 Xijing Liu
Xijing Liu Jianmin Wang1,2,3
Jianmin Wang1,2,3 Ting Hu
Ting Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 29 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.950271
This article is part of the Research Topic Prenatal Genetic Screening, Diagnosis and Treatment View all 10 articles
Introduction: Campomelic dysplasia (CD) is a rare autosomal dominant skeletal malformation syndrome characterized by shortness and bowing of the lower extremities with or without XY sex reversal. Diagnosis using ultrasonography is most often made in the latter half of pregnancy. Intragenic heterozygous mutations in SOX9 are responsible for most cases of CD. CD caused by SOX9 deletion is a rare condition.
Case presentation: We present a single case report of an individual with cystic hygroma accompanied by CD, which was detected by ultrasound in the first trimester. Chromosomal microarray analysis (CMA) was performed to determine copy number variants, whereas whole exome sequencing (WES) was performed to elucidate single-nucleotide variants. Chorionic villus sampling was performed to enable such analyses. Ultimately, CMA detected a 606 kb deletion in the 17q24.3 region with only one protein-coding gene (SOX9). However, no mutation in the SOX9 protein-coding sequence was detected by WES.
Conclusion: When cystic hygroma is detected, prenatal diagnoses for skeletal dysplasia by ultrasound are likely to be confirmed in the first trimester. We propose a comprehensive prenatal diagnostic strategy that combines CMA and WES to diagnose fetuses with cystic hygroma accompanied by skeletal dysplasia.
Campomelic dysplasia (CD, MIM: #114290) is a rare autosomal dominant skeletal malformation syndrome characterized by an under-mineralized skeleton with shortness and bowing of lower extremities and hypoplasia of scapular and pelvic bones. Moreover, other notable anatomical characteristics include a small chest, 11 pairs of ribs, clubfeet, a cleft palate, and micrognathia (Mansour et al., 1995; Jain and Sen, 2014). Two-thirds of affected males showed XY sex reversal or a lesser degree of genital defects (Mansour et al., 1995). CD is a semi-lethal disorder because most patients die during the neonatal period secondary to respiratory distress (Mansour et al., 2002).
SOX9 (SRY-related HMG-box gene 9, OMIM:608160), located in chromosome 17q24.3, is a transcription factor that plays a critical role in the development of skeletal and reproductive systems by inducing chondrocyte differentiation and anti-Mullerian hormone expression (Wagner et al., 1994; Olney et al., 1999). Most cases of CD are caused by intragenic heterozygous mutations in SOX9, including missense, nonsense, frameshift, and splice mutations (Unger et al., 1993). A second mechanism for CD is balanced chromosomal rearrangements, including translocations, inversions, and deletions, which do not affect SOX9 but cause interruptions upstream or downstream of SOX9 (Unger et al., 1993; Gordon et al., 2009; Lecointre et al., 2009). Additionally, a few CDs are caused by large deletions covering SOX9 (Olney et al., 1999; Pop et al., 2004; Smyk et al., 2007; Kayhan et al., 2019).
CD might be suspected after shortened lower limbs and femoral angulation are detected by prenatal ultrasonography, although observations are often made in the latter half of pregnancy. Thus, antenatal diagnosis of CD is difficult during the first trimester (Tongsong et al., 2000). We report an individual with cystic hygroma accompanied by CD in the first trimester and confirm that such a phenotype resulted from SOX9 deletion.
A 27-year-old nulliparous woman was referred to the Prenatal Diagnosis Center of West China Second University Hospital of Sichuan University (a tertiary referral center) for consultation and sonography concerning a cystic hygroma discovered at 13 weeks and 4 days. The crown-rump length was 60.9 mm and a cystic hygroma measuring 18 × 6 × 14 mm was confirmed (Figures 1A,B). The couple has no history of consanguinity. At 14 weeks and 6 days, a detailed fetal anomaly scan revealed severe short and bowed femurs (<first centile), sagittal anterior-angulated tibiae and fibula (fifth centile), unilateral equinovarus clubfoot, and micrognathia (Figures 1C–G). The biparietal diameter was 27.9 mm, while the thorax and upper limbs appeared normal. The fetus was found to have female external genitalia. A presumptive diagnosis of skeletal dysplasia was made. However, a precise diagnosis was difficult to achieve based on limited ultrasound findings. Chorionic villus sampling was performed to enable chromosomal microarray analysis (CMA) and whole exome sequencing (WES).
