 Xianxian Liu
Xianxian Liu Jiusheng Zheng
Jiusheng Zheng Siming Xin
Siming Xin- Key Laboratory of Women’s Reproductive Health of Jiangxi Province, Jiangxi Provincial Maternal and Child Health Hospital, Nanchang, Jiangxi, China
Background: Intrahepatic cholestasis of pregnancy (ICP) is associated with a high incidence of fetal morbidity and mortality. Therefore, revealing the mechanisms involved in ICP and its association with fetal complications is very important.
Methods: Here, we used a whole-exome sequencing (WES) approach to detect novel mutations of organic anion transporting polypeptide (OTAP) genes, ATP-binding cassette transporter (ABC) genes, and receptor genes associated with ICP in 249 individuals and 1,029 local control individuals. Two available tools, SIFT and PolyPhen-2, were used to predict protein damage. Protein structuremodeling and comparison between the reference and modified protein structures were conducted by SWISS-MODEL and Chimera 1.14rc software, respectively.
Results: A total of 5,583 mutations were identified in 82 genes related to bile acid transporters and receptors, of which 62 were novel mutations. These novel mutations were absent in the 1,029 control individuals and three databases, including the 1,000 Genome Project (1000G_ALL), Exome Aggregation Consortium (ExAC), and Single-Nucleotide Polymorphism Database (dbSNP). We classified the 62 novel loci into two groups (damaging and probably damaging) according to the results of SIFT and PolyPhen-2. Out of the 62 novel mutations, 24 were detected in the damaging group. Of these, five novel possibly pathogenic variants were identified that were located in known functional genes, including ABCB4 (Ile377Asn), ABCB11 (Ala588Pro), ABCC2 (Ile681Lys and Met688Thr), and NR1H4 (Tyr149Ter). Moreover, compared to the wild-type protein structure, ABCC2 Ile681Lys and Met688Thr protein structures showed a slight change in the chemical bond lengths of ATP-ligand binding amino acid side chains. The combined 32 clinical data points indicate that the mutation group had a significantly (p = 0.04) lower level of Cl ions than the wild-type group. Particularly, patients with the 24 novel mutations had higher average values of alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), total bile acids (TBA), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) than patients with the 38 novel mutations in the probably damaging group and the local control individuals.
Conclusion: The present study provides new insights into the genetic architecture of ICP involving these novel mutations.
Introduction
Intrahepatic cholestasis of pregnancy (ICP) is the most common pregnancy-specific liver disease, and it is characterized by skin pruritus, predominantly on the palms and soles and elevated levels of total serum bile acids (≥10 μmol/L) and liver enzymes (Ovadia and Williamson, 2016). This disease typically commences in the late second or third trimester of pregnancy and resolves rapidly after delivery in the early postpartum period (Rook et al., 2012; Puljic et al., 2015). The incidence of ICP was reported to range from 0.2 to 15.6% depending on ethnicities and geographic locations (Stulic et al., 2019). In particular, the incidence rate is higher in the Scandinavian, Bolivian, and Chilean populations (Reyes et al., 1979; Saleh and Abdo, 2007; Rook et al., 2012; Puljic et al., 2015; Simjak et al., 2015). The recurrence rate of ICP during the next pregnancy can be as high as 40–60% (Ovadia and Williamson, 2016).
ICP is linked with a higher risk of spontaneous preterm birth, fetal asphyxia, meconium-stained amniotic fluid, low Apgar scores, and fetal death (Glantz et al., 2004; Geenes et al., 2014; Williamson and Geenes, 2014). Many researchers found that pregnant women with serum bile acid levels of 100 μmol/L or more have a significantly increased risk of stillbirth (Kawakita et al., 2015; Ovadia et al., 2019). In addition, ICP is associated with a disrupted metabolic profile and causes metabolic disease. Thus, it has long-term consequences for the health of the mother and child (Papacleovoulou et al., 2013; Wikstrom Shemer et al., 2015). Therefore, understanding the underlying genetic factors of ICP disease is of great significance to patient diagnosis and treatment.
