatal findings and the parents’ decision after genetic counseling based on the genetic tests in 21 cases.
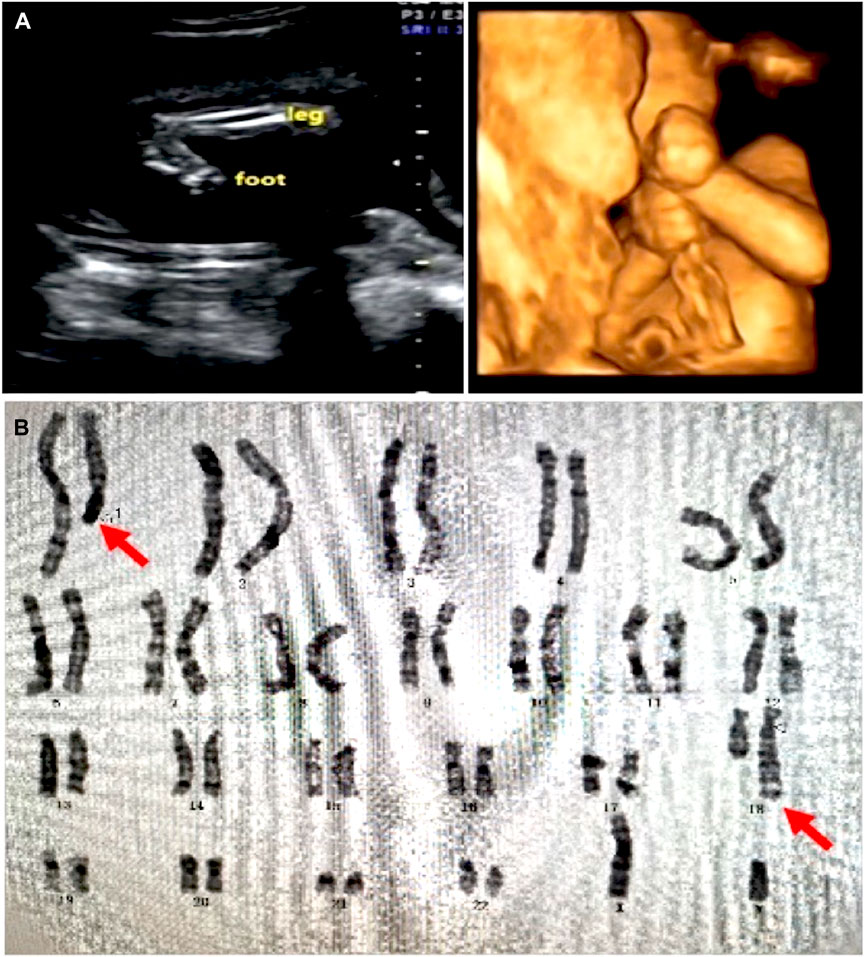
FIGURE 1. Ultrasound and karyotyping results in case 12. (A) Ultrasound showed significant macrocephaly, bilateral talipes calcaneovalgus, and clenching hand. (B) Karyotyping showed a balanced translocation 46,XN,t(1; 18) (q25; q23)dn. The original report of karyogram was unavailable. Red arrows showed the mutually translocated chromosomal fragments.
The average and median depth coverage of WGS was 33 ×, and the percentage of coverage over 10 × and 20 × was 88% and 97%, respectively. Five balanced chromosome translocations occurred in centromeric or pericentromeric heterochromatic regions (due to low quality base calls with masked substitutes “N”s, i.e., undetermined bases, as known as N-masked) were not detected by WGS. WGS detected BCAs in 13 cases out of the remaining 16 fetuses, including 11 balanced chromosome translocations and two inversions. (Table 1)., WGS refined the sub-bands of chromosomal variants detected by karyotyping in the 14 cases, including two inversions. (Table 1). Sanger sequencing confirmed all, except two cases due to insufficient DNA specimen, breakpoints detected by WGS. It should be noted that, in case 21, insertion of 7bp at the breakpoint was found only by Sanger sequencing (Supplementary Figure S1), which casts doubt on the stringency of the definition of balanced translocation actually found clinically. Unfortunately, due to insufficient DNA samples from the parents, Sanger sequencing could not be added to confirm whether the insertion was inherited from the mother or de novo. In summary, no genomic imbalance was detected by WGS analysis in all the subjects.