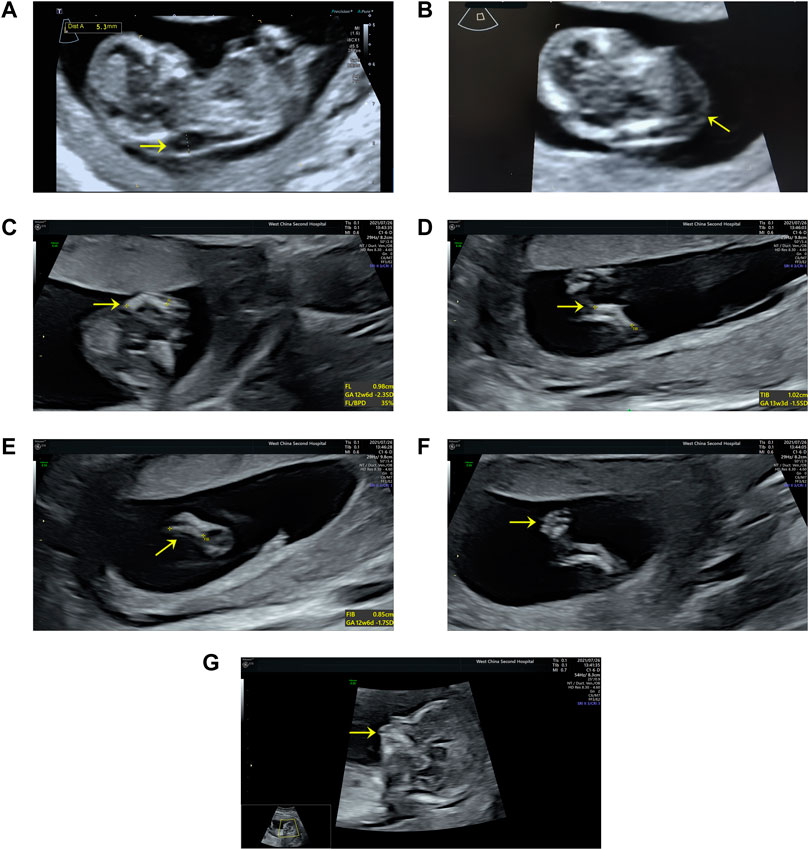
FIGURE 1. Ultrasound findings: (A) Cystic hygroma measuring 18 × 6 × 14 mm (CRL:60.9 mm) at the mid-sagittal plane. (B) Cystic hygroma in a transverse view. (C) Short and bowed femurs (<first centile). (D) Sagittal anterior angulated tibiae (fifth centile). (E) Sagittal anterior angulated fibula (fifth centile). (F) Unilateral equinovarus clubfoot. (G) Micrognathia.
The study was approved by the Medical Ethics Committee of the West China Second University Hospital of Sichuan University in China.
We conducted the chromosomal microarray analysis using CytoScan 750 K Array (Affymetrix, Santa Clara, CA, United States). The samples were prepared according to the manufacturer’s instructions. The array scan data were visualized using the Chromosome Analysis Suite v4.1 software. The GRCh38 genome was used for annotation. The pathogenicity of copy number variants (CNVs) was based on the technical standards of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) (Riggs et al., 2020).
The Nano WES Human Exome V1 (Berry Genomics, Beijing, China) was used to capture the sequences. The enriched library was sequenced on a NovaSeq 6,000 with 150 paired-end reads. The reads were mapped to a human reference genome (GRCh38) using BWA (v0.7.15). Variant calling was performed using the Verita Trekker (v1.2.0.2). After filtering the variants with the classic population frequency databases, we rated the pathogenicity of the remaining mutations according to the ACMG guidelines, as previously described (Chen et al., 2022).
CMA was performed following chorionic villus sampling, which revealed a 606 kb deletion [arr [GRCh38] 17q24.3 (71,620,225_72,226,459)x1] in the 17q24.3 region (Figure 2A). The deletion contained one protein-coding gene (SOX9) and five other genes (LINC01152, LINC02097, MYL6P5, ROCR, and SOX9-AS1), which extended from 501 kb upstream to 100 kb downstream of SOX9 (Figure 2B). The deletion overlapped only 9.3% of the SOX9 upstream enhancer region, which is identified as a haploinsufficiency (HI) genomic region (ClinGen database (http://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/)). The sex chromosome of the fetus was XX. This deletion was not detected in the couple, suggesting that a de novo event had occurred. The CNVs were classified as “Pathogenic” according to ACMG and ClinGen Technical standards (Riggs et al., 2020). Sequentially, a trio-WES was performed for genetic factors of skeletal dysplasia in addition to SOX9 deletion, and yet the analysis yielded negative results.