The etiology of ICP is complex and not fully understood. The pathogenesis of ICP is multifactorial and is related to the interactions among genetic, hormonal, immunological, and environmental factors (Sampson, 1927; Xiao et al., 2021). The familial clustering analysis in pedigree studies showed a high incidence of these factors in the mothers and sisters of patients with ICP. There are population-specific risk differences and a high recurrence rate in these individuals, which might indicate the importance of genetic components in ICP (Reyes et al., 1976; Eloranta et al., 2001; Dixon and William, 2008). Previous studies have demonstrated that OTAP genes, ABC genes, and receptor genes play a pivotal role in the control of bile acid homeostasis (Van Mil et al., 2007; Kakizaki et al., 2011; Yan et al., 2015; Ahmad and Haeusler, 2019; Turro et al., 2020). Yan et al., 2015 confirmed the role of SLCO1B3 in bile acid transport. Ontsouka et al., 2021 reported that placental SLC10A2, SLCO4A1, and ABCC2 mRNA levels were positively correlated with bile acid concentrations in ICP patients (). Therefore, considering that women with ICP exhibited elevated serum bile acid levels and that OTAP abnormalities can result in altered bile acid levels, we speculated that mutations in OTAP genes might be present in women with ICP. In addition, mutations in ABCB4, ABCB11, and FXR were confirmed to be associated with ICP disease (Van Mil et al., 2007; Castano et al., 2010; Dixon et al., 2014; Dixon et al., 2017; Turro et al., 2020; Wolski et al., 2020). To the best of our knowledge, until now, most studies have mainly focused on functionally known genes, such as ABCB4, ABCB11, ABCC2, FXR, ATP8B1, and TJP2, that confer susceptibility to ICP (Dixon et al., 2014; Dixon et al., 2017). However, the roles of other bile acid receptor genes, such as CAR, AhR, VDR, and PXR, seem to be less studied. Accumulating evidence suggests that the role of these receptor genes as promising drug targets in cholestasis is now emerging since they play a crucial role in the regulation of bile acid synthesis, detoxification, and transport (Ahmad and Haeusler, 2019). However, to date, only a few receptor mutations associated with ICP disease have been identified (Van Mil et al., 2007; Castano et al., 2010; Lai et al., 2022). Therefore, it is necessary to systematically identify ICP-associated bile acid transporter and receptor gene susceptibility mutations.
In past decades, most of the genetic mutations have been found in families or sequenced in a limited number of individuals with sporadic ICP, and this makes the identification of rare variants challenging. However, the 1000 Genomes Project revealed that rare variants constitute the majority of polymorphic sites in human populations and have higher effect sizes on human diseases than common variants (Genomes Project et al., 2012; Wang et al., 2021). Fortunately, by taking advantage of high-throughput sequencing in a large population, whole-genome sequencing (WGS)/WES has been shown to be a useful and efficient tool for identifying potentially pathogenic rare mutations in rare diseases, such as ICP (Turro et al., 2020). Previously, we performed WES to identify the novel gene ANO8 as a gene risk factor for ICP and novel mutations in ABC transporter genes that were associated with ICP disease in 151 ICP samples (Liu et al., 2020; Liu et al., 2021). We would like to further conduct analyses of comprehensive sequencing of the bile acid transporter and receptor gene series in a larger cohort of ICP cases.
Given the aforementioned background, we aimed to systematically identify mutations associated with bile acid transporters and receptor genes in 249 ICP individuals. Our particular focus will be on new functional mutations and their relationship with clinical data and pregnancy outcomes in this study.
Materials and methods
Patients and clinical data
We recruited a total of 249 pregnant women who did not have other liver diseases and were diagnosed with ICP disease between 2018 and 2022. These peripheral blood samples were collected from the Department of Obstetrics, Jiangxi Provincial Maternal and Child Health Hospital in Nanchang, China. Additionally, thirty-five available clinical characteristics, including basic patient information, were recorded. This basic information included the age at diagnosis, body mass index (BMI), gestational age, gravidity, and parity. It also included the four main serum biochemical indexes of maternal and neonatal data, such as the ion concentrations of K, Na, Cl, Ca, P, and Mg. The basic information also included routine blood tests, such as white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts and red blood cell distribution width SD (RDW-SD), and liver function indexes, including ALT, AST, γ-glutamyl transpeptidase (GGT), ALP, TBA, cholyglycine (CG), total bilirubin (TBIL), direct bilirubin (DBIL), and indirect bilirubin (IDBIL) levels. Additionally, it included lipid-related indices, including total cholesterol (CHOL), triglyceride (TG), HDL, LDL, and uric acid (UA) levels, and outcomes of pregnant women and newborn babies, including newborn birth weight, Apgar score, bleeding amount, preterm birth, meconium staining amniotic fluid (MSAF), and intrauterine fetal death. The measurement of these blood biochemical variables can be found in our previous studies (Liu et al., 2020; Liu et al., 2021). In brief, the routine blood tests were determined by a Sysmex-xn-2000 automated blood cell analyzer (Sysmex Corporation, Japan). The other serum biochemical indices were examined by an AU5800 automatic biochemical analyzer (Beckman Coulter, Inc., United States). A summary of the statistics for all the clinical features in 249 samples is shown in Table 1. The clinical data from 151 of these samples were described in our previous study (Liu et al., 2020; Liu et al., 2021). Each participating woman gave written informed consent.