Thirteen genes were found disrupted, in which the CACNA1E (in case 12) and STARD7 (in case 17) genes are associated with the developmental and epileptic encephalopathy (MIM: #618285) and the familial adult myoclonic epilepsy (MIM: #607876), respectively. In addition, TADs were found interrupted in six cases. (Table 1).
Except for two case of insufficient DNA specimen, Sanger sequencing was performed in 11 subjects (Supplementary Figure S1). Compared with WGS results, Sanger sequencing verified that four breakpoints were identical and six with differences within 10 bp. Only one breakpoint, in case 18, showed difference in 16 base pairs. (Table 1; Supplementary Figure S1).
Case 12 was a 27-year-old female at 18-weeks pregnant, who had one previous abortion (not for medical reasons). In the first trimester of this gestation, the woman had no history of exposure to toxic substances and inappropriate medication. The pregnant woman and her husband were healthy and they had no family history. At 22 weeks pregnant, ultrasound showed her fetus had significant macrocephaly with bilateral cerebral ventriculomegaly (12/12 mm), bilateral talipes calcaneovalgus, and clenching hand. (Figure 1A). By amniocentesis, karyotyping showed the fetus had de novo 46,XN,t(1; 18) (q25; q23)dn (Figure 1B), while the chromosome microarray analysis (CMA) was negative, thereby singleton WGS was performed and the result showed the balanced translocation with breakpoints at chr1:181, 523, 009 (q25.3) and chr18:73, 066, 267 (q22.3) that confirmed by Sanger sequencing, chr1:181, 523, 013 and chr18:73, 066, 267, respectively. (Figure 2).

FIGURE 2. Sanger sequencing confirmation of the balanced chromosomal abnormalities (BCAs) breakpoints in Case 12 and 16. The comparison of sequences detected by WGS and Sanger sequencing in the cases is listed in Table 1. PCR primers used for Sanger sequencing as following: Case 12 F-TGGGAGGGATAAAGCGACTTG, R-CCTCCCATGACATGTAGGGATTAT, case 16 F-AACAGACTCTCCACAACAGAGAAC, R-ACTCGCCCTCCGAGTGAAAGGAG.
Breakpoints chr1:181, 523, 009 or 181, 523, 013 locates on intron 3 of CACNA1E and the translocation caused a deletion of the downstream exons in the CACNA1E gene. CACNA1E-associated epileptic encephalopathy 69 (MIM: #618285) is an autosomal dominant inherited disease characterized by inborn errors as macrocephaly and joint contractures, which matches the ultrasound findings in the fetus. According to the diagnosis, the couple chose to terminate their pregnancy at 22 + weeks gestation. The terminated fetus appeared severely bent and deformed limbs and, however, the couple did not authorize the biopsy.
The woman was a balanced translocation carrier and she had 46,XN,t(9; 10) (q32; p13)mat detected by karyotyping. Her first baby had congenital cranial hypoplasia, ventricular septal defect (VSD), and ASD, who died at age 8 months. In this pregnancy, amniotic fluid karyotyping showed the fetus had 46,XN,t(9; 10) (q32; p13)mat and no significant CNV was found by the previous CMA analysis. WGS reveal the translocation breakpoints at chr9:127, 657, 832 and chr10:3,710,364 that both locate on chromosomal regions without a known functional annotation (Figure 2). The couple were healthy and had no history of harmful exposure and family history. Based on the description of signs in her first baby, it is rational to speculate the previous baby with 9q34.3 terminal/subtelomeric deletion syndrome, or Kleefstra syndrome 1 (KS1, MIM: # 610,253), caused by heterozygous EHMT1 defect, however, the previous results of genetic tests in her first baby were not retrievable. The core phenotype of KS1, including brachycephaly or microcephaly, conotruncal heart defects, severe neurodevelopmental, and behavioral impairment, explains the findings in the first baby of the woman. However, none of the signs of KS1 were found in the second gestation tests and it could not be explained by the balanced translocation. Based on the above findings, the balanced translocation was considered benign.
After genetic counseling, the couple decided to continue the pregnancy and their boy was normal at age two years.