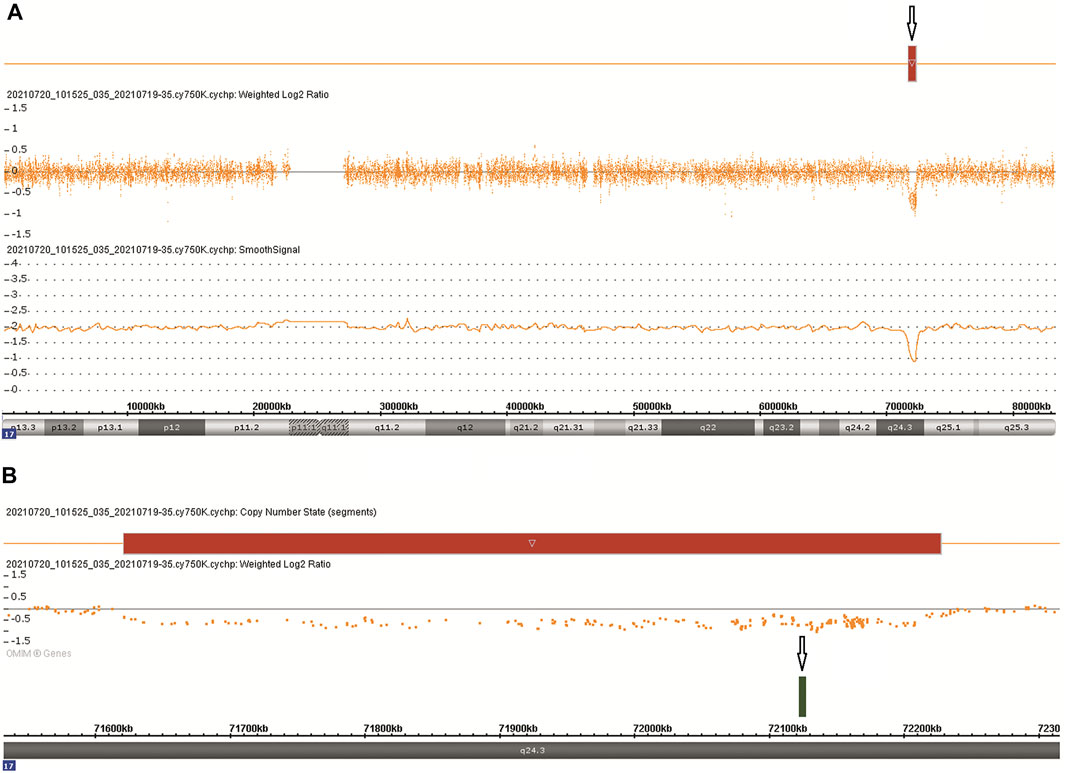
FIGURE 2. Chromosomal microarray findings: (A) A 606 kb deletion in the 17q24.3 region. (B) The deletion contains one OMIM gene SOX9.
The diagnosis of CD was confirmed based on the SNP-array results and fetal ultrasound findings.
We report a case of CD with cystic hygroma detected in the first trimester by ultrasonography, and in which further testing confirmed the presence of SOX9 deletion. Commonly, prenatal diagnosis of skeletal dysplasia is performed by ultrasonography during the second trimester. Recently, Kayhan et al. (2019) reported a 46, XY prenatal case of CD with SOX9 deletion, characterized by micrognathia, bowed limbs, clubfeet, and female genitalia in the second trimester. However, ultrasound in the first trimester appeared normal, and no apparent anomalies, such as cystic hygroma or increased nuchal translucency, were observed. However, a cystic hygroma or increased nuchal translucency during the first 3 months has been reported to be a relatively frequent sign in fetuses subsequently diagnosed with skeletal dysplasia (Massardier et al., 2008; Zhen et al., 2015). As in our case, a detailed fetal anomaly scan was performed sequentially when cystic hygroma was detected, and skeletal dysplasia was confirmed. Kenkhuis et al. (2018) concluded that it is possible to detect approximately half of the prenatally detectable structural anomalies with an early scan performed in the first trimester by competent fetal sonographers. Thus, a detailed ultrasonography should be immediately performed when cystic hygroma or increased nuchal translucency is detected in the first trimester.