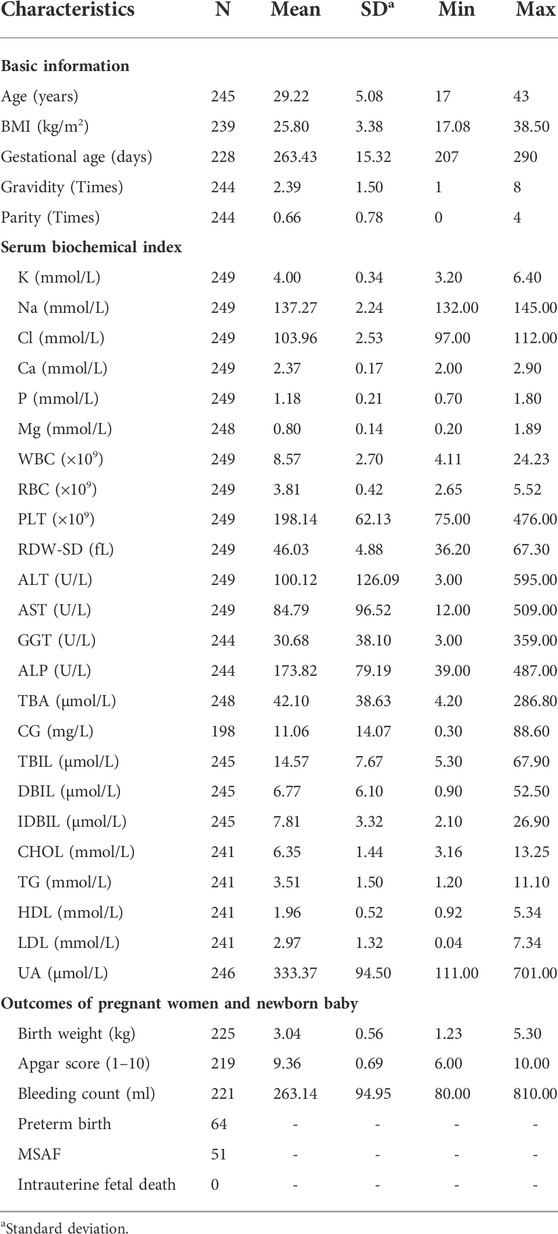
TABLE 1. Descriptive statistics of thirty-five clinical characteristics of 249 ICP patients.
Whole-exome sequencing
A total of 249 genomic DNA samples were extracted from peripheral blood by an Axy Prep Blood Genomic DNA Mini Prep Kit (Item No. 05119KC3, Axygen Scientific, Inc., Union City, CA, United States) and dissolved in Tris-EDTA buffer. The quality and concentrations of the DNA samples were determined by a Nanodrop-1000 spectrophotometer (Thermo Fisher, United States). The integrity of the DNA samples was examined by gel electrophoresis. After the quality control analysis, a total of 249 qualified DNA samples were subjected to exome sequencing following the standard manufacturer’s protocol. The detailed procedures were conducted as previously described (Liu et al., 2020; Liu et al., 2021). Briefly, 1 µg of genomic DNA was randomly sheared into short fragments (150–250 bp) using Covaris technology (Woburn MA). The prepared DNA fragments were amplified by ligation-mediated PCR, purified, and hybridized to the BGI Exon Kit V4 (BGI, Shenzhen, China) for enrichment. The enriched library was then loaded on BGISEQ-500 (BGI, Shenzhen, China) platforms and subjected to high-throughput sequencing. Additionally, whole-exome sequencing was also conducted in 1,029 control individuals. These control samples without ICP or liver disease were recruited as negative control individuals in the same periods. We collected these local control individuals to compare the candidate locus frequency differences between cases and local control individuals.
Variant calling, annotations, filtering, and prioritization
The bioinformatics analysis was also previously described (Liu et al., 2020; Liu et al., 2021). In the aggregate, reads from the BGISEQ-500 machine containing sequencing adapters and low-quality reads (read depth <15 and genotype quality score <20) were removed. The clean reads were aligned to the human reference genome (UCSC Genome Browser GRCh37/hg19) using Burrows–Wheeler Aligner (BWA) software (Li and Durbin, 2009). Then, variant calls and annotations were conducted by the genome Analysis Toolkit (GATK) and ANNOVAR tool (Mckenna et al., 2010; Wang et al., 2010), respectively. After that, we removed variants with a minor allele frequency (MAF) > 0.05 in the 1,029 control individuals, 1000G_ALL (http://www.internationalgenome.org/), ExAC (http://exac.broadinstitute.org/), and dbSNP (https://www.ncbi.nlm.nih.gov/snp/) databases. Then, variants, such as missense, nonsense, loss or gain of function mutations of OTAP genes, ABC genes, and receptor genes, were included in the subsequent analysis. In particular, we concentrated on novel variants that were filtered by National Center for Biotechnology Information (NCBI) and Ensembl. Additionally, SIFT and PolyPhen-2 tools were applied to annotate and predict the functions of the mutation variants. These were prioritized with attention given to novel variants that would likely have functional effects. For instance, a variant was highlighted when it was a loss- or gain-of-function variant or predicted to be simultaneously deleterious by SIFT and PolyPhen-2 software. According to the prediction results, predictions were defined as damaging when the two prediction software results both reached the damaging level. The other variants were assigned to the probably damaging group.
Statistical analysis
We performed the sapply function to calculate descriptive statistics for 35 clinical data points. The t test method was performed to analyze the potential significant differences between gene mutations and wild-type genotypes for clinical characteristics. The result was considered significant when the p value was below 0.05. Fisher’s test was used to test the significance of the frequencies of the 249 ICP patients and 1,029 control individuals. In addition, comparisons of the average value differences in six clinical parameters, including ALT, AST, ALP, TBA, HDL, and LDL, among the three groups were performed by one-way ANOVA. The pie function was used to draw the percentages of the types of OTAP, ABC, and receptor gene mutations. Logistic regression analysis was performed to assess the relationship between the clinical parameters (age, gestational age, BMI, gravidity, and parity) and the mutations. All the aforementioned analyses were performed by R software.