Case 17 was enrolled because the prenatal screening in her second trimester revealed a high risk of Down syndrome. Both the husband and wife were healthy and had no family history of genetic disease (but could not recall whether a family member had transient benign epilepsy). This was the first child of the couple, and there was no history of exposure to toxic substances or drugs in the first trimester of pregnancy. At 20 weeks of gestation, her routine prenatal testing was normal; prenatal screening by the quad test revealed a possible high risk of Down syndrome. Karyotype analysis showed no abnormalities in chromosome number but paternal inherited balanced translocation of chromosomes 2 and 21; fetal amniotic fluid WGS showed that one of the breakpoints of this balanced translocation was located within the STARD7 gene (Table 1), which lead to a heterozygous deletion of the last exon. STARD7 pathogenic variants cause an autosomal dominant familial adult myoclonic epilepsy (MIM: #607876), which is characterized as epilepsy that can occur as early as puberty, but no other central nervous systemic abnormality or phenotype have been reported in infancy (De Fusco et al., 2014). However, in all reports of STARD7-related epilepsy, all cases are due to the expansion of the 5 bp repeat in intron 1, and other STARD7 pathogenic variants have never been reported in patients(Corbett et al., 2019). After informing the couple of the test results, the couple decided to continue the pregnancy.
We demonstrated WGS being an efficient method for breakpoint mapping with a high resolution in detecting and interpreting BCAs compared to sequential and multiple tests commonly performed clinically. However, it does not always permit a phenotype correlation in de novo BCAs carriers while the breakpoints do not disrupt known disease-causative genes. In addition, a short-read, paired-end sequencing method cannot detect breakpoints of low-level mosaicism, Robertsonian translocations, small supernumerary marker chromosomes and ones in repetitive sequences cannot be detected (Hochstenbach et al., 2021). It is estimated that 7.6% of breakpoints were localized within genomic segments that cannot be confidently mapped by short-read sequencing (Redin et al., 2017). In this study, translocation breakpoints of 13 BCAs, including all inversions (2/2), were identified by WGS, in which 10 breakpoints were confirmed by Sanger sequencing with differences of <10 bp (Table 1). In the 12 BCAs, two cases (case 12 and 17) were identified interrupting known disease-causative genes (CACNA1E and STARD7, respectively). Eight (case 7, 11, 15, 16, 18, 19, 20, and 21) with genes disrupted by breakpoints were classified as benign or likely benign according to the ACMG guidelines. However, as an interpretation tool for large fragment variants, the ACMG guidelines for CNVs are difficult to be used to determine breakpoints (Brandt et al., 2020; Riggs et al., 2020), especially large fragment genetic variants that may be caused by intragenic breakpoints. So, we, by reviewing the protein function of these eight genes, found a potential association between PDCL defect and the phenotype in case 11. TAD disruption was found in six cases, case 14, 7, 11, 12, 16, and 17. For the three cases with negative results, case 4–6, we speculated that this may be due to the limits of the short-read sequencing, resulting in insufficient coverage depth of these breakpoint regions. For some balanced translocation breakpoints that are highly suspected of being pathogenic, we suggest additional sequencing methods for further confirmation, such as high-throughput sequencing based on a long-read method.
The results of WGS led to improved clinical utility by giving a definitive molecular diagnosis in two cases. In case 12, the diagnosis of CACNA1E-associated epileptic encephalopathy 69 (MIM: #618285) by WGS analysis explained the abnormalities by ultrasonography screening. In another converse situation, case 15, for example, WGS analysis of the breakpoints of 46,XN,t(9; 10) (q32; p13)mat was key to differentially diagnose with 9q34.3 terminal/subtelomeric deletion syndrome, the putative cause of a severe disease in the previous died child of the family. The results of case 15 show that in addition to the detection of candidate causative genes/chromosomes, WGS is ideal for excluding suspicious findings, especially in scenario applications of genetic counseling and family decision-making.
Phenotype-association of most of the interrupted genes are unknown, however, PDCL found in case 11 with situs inversus piqued our interest. Human phosducin-like protein (PDCL) is a chaperone of heterotrimeric G proteins and it, together with phosducin, a phosphoprotein, have been demonstrated to regulate G-protein signaling. (Thibault et al., 1999; Lukov et al., 2005). As far as we know, PDCL variants or functional defects were never found in patients with Mendelian disorders, however, in mice, Pdcl plays a role in regulating ciliary function by being a positive regulator of hedgehog signaling (Pusapati et al., 2018). Situs inversus is quite common in patients with ciliary abnormalities in human (Pennekamp et al., 2015) and PDCL defects found in this study may provide clues for future discovery of the new disease-causing gene.