CD is an autosomal dominant disease usually caused by a heterozygous pathogenic variant of SOX9 (Unger et al., 1993). Most CD cases show heterozygous de novo mutations in the coding region of SOX9. A small number of cases have a heterozygous interstitial deletion or reciprocal translocation of 17q24.3-q25.1, which involves SOX9 or its regulatory region (Unger et al., 1993; Gordon et al., 2009; Lecointre et al., 2009). Currently, the haploinsufficiency of SOX9 is disputable. In the last few decades, loss-of-function mutations in one of the three SOX9 exons have been identified in CD. Therefore, it has been assumed that the disease results from the haploinsufficiency of SOX9 (Unger et al., 1993; Ninomiya et al., 2000). However, the research conducted by Csukasi et al. (2019) suggested a dominant-negative nature to SOX9 mutations in CD. As isolated SOX9 deletion is rare, the evidence for SOX9 haploinsufficiency is insufficient.
Currently, only four CD cases have been reported with the complete deletion of the SOX9 gene (Olney et al., 1999; Pop et al., 2004; Smyk et al., 2007; Kayhan et al., 2019). Olney et al. (1999) first reported an infant with CD who has an interstitial deletion from 17q23.3 to q24.3 by karyotyping, whereas Pop et al. (2004) reported a male CD patient with a de novo deletion of at least 4.0 Mb, and Smyk et al. (2007) reported a female CD patient with a paternally inherited deletion of ∼4.7 Mb in size. Interestingly, all the three cases completely overlapped the SOX9 upstream enhancer region that is positioned 2 Mb 5’ to SOX9, and which is associated with the Pierre Robin sequence (PRS, MIM #261800). PRS, a partial phenotype of CD, is characterized by micrognathia, cleft palate, and glossoptosis (Lecointre et al., 2009). Thus, the evidence of haploinsufficiency in SOX9 deletion-induced CD remains elusive. Kayhan et al. (2019) performed a prenatal array comparative genomic hybridization on a 46, XY CD fetus and revealed an 886 kb deletion in the 17q24.3 region, which included the entire SOX9 gene. Although the deletion also overlapped approximately 29.3% of the SOX9 upstream enhancer region, it is difficult to confirm that SOX9 deletion is solely responsible for the CD phenotype.
The genotype–phenotype correlation of SOX9 remains unclear. Although loss-of-function mutations have been identified in some CD cases, functional studies suggest a dominant-negative phenotype (Wagner et al., 1994; Ninomiya et al., 2000; Jain and Sen, 2014; Csukasi et al., 2019). The advent of CMA has revealed CNVs, including both the coding and regulatory regions of SOX9 (Croft et al., 2018). The utility of WES elucidated single-nucleotide variants of genetic disorders that were not detectable by CMA. However, deletions or duplications associated with cis-regulatory elements upstream of SOX9 were not detected by the algorithms (Mansour et al., 2002). Therefore, we performed CMA combined with WES as an unbiased approach when cystic hygroma and skeletal dysplasia were detected. In our case, the phenotype of the fetus revealed by ultrasonography was consistent with the loss of SOX9, providing an increasing evidence of SOX9 haploinsufficiency.
With the advancements in molecular detection technology, the prenatal diagnosis of fetuses at risk for genetic disorders has rapidly increased in recent years. The choice of application of CMA and WES deserves consideration. When a fetal anomaly is observed, we support the recommendations released by the International Society for Prenatal Diagnosis (Van den Veyver et al., 2022), which contends that, if no genetic diagnosis is found after CMA, a fetus with a major single anomaly or multiple organ system anomalies will benefit from WES or other genome sequencing methods. If the anomaly “pattern” strongly suggests a single genetic disorder with no prior genetic testing, CMA should be run before or parallel to WES. In this case, a cystic hygroma was first observed, and CMA was the prior genetic test. After a detailed ultrasonography, CD was highly suspected, and whether WES could have been a prior genetic test is worth considering, as CD is a disorder mainly caused by SOX9 heterozygous de novo mutations.
In clinical practice, when a cystic hygroma or increased nuchal translucency is detected using a detailed ultrasonography, prenatal diagnoses for skeletal dysplasia are likely to be confirmed in the first trimester. Since a meta-analysis has found that the highest exome sequencing diagnostic yields occurred in fetuses with skeletal abnormalities (53% [95% CI 42–63%], p < 0.0001) (Mellis et al., 2022), the prenatal diagnostic strategy for fetuses with cystic hygroma accompanied by skeletal dysplasia requires more clinical experience.