Sanger sequencing
To validate putative mutations, Sanger sequencing was conducted. We selected 12 interesting novel mutations from WES analyses to be confirmed. The twelve mutations were selected for sequencing mainly based on functional prediction results (damaging group) and the classification of the members of the OTAP, ABC, and receptor gene superfamilies. The primers flanking the 12 candidate loci were designed based on the reference genome of the human genome from GenBank in NCBI using Primer Premier 5 software. The details of the PCR primers, their optimum annealing temperatures, and the fragment lengths of the amplified products are shown in Supplementary Table S1.
Evolutionary conservation analysis
The evolutionary conservation analysis of the amino acids encoded by the new functional sites in OTAP genes, ABC genes, and receptor genes in the damaging group was performed among vertebrates, including gibbons, gorillas, goats, macaques, mice, cats, cows, horses, pigs, and sheep, using genomic alignments of the Ensembl Genome Browser.
Protein structure modeling
There are two steps to complete the protein structure modeling. First, the reference and modified (ABCC2 Ile681Lys and Met688Thr) protein sequences were submitted to SWISS-MODEL (https://swissmodel.expasy.org/) to build the model. Then, the reference and modified protein models were compared simultaneously using the Chimera 1.14rc package.
Results
Clinical presentation
During the study period, 249 cases of ICP were diagnosed. Of these 249 women, fourteen women (5.62%, 14/249) had >1 ICP-affected pregnancy. Among them, two had a history of ICP stillbirths. One and 51 pregnant women presented with fetal distress and MSAF, respectively. In addition, 227 women of the 249 sampled women delivered their babies. Out of the 227 women, 173 individuals (76.21%, 173/227) gave birth by cesarean section, 53 (23.35%, 53/227) gave birth by vaginal delivery, and one (0.44%, 1/227) gave birth by vaginal delivery with forceps. Sixty-four (28.44%, 64/225) babies were born prematurely, and 32 (14.22%, 32/225) babies weighed less than 2.5 kg. No indications of fetal death were observed in utero.
Whole-exome sequencing results of the variants of organic anion transporting polypeptide, ATP-binding cassette transporter, and receptor genes in 249 intrahepatic cholestasis of pregnancy samples
To survey the novel possible pathogenic variants of OTAP genes, ABC genes, and receptor genes in ICP, we sequenced whole exomes from 249 individuals in the study cohort. In total, we identified 5,583 genetic variants in 82 genes (Supplementary Table S2) that were associated with bile acid transporters and receptors. These included 874 variants in 16 OTAP genes, 4,079 variants in 44 ABC genes, 483 variants in 20 receptors, and 147 variants in the ATP8B1 and TJP2 genes (Figure 1A). These types of variants included 3,854 intron variants, 735 missense variants, 531 synonymous variants, 185 5/3 prime UTR variants, 135 splice variants, 106 upstream/downstream gene variants, and 29 start lost/stop gained variants, and 8 structural interaction variants. The percentage of these types of variants is shown in Figure 1B. When MAF was controlled at 0.05, a total of 449 variants were conserved for further analysis (Supplementary Table S3, Table 2). Out of these 449 variants, 62 were novel variants. These included 13 novel variants in OTAP genes, 36 novel variants in ABC genes, 9 novel variants in receptor genes, and 2 novel variants in the ATP8B1 and TJP2 genes (Table 2).

FIGURE 1. The distribution and numbers of genes and genetic variants from the WES data for ICP. (A) The total number of genes, identified SNPs, and novel SNPs in OTAP genes, ABC genes, receptor genes, and other genes are summarized. (B) The percentage of the types of genetic variants in the four gene series.