Another particular concern is case 2, which was reported with a de novo TAD interruption on 4q35 (Table 1; Supplementary Figure S1), causative for Facio-Scapulo Humeral dystrophy (FSHD) (Sarfarazi et al., 1992; Lemmers et al., 2010). FSHD is characterized as adult- (90%) or childhood-onset (10%) progressive skeletal muscle disorder with a highly variable phenotype (van den Boogaard et al., 2016; Johnson and Ankala, 2020; Schatzl et al., 2021), which is not possibly detected by ultrasound screening in fetus. TADs are fundamental units of three-dimensional (3D) nuclear organization, at the sub-megabase scale, chromatin domains with high interaction frequency and relatively isolated from neighbor regions (TAD boundaries) form TADs (Lieberman-Aiden et al., 2009; Dixon et al., 2016). More recently, human development and diseases due to TAD disruptions received attention from researchers (Tena and Santos-Pereira, 2021). However, we have limited resources and framework for integrating the TADs and standard guidelines and tools for TAD analysis, for now, are not available. Therefore, long-term follow-ups, especially in case 2, are needed.
Although it is now difficult to reach a definitive diagnosis in the prenatal setting, a negative WGS result can also benefit genetic counseling by giving additional reassurance. In this study, WGS results reduced anxiety of the family found high-risk for trisomy or chromosomal aneuploidies by previous screening when interpreting BCA findings.
There are limitations to this study. Firstly, prenatal or postnatal follow-up was incomplete in most cases. This may be because these families that chose to continue the pregnancy after genetic counseling have normal newborn children, or because the WGS breakpoint suggests an association with a late-onset disease with mild phenotype, such as in case 17. In addition, integrating disruptions of TADs is still a big challenge. Secondly, cases with inherited BCAs were not excluded in this study, in which a prenatal diagnosis was considered to be made based on family history. Thirdly, the sample size of this study is small and only one with significant ultrasound abnormality. Considering of a few previously published articles using short-read WGS in prenatal studies (Redin et al., 2017; Yu et al., 2020), our study provides useful information to assess the application of WGS in prenatal diagnosis in fetuses with BCAs. Finally, it is reported that low-pass (one-fold coverage) WGS has a certain robustness for the detection of BCA (Liang et al., 2017; Dong et al., 2018), however, in the setting of clinical prenatal diagnosis, the comparison of 30X and low-pass WGS for BCA breakpoint detection is still lacking in larger studies. We hope to conduct cohort studies of these two WGS techniques in more prenatal diagnosis cases of BCA in the future, so as to discuss whether there is consistent reliability while reducing testing costs.
This study demonstrated the advantages of WGS over prenatal diagnosis on the detection of BCAs. The strategy of BCA breakpoints detecting is available by WGS, which gives additional information for prenatal diagnosis. Although, for now, only the case with abnormality revealed by routine prenatal screening reached a definitive diagnosis, WGS results give physicians potential opportunities to improve the genetic counseling and perinatal care, and further reduced anxiety in the families coming for medication.
The data presented in the study are uploaded in the Supplementary files, accessible via the link: https://www.frontiersin.org/articles/10.3389/fgene.2022.951829/full#supplementary-material.
The studies involving human participants were reviewed and approved by Guangzhou Women and Children’s Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
FF and RL designed this study. XD, QY, DW, and XY. designed and carried out the genetic sequencing used in this study. MP, KX, and LZ wrote the manuscript. YZ, FL, XJ, and FL. C collected data and carried out bioinformatics and statistical analysis. DL, and KX. created figures and tables. CL directed the overall research. WG reanalyzed the data. All authors read and reviewed the final manuscript.
This work was supported by the Subproject of the National Key R&D Program (2021YFC2701002), the National Natural Science Foundation of China (81801461, 81873836, 81771594, 81671474, 81501267), the Natural Science Foundation of Guangdong Province (2019A1515012034), and the Project of Guangzhou Science and Technology Bureau (202201020643).
We are indebted to the parents who participated in this study.
Authors KX and WG are employed by Beijing Chigene Translational Medical Research Center Co. Ltd..