After receiving the genetic and ultrasound reports, diagnosis was established, and the couple decided to terminate the pregnancy. The couple provided written informed consent to participate in this study. Written informed consent was obtained from each individual for the publication of potentially identifiable images or data included in this article.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of the West China Second University Hospital, Sichuan University, China. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XL wrote the manuscript and analyzed the CMA data. JW collected the information of the patient and performed the CMA experiment. MY performed the WES experiment and analyzed the WES data. TT performed the ultrasound examination and revised the manuscript. TH designed the research and revised the manuscript.
This work was supported by the National Key Research and Development Program of China (2021YFC1005304) and Sichuan Province Science and Technology Support Program (2022YFS0078).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Chen, J., Xiang, Q., Xiao, X., Xu, B., Xie, H., Wang, H., et al. (2022). Carrying both COL1A2 and FBN2 gene heterozygous mutations results in a severe skeletal clinical phenotype: an affected family. BMC Med. Genomics 15 (1), 154. Epub 2022/07/09 PubMed PMID: 35804365; PubMed Central PMCID: PMCPMC9270787. doi:10.1186/s12920-022-01296-8
Croft, B., Ohnesorg, T., and Sinclair, A. H. (2018). The role of copy number variants in disorders of sex development. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation. Sex. Dev. 12 (1-3), 19–29. Epub 2017/11/18 PubMed PMID: 29145200. doi:10.1159/000481896
Csukasi, F., Duran, I., Zhang, W., Martin, J. H., Barad, M., Bamshad, M., et al. (2019). Dominant-negative SOX9 mutations in campomelic dysplasia. Hum. Mutat. 40 (12), 2344–2352. Epub 2019/08/08 PubMed PMID: 31389106; PubMed Central PMCID: PMCPMC7608528. doi:10.1002/humu.23888
Gordon, C. T., Tan, T. Y., Benko, S., Fitzpatrick, D., Lyonnet, S., and Farlie, P. G. (2009). Long-range regulation at the SOX9 locus in development and disease. J. Med. Genet. 46 (10), 649–656. Epub 2009/05/29 PubMed PMID: 19473998. doi:10.1136/jmg.2009.068361
Jain, V., and Sen, B. (2014). Campomelic dysplasia. J. Pediatr. Orthop. B 23 (5), 485–488. Epub 2014/05/08 PubMed PMID: 24800790. doi:10.1097/bpb.0000000000000058
Kayhan, G., Calis, P., Karcaaltincaba, D., and Tug, E. (2019). Prenatal diagnosis of campomelic dysplasia due to SOX9 deletion. J. Obstet. Gynaecol. 39 (8), 1175–1176. Epub 2019/06/27 PubMed PMID: 31234679. doi:10.1080/01443615.2019.1601165
Kenkhuis, M. J. A., Bakker, M., Bardi, F., Fontanella, F., Bakker, M. K., Fleurke-Rozema, J. H., et al. (2018). Effectiveness of 12-13-week scan for early diagnosis of fetal congenital anomalies in the cell-free DNA era. Ultrasound Obstet. Gynecol. 51 (4), 463–469. Epub 2017/04/12 PubMed PMID: 28397377. doi:10.1002/uog.17487
Lecointre, C., Pichon, O., Hamel, A., Heloury, Y., Michel-Calemard, L., Morel, Y., et al. (2009). Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am. J. Med. Genet. A 149a (6), 1183–1189. Epub 2009/05/19 PubMed PMID: 19449405. doi:10.1002/ajmg.a.32830
Mansour, S., Hall, C. M., Pembrey, M. E., and Young, I. D. (1995). A clinical and genetic study of campomelic dysplasia. J. Med. Genet. 32 (6), 415–420. Epub 1995/06/01 PubMed PMID: 7666392; PubMed Central PMCID: PMCPMC1050480. doi:10.1136/jmg.32.6.415
Mansour, S., Offiah, A. C., McDowall, S., Sim, P., Tolmie, J., and Hall, C. (2002). The phenotype of survivors of campomelic dysplasia. J. Med. Genet. 39 (8), 597–602. PubMed PMID: 12161603; PubMed Central PMCID: PMCPMC1735206. doi:10.1136/jmg.39.8.597