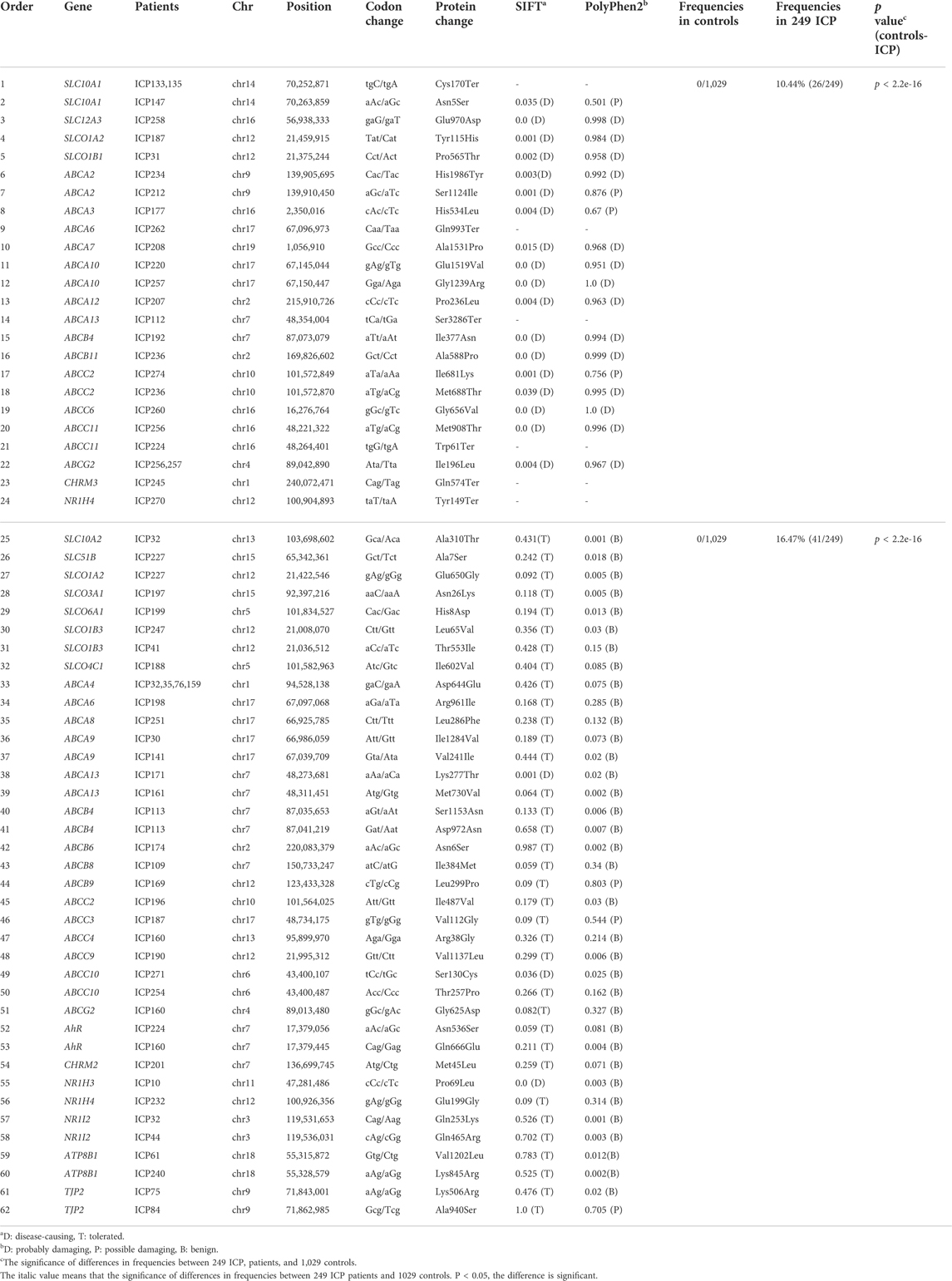
TABLE 2. Sixty-two novel mutations were identified in 249 Han Chinese patients with ICP disease.
These 62 novel mutations were divided into two groups, damaging and probably damaging, according to the prediction results (Table 2). The damaging group had 24 novel mutations, including five in OTAPs, seventeen in ABC genes, and two in receptor genes. For OTAP genes, five novel variants (SLC10A1 Cys170Ter, Asn5Ser, SLC12A3 Glu970Asp, SLCO1A2 Tyr115His, and SLCO1B1 Pro565Thr) were first detected. Seventeen novel mutations were identified in ABC series genes, including 9 in ABCA genes (ABCA2 His1986Tyr and Ser1124Ile, ABCA3 His534Leu, ABCA6 Gln993Ter, ABCA7 Ala153Pro, ABCA10 Glu1519Val, and Gly1239Arg, ABCA12 Pro236Leu, ABCA13 Ser3286Ter), 2 in ABCB genes (ABCB4 Ile377Asn and ABCB11 Ala588Pro), 5 in ABCC genes (ABCC2 Ile681Lys and Met688Thr, ABCC6 Gly656Val, ABCC11 Met908Thr, and Trp61Ter), and 1 in an ABCG gene (ABCG2 Ile196Leu). In addition, two mutations in receptor genes (CHRM3 Gln574 and NR1H4 Tyr149Ter) were also detected. The frequency of these mutations in the 249 ICP individuals reached 10.44% (26/249). The difference in the frequency of these mutations between the 249 ICP cases and 1,029 control individuals was significant (p < 2.2e-16).
In addition, 38 novel mutations were assigned to the probably damaging group, including 8 in OTAP genes (SLC10A2 Ala310Thr, SLC51B Ala7Ser, SLCO1A2 Glu650Gly, SLCO3A1 Asn26Lys, SLCO6A1 His8Asp, SLCO1B3 Leu65Val and Thr553Ile, and SLCO4C1 Ile602Val), 7 in ABCA genes (ABCA4 Asp644Glu, ABCA6 Arg961Ile, ABCA8 Leu286Phe, ABCA9 Ile1284Val, Val241Ile, and ABCA13 Lys277Thr and Met730Val), 5 in ABCB genes (ABCB4 Ser1153Asn and Asp972Asn, ABCB6 Asn6Ser, ABCB8 Ile384Met, and ABCB9Leu299Pro), 6 in ABCC genes (ABCC2 Ile487Val, ABCC3 Val112Gly, ABCC4 Arg38Gly, ABCC9 Val1137Leu, ABCC10 Ser130Cys, and Thr257Pro), and 1 in an ABCG gene (ABCG2 Gly625Asp). These 62 novel mutations were absent in the 1,029 control individuals and the 1000 Genomes Project, ExAC, and dbSNP databases.