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.951829/full#supplementary-material
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39 (11), 1517–1524. doi:10.1002/humu.23626 | |
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21 (6), 974–984. doi:10.1101/gr.114876.110 | |
Blake, J., Riddell, A., Theiss, S., Gonzalez, A. P., Haase, B., Jauch, A., et al. (2014). Sequencing of a patient with balanced chromosome abnormalities and neurodevelopmental disease identifies disruption of multiple high risk loci by structural variation. PLoS One 9 (3), e90894. doi:10.1371/journal.pone.0090894 | |
Brandt, T., Sack, L. M., Arjona, D., Tan, D., Mei, H., Cui, H., et al. (2020). Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 22 (2), 336–344. doi:10.1038/s41436-019-0655-2 | |
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560 | |
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Källberg, M., et al. (2016). Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32 (8), 1220–1222. doi:10.1093/bioinformatics/btv710 | |
Corbett, M. A., Kroes, T., Veneziano, L., Bennett, M. F., Florian, R., Schneider, A. L., et al. (2019). Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 10 (1), 4920. doi:10.1038/s41467-019-12671-y | |
De Fusco, M., Vago, R., Striano, P., Di Bonaventura, C., Zara, F., Mei, D., et al. (2014). The α2B-adrenergic receptor is mutant in cortical myoclonus and epilepsy. Ann. Neurol. 75 (1), 77–87. doi:10.1002/ana.24028 | |
Dixon, J. R., Gorkin, D. U., and Ren, B. (2016). Chromatin domains: The unit of chromosome organization. Mol. Cell 62 (5), 668–680. doi:10.1016/j.molcel.2016.05.018 | |
Dolzhenko, E., Deshpande, V., Schlesinger, F., Krusche, P., Petrovski, R., Chen, S., et al. (2019). ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35 (22), 4754–4756. doi:10.1093/bioinformatics/btz431 | |
Dong, Z., Ye, L., Yang, Z., Chen, H., Yuan, J., Wang, H., et al. (2018). Balanced chromosomal rearrangement detection by low-pass whole-genome sequencing. Curr. Protoc. Hum. Genet. 96, 8 18 11–18. doi:10.1002/cphg.51 | |
Eisfeldt, J., Nilsson, D., Andersson-Assarsson, J. C., and Lindstrand, A. (2018). Amycne: Confident copy number assessment using whole genome sequencing data. PLoS One 13 (3), e0189710. doi:10.1371/journal.pone.0189710 | |
Geoffroy, V., Herenger, Y., Kress, A., Stoetzel, C., Piton, A., Dollfus, H., et al. (2018). AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 34 (20), 3572–3574. doi:10.1093/bioinformatics/bty304 | |
Gianfrancesco, F., Esposito, T., Penco, S., Maglione, V., Liquori, C. L., Patrosso, M. C., et al. (2008). ZPLD1 gene is disrupted in a patient with balanced translocation that exhibits cerebral cavernous malformations. Neuroscience 155 (2), 345–349. doi:10.1016/j.neuroscience.2008.05.030 | |
Hochstenbach, R., Liehr, T., and Hastings, R. J. (2021). Chromosomes in the genomic age. Preserving cytogenomic competence of diagnostic genome laboratories. Eur. J. Hum. Genet. 29 (4), 541–552. doi:10.1038/s41431-020-00780-y | |
Jacobs, P. A., Browne, C., Gregson, N., Joyce, C., and White, H. (1992). Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J. Med. Genet. 29 (2), 103–108. doi:10.1136/jmg.29.2.103 | |
Johnson, N. E., and Ankala, A. (2020). Evolving genetic heterogeneity of facioscapulohumeral muscular dystrophy. Neurology 94 (23), 1011–1012. doi:10.1212/WNL.0000000000009580 | |
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., et al. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30 (1), 61–68. doi:10.1002/humu.20814 | |
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). Lumpy: A probabilistic framework for structural variant discovery. Genome Biol. 15 (6), R84. doi:10.1186/gb-2014-15-6-r84 | |
Lemmers, R. J., van der Vliet, P. J., Klooster, R., Sacconi, S., Camano, P., Dauwerse, J. G., et al. (2010). A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329 (5999), 1650–1653. doi:10.1126/science.1189044 | |
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi:10.1093/bioinformatics/btp698 | |