Massardier, J., Roth, P., Michel-Calemard, L., Rudigoz, R. C., Bouvier, R., Dijoud, F., et al. (2008). Campomelic dysplasia: echographic suspicion in the first trimester of pregnancy and final diagnosis of two cases. Fetal diagn. Ther. 24 (4), 452–457. Epub 2008/11/27 PubMed PMID: 19033726. doi:10.1159/000176299
Mellis, R., Oprych, K., Scotchman, E., Hill, M., and Chitty, L. S. (2022). Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 42 (6), 662–685. Epub 2022/02/17 PubMed PMID: 35170059. doi:10.1002/pd.6115
Ninomiya, S., Yokoyama, Y., Teraoka, M., Mori, R., Inoue, C., Yamashita, S., et al. (2000). A novel mutation (296 del G) of the SOX90 gene in a patient with campomelic syndrome and sex reversal. Clin. Genet. 58 (3), 224–227. Epub 2000/11/15 PubMed PMID: 11076045. doi:10.1034/j.1399-0004.2000.580310.x
Olney, P. N., Kean, L. S., Graham, D., Elsas, L. J., and May, K. M. (1999). Campomelic syndrome and deletion of SOX9. Am. J. Med. Genet. 84 (1), 20–24. Epub 1999/04/23. PubMed PMID: 10213041. doi:10.1002/(sici)1096-8628(19990507)84:1<20::aid-ajmg5>3.0.co;2-n
Pop, R., Conz, C., Lindenberg, K. S., Blesson, S., Schmalenberger, B., Briault, S., et al. (2004). Screening of the 1 Mb SOX9 5' control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J. Med. Genet. 41 (4), e47. Epub 2004/04/03 PubMed PMID: 15060123; PubMed Central PMCID: PMCPMC1735745. doi:10.1136/jmg.2003.013185
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of medical genetics and genomics (ACMG) and the clinical genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. Epub 2019/11/07 PubMed PMID: 31690835; PubMed Central PMCID: PMCPMC7313390. doi:10.1038/s41436-019-0686-8
Smyk, M., Obersztyn, E., Nowakowska, B., Bocian, E., Cheung, S. W., Mazurczak, T., et al. (2007). Recurrent SOX9 deletion campomelic dysplasia due to somatic mosaicism in the father. Am. J. Med. Genet. A 143a (8), 866–870. Epub 2007/03/14 PubMed PMID: 17352389. doi:10.1002/ajmg.a.31631
Tongsong, T., Wanapirak, C., and Pongsatha, S. (2000). Prenatal diagnosis of campomelic dysplasia. Ultrasound Obstet. Gynecol. 15 (5), 428–430. Epub 2000/09/08 PubMed PMID: 10976487. doi:10.1046/j.1469-0705.2000.00126.x
Unger, S., Scherer, G., and Superti-Furga, A. (1993). “Campomelic dysplasia,” in GeneReviews(®). M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and G. Mirzaa. Editors (Seattle (WA): University of Washington, Seattle).
Van den Veyver, I. B., Chandler, N., Wilkins-Haug, L. E., Wapner, R. J., and Chitty, L. S.ISPD Board of Directors (2022). International society for prenatal diagnosis updated position statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat. Diagn. 42 (6), 796–803. Epub 2022/05/19 PubMed PMID: 35583085. doi:10.1002/pd.6157
Wagner, T., Wirth, J., Meyer, J., Zabel, B., Held, M., Zimmer, J., et al. (1994). Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79 (6), 1111–1120. Epub 1994/12/16 PubMed PMID: 8001137. doi:10.1016/0092-8674(94)90041-8
Keywords: campomelic dysplasia, cystic hygroma, SOX9, ultrasound, chromosomal microarray analysis
Citation: Liu X, Wang J, Yang M, Tian T and Hu T (2022) Case report: Cystic hygroma accompanied with campomelic dysplasia in the first trimester caused by haploinsufficiency with SOX9 deletion. Front. Genet. 13:950271. doi: 10.3389/fgene.2022.950271
Received: 22 May 2022; Accepted: 29 July 2022;
Published: 29 August 2022.
Edited by:
Walter Erwin Kaufmann, Emory University, United StatesReviewed by:
Xiaofan Zhu, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2022 Liu, Wang, Yang, Tian and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Tian, MTU4MTYzOTIxQHFxLmNvbQ==; Ting Hu, aHV0aW5nNDEyM0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.