Sanger sequencing to validate novel mutations
Sanger sequencing was used to confirm the 12 possibly pathogenic mutations in the damaging group. The results (Figure 2) were all consistent with WES.

FIGURE 2. Sanger sequencing to validate the novel mutations in the OTAP genes, ABC genes, and receptor genes. The box represents the mutation site.
Evolutionary conservation analysis
The Evolutionary conservative analysis results showed that most mutations belonging to the damaging group were highly conserved among vertebrate species, including mice, cats, cows, horses, pigs, and sheep (Figure 3 and Supplementary Figure S1).

FIGURE 3. The evolutionary conservation analysis of the mutations in the damaging group among vertebrates, including chimpanzees, gorillas, rats, dogs, pigs, and cows. The amino acids in the red horizontal line were highly conserved.
Comparison of the protein structural model of the ABCC2 Ile681Lys and Met688Thr mutations
The ABCC2 gene encodes a member of the superfamily of ATP-binding cassette transporters, which bind and hydrolyze ATP to enable the active transport of various substrates, including many drugs, toxicants, and endogenous compounds (bile acid, bile salt, bilirubin, etc.), across the cell membrane (Kamisako et al., 1999; Hayashi et al., 2005). ABCC2 has two ATP binding cassette domains, ATP 1 and ATP 2, which are located at positions 671–678 and 1,334–1,341, respectively (Figure 4A). In the present study, we identified two novel missense mutations at positions 681 (Ile681Lys) and 688 (Met688Thr). These two mutations have been predicted to be damaging to protein function according to the prediction results of SIFT and PolyPhen2 software.

FIGURE 4. Effects of ABCC2 Ile681Lys and Met688Thr variants on the protein sequence and structure. (A) Distribution of ABCC2 variants. ABCC2 is a 1545-amino acid protein containing two ATP binding cassette domains. Schematic representation of ABCC2 NM_000,392.4 cDNA and protein showing the locations of the two novel possible pathogenic variants, Ile681Lys and Met688Thr, detected in two out of 249 ICP patients. Effects of ABCC2 Ile681Lys (B) and Met688Thr variants (C) on the protein structure. The three-dimensional models of reference and modified (Ile681Lys and Met688Thr) ABCC2 are shown in gold and blue rounded structures, respectively. The enlarged portion shows that the two ATP binding cassette regions have small changes in the chemical bond lengths. (A) The ATP binding cassette region.
To further investigate the possible effect of these two mutations on the ABCC2 protein structure, the reference and modified protein structures (Ile681Lys and Met688Thr) were compared simultaneously using UCSF Chimera 1.14rc. The results showed that the 3D model of the Ile681Lys and Met688Thr mutations both had a slight change in the chemical bond lengths of ATP-ligand binding amino acid side chains at positions Ser678, Gln706, Ser1342, and Gln1382 (Figure 4B, C).
Correlations between the mutations and clinical data
In the mutation group, 15 pregnant women presented with preterm birth (32.61%, 15/46), and 7 pregnant women presented with MSAF (15.22%, 7/46). The descriptive statistics of 32 other clinical characteristics for patients with ICP with the 62 novel mutations are shown in Table 3. Regardless of whether the difference was significant, the mutation group tended to be associated with higher age and BMI and K, Na, Ca, P, Mg, WBC, RDW-SD, TBA, CHOL, TG, HDL, and LDL levels and lower gestational age and birth weight. It is worth noting that the mutation group (103.34 mmol/L) had a significantly (p = 0.04) lower Cl concentration than the wild-type (104.14 mmol/L) group. The associations between the clinical parameters (age: odds ratio (OR) = 1.008; 95%; confidence intervals (CI): 0.950–1.069; gestational age (OR = 0.991; 95% CI: 0.972–1.010); BMI (OR = 1.006, 95% CI: 0.919–1.102); gravidity (OR = 0.899, 95% CI: 0.726–1.114); parity (OR = 0.943, 95% CI: 0.636–1.398)) and the mutations were determined by logistic regression analysis.

TABLE 3. Descriptive statistics of 32 clinical features of ICP individuals associated with/without 62 novel mutationsa.
In addition, we found that patients with the 24 mutations in the damaging group (A) had higher ALT, AST, ALP, TBA, HDL, and LDL levels than patients with mutations in the probably damaging group (B) and the 414 local control individuals without ICP (C) (Figure 5). In particular, the levels of ALT, AST, and TBA were significantly different among the three groups (p < 0.05).

FIGURE 5. Effects of bile acid transporter and receptor gene mutations on biochemical indices. The difference in the average levels of ALT, AST, ALP, TBA, HDL, and LDL in the following groups: (A) 24 novel mutations in the damaging group, (B) 38 novel mutations in the probably damaging group, and (C) healthy control individuals without ICP disease. The means of different groups in rows with different superscript letters are significantly (p < 0.05) different from each other.