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352 | |
Liang, D., Wang, Y., Ji, X., Hu, H., Zhang, J., Meng, L., et al. (2017). Clinical application of whole-genome low-coverage next-generation sequencing to detect and characterize balanced chromosomal translocations. Clin. Genet. 91 (4), 605–610. doi:10.1111/cge.12844 | |
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 (5950), 289–293. doi:10.1126/science.1181369 | |
Lukov, G. L., Hu, T., McLaughlin, J. N., Hamm, H. E., and Willardson, B. M. (2005). Phosducin-like protein acts as a molecular chaperone for G protein betagamma dimer assembly. EMBO J. 24 (11), 1965–1975. doi:10.1038/sj.emboj.7600673 | |
Moller, R. S., Kubart, S., Hoeltzenbein, M., Heye, B., Vogel, I., Hansen, C. P., et al. (2008). Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 82 (5), 1165–1170. doi:10.1016/j.ajhg.2008.03.001 | |
Pennekamp, P., Menchen, T., Dworniczak, B., and Hamada, H. (2015). Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia 4 (1), 1. doi:10.1186/s13630-014-0010-9 | |
Pusapati, G. V., Kong, J. H., Patel, B. B., Krishnan, A., Sagner, A., Kinnebrew, M., et al. (2018). CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 44 (1), 113–129. doi:10.1016/j.devcel.2017.12.003 | |
Redin, C., Brand, H., Collins, R. L., Kammin, T., Mitchell, E., Hodge, J. C., et al. (2017). The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 49 (1), 36–45. doi:10.1038/ng.3720 | |
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30 | |
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8 | |
Sarfarazi, M., Wijmenga, C., Upadhyaya, M., Weiffenbach, B., Hyser, C., Mathews, K., et al. (1992). Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: Combined analysis of an international consortium. Am. J. Hum. Genet. 51 (2), 396–403. |
Schatzl, T., Kaiser, L., and Deigner, H. P. (2021). Facioscapulohumeral muscular dystrophy: Genetics, gene activation and downstream signalling with regard to recent therapeutic approaches: An update. Orphanet J. Rare Dis. 16 (1), 129. doi:10.1186/s13023-021-01760-1 | |
Tena, J. J., and Santos-Pereira, J. M. (2021). Topologically associating domains and regulatory landscapes in development, evolution and disease. Front. Cell Dev. Biol. 9, 702787. doi:10.3389/fcell.2021.702787 | |
Thibault, C., Feng Wang, J., Charnas, R., Mirel, D., Barhite, S., and Miles, M. F. (1999). Cloning and characterization of the rat and human phosducin-like protein genes: Structure, expression and chromosomal localization. Biochim. Biophys. Acta 1444 (3), 346–354. doi:10.1016/s0167-4781(99)00006-8 | |
van den Boogaard, M. L., Lemmers, R., Balog, J., Wohlgemuth, M., Auranen, M., Mitsuhashi, S., et al. (2016). Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am. J. Hum. Genet. 98 (5), 1020–1029. doi:10.1016/j.ajhg.2016.03.013 | |
Wang, Y., Song, F., Zhang, B., Zhang, L., Xu, J., Kuang, D., et al. (2018). The 3D genome browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19 (1), 151. doi:10.1186/s13059-018-1519-9 | |
Warburton, D. (1991). De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: Clinical significance and distribution of breakpoints. Am. J. Hum. Genet. 49 (5), 995–1013. |
Keywords: Balanced chromosomal abnormalities, prenatal diagnosis, whole-genome sequencing, CACNA1E, PDCL
Citation: Fu F, Li R, Dang X, Yu Q, Xu K, Gu W, Wang D, Yang X, Pan M, Zhen L, Zhang Y, Li F, Jing X, Li F, Li D and Liao C (2022) Prenatal diagnosis of 21 fetuses with balanced chromosomal abnormalities (BCAs) using whole-genome sequencing. Front. Genet. 13:951829. doi: 10.3389/fgene.2022.951829
Received: 24 May 2022; Accepted: 24 August 2022;
Published: 15 September 2022.
Edited by:
Nallasivam Palanisamy,Henry Ford Health System, United StatesReviewed by:
Anita Kan, Queen Mary Hospital, Hong Kong, SAR ChinaCopyright © 2022 Fu, Li, Dang, Yu, Xu, Gu, Wang, Yang, Pan, Zhen, Zhang, Li, Jing, Li, Li and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Fu, ZnV5aW5neWkyMDA4QDE2My5jb20=; Can Liao, Y2FubGlhbzYwMDhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.