Discussion
To the best of our knowledge, this study is the largest analysis to use WES technology to identify potential novel possibly pathogenic mutations in OTAP genes, ABC genes, and receptor family genes involved in bile acid transporters and receptors. We identified 62 novel mutations in 82 genes in 249 ICP patients.
The present study has 3 major strengths. First, many previous studies that have identified the genetic loci contributing to ICP disease were mainly conducted by sequencing a single gene in a small number of individuals or a disease-affected family. This can omit some potentially pathogenic mutations. Recently, WES technology has proved to be a powerful new approach for detecting low-frequency (0.01 ≤ MAF <0.05) and rare (MAF <0.01) variations that affect human diseases, such as spontaneous preterm birth (Huusko et al., 2018) and fetal hydrocephalus (Sun et al., 2019). Using this method, we have also successfully revealed ANO8 as a genetic risk factor for ICP (Liu et al., 2020) and identified 42 novel mutations in ABC transporter genes that are associated with ICP in 151 samples (Liu et al., 2021). Moreover, by using WES, our present study also successfully identified novel candidate potential disease-causing loci in 82 genes that are currently known to have functions related to bile acid transporters and receptors. Second, this is the first systematic identification of OTAP, ABC, and receptor gene mutations from a relatively large sample of ICP patients in China (n = 249). Third, we collected relatively complete clinical data from these patients to support the potential association between mutations and clinical data and to provide further insight into the mechanisms of ICP disease. Certainly, our study also has limitations. First, most of the patient samples were collected from Jiangxi Province, which may limit the applicability of the generality of these results. However, this study is still meaningful because the incidence of ICP in Jiangxi reached as high as 3–5%. In addition, in this study, we only collected a simple sample of subjects for WES. However, genotypic information from parents provided by trio-based WES enabled the detection of a high percentage of de novo variants inside the ICP disease cohort. However, considering the number (249) of ICP samples, the focus of the analysis was extended beyond well-known ICP-related genes and bioinformatics analyses, thus making it possible to find new candidates. In addition, this study identified some interesting novel pathogenic loci that were associated with ICP; however, the causality between loci and ICP disease needs to be verified by further functional experiments.
The OTAP family is a type of bidirectional transport family of multispecific membrane carriers that can transport many types of endogenous and exogenous substances, such as bile acids and steroid hormones. This implies that OTAP abnormalities can affect bile acid levels (Dawson et al., 2005). Previous studies have focused more on the effect of the expression of OTAPs, such as SLC10A2 and SLCO4A1, on maternal bile acid levels (Yan et al., 2015; Ontsouka et al., 2021). Our present study identified 13 new mutations, including SLC10A2 and Ala310Thr, in the OTAP family from 249 ICP patients. This result extends the role of OTAPs in ICP. These mutations affect a specific pathway/mechanism of ICP and need further experimental verification.
Compared to solute carriers and receptors, most of the reports about the genetic susceptibility to ICP have thus far mainly focused on the ABC gene series. Since the ABCB4 gene mutation was first reported to be associated with ICP in a Caucasian population in 1999, more heterozygous mutant alleles of functionally known genes, including ABCB4, ABCB11, ABCC2, ATP8B1, and TJP2, have been described (Jacquemin et al., 1999; Bacq et al., 2009; Dixon et al., 2014; Dixon et al., 2017; Piatek et al., 2018; Aydin et al., 2020). In this study, we extended the analysis across all ABC genes and revealed genetic mutations, including ABCB4 Ile377Asn, Ser1153Asn and Asp972Asn, ABCB11 Ala588Pro, ABCC2 Ile681Lys, Met688Thr, and Ile487Val. Among them, the 3D model construction analysis indicated that the ABCC2 Ile681Lys and Met688Thr mutations altered the amino side chains. This change could affect the binding efficiency of the ATP molecule and further change the transport function. This effect is similar to the effect of the ABCC2 Ser1342Tyr mutation that we previously identified (Liu et al., 2021). Consistent with this result, Corpechot et al. 2020 reported the genetic contribution of ABCC2 to inherited cholestatic disorders, indicating that ABCC2 is more closely related to ICP disease. The results of this study not only confirmed the functionally known genes but also expanded the role of new mutations in other ABC genes. This deepens our understanding of the pathogenesis of ABC genes in ICP.
In recent years, an accumulating body of evidence has demonstrated that nuclear receptors are key regulators of various processes, including the metabolism of xeno- and endobiotics such as bile acids and drugs (Ahmad and Haeusler, 2019). These receptors are generally considered to be therapeutic targets for cholestatic liver disease (Van Mil et al., 2007; Manna and Williamson, 2021). Recent studies have also made significant progress in identifying the genetic mutations that contribute to ICP disease. For example, functional variants in FXR were implicated as a genetic predisposition for ICP in a European population (Van Mil et al., 2007; Zimmer et al., 2009). Additionally, we also found two missense mutations (Ser145Phe and Met185Leu) that were implicated in individual susceptibility to ICP disease (Lai et al., 2022). Our present study detected two novel mutations, including the nonsense mutation Tyr149Ter and the missense mutation Glu199Gly, which might contribute to the development of ICP. Of course, the mechanisms that affect ICP need to be further validated. In addition to FXR, the common gene variants of PXR have also demonstrated genetic susceptibility to ICP. Castano et al. 2010 identified that PXR polymorphisms (rs2461823 G allele) are significantly associated with ICP and adverse pregnancy outcomes, including low birth weight and Apgar score, by comparing a total of 101 ICP patients and 171 control individuals. In this study, two missense mutations (Gln253Lys and Gln465Arg) were identified in the PXR gene. Furthermore, we also first identified other receptor genes with novel mutations that are susceptible to ICP. These results expand the receptor gene mutation datasets and provide more available targets for the treatment of ICP. To our knowledge, this is the first study in which whole-exome sequencing has been performed to systematically identify mutations in the receptor genes in ICP.
In recent decades, bile acid signaling molecules have been shown to activate bile acid receptors in the nucleus and membrane of cells, thereby regulating lipids, glucose, and metabolism in the liver (Lefebvre et al., 2009; Chiang, 2013; Ahmad and Haeusler, 2019). This implies that the accumulation of TBA might lead to lipid abnormalities. Consistent with this result, our results showed that the mutation group had higher CHOL, TG, HDL, and LDL levels than the wild-type group (Table 3). The average levels of HDL and LDL were higher in the group with the 24 damaging mutations than in the group with the 38 probably damaging mutations and the healthy group with no mutations (Figure 5). This finding suggests that the effect size of the mutations of the damaging group was higher than that of the probably damaging group for the lipid indices.
Conclusion
In conclusion, this is the first study to conduct WES to reveal the genetic variants in bile acid transporters and receptor genes that are associated with ICP disease. We identified 62 novel potentially pathogenic mutations in 82 genes in 249 ICP patients. In particular, out of the 62 novel mutations, 24 were classified as damaging. Functional validation and experimental verification of these 62 novel mutations need to be further investigated. Our findings provide new insights into the genetic basis of ICP disease and suggest potential candidate variants for ICP clinical treatment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Jiangxi Provincial Maternal and Child Health Hospital in China. The patients/participants provided their written informed consent to participate in this study.
Author contributions
XL performed the experiments, analyzed the data, prepared the figures, and drafted the manuscript. JZ, SX, YZe, and XW collected the samples. XZ, HL, and YZo performed the experiments, analyzed the data, and revised the manuscript. All authors read and approved the final manuscript.
Funding
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (No. 82160298) and the National Science Foundation of Jiangxi Province (No. 20202BABL216010) and the Science and Technology Plan of Jiangxi Provincial Health Commission (No. 202130764).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.941027/full#supplementary-material
References
Ahmad, T. R., and Haeusler, R. A. (2019). Bile acids in glucose metabolism and insulin signalling - mechanisms and research needs. Nat. Rev. Endocrinol. 15, 701–712. doi:10.1038/s41574-019-0266-7
Aydin, G. A., Ozgen, G., and Gorukmez, O. (2020). The role of genetic mutations in intrahepatic cholestasis of pregnancy. Taiwan. J. Obstet. Gynecol. 59, 706–710. doi:10.1016/j.tjog.2020.07.014
Bacq, Y., Gendrot, C., Perrotin, F., Lefrou, L., Chretien, S., Vie-Buret, V., et al. (2009). ABCB4 gene mutations and single-nucleotide polymorphisms in women with intrahepatic cholestasis of pregnancy. J. Med. Genet. 46, 711–715. doi:10.1136/jmg.2009.067397
Castano, G., Burgueno, A., Fernandez Gianotti, T., Pirola, C. J., and Sookoian, S. (2010). The influence of common gene variants of the xenobiotic receptor (PXR) in genetic susceptibility to intrahepatic cholestasis of pregnancy. Aliment. Pharmacol. Ther. 31, 583–592. doi:10.1111/j.1365-2036.2009.04210.x
Chiang, J. Y. (2013). Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212. doi:10.1002/cphy.c120023
Corpechot, C., Barbu, V., Chazouilleres, O., Broue, P., Girard, M., Roquelaure, B., et al. (2020). Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited cholestatic disorders. Liver Int. 40, 163–174. doi:10.1111/liv.14260
Dawson, P. A., Hubbert, M., Haywood, J., Craddock, A. L., Zerangue, N., Christian, W. V., et al. (2005). The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J. Biol. Chem. 280, 6960–6968. doi:10.1074/jbc.M412752200
Dixon, P. H., Sambrotta, M., Chambers, J., Taylor-Harris, P., Syngelaki, A., Nicolaides, K., et al. (2017). An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 7, 11823. doi:10.1038/s41598-017-11626-x
Dixon, P. H., Wadsworth, C. A., Chambers, J., Donnelly, J., Cooley, S., Buckley, R., et al. (2014). A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am. J. Gastroenterol. 109, 76–84. doi:10.1038/ajg.2013.406
Dixon, P. H., and Williamson, C. (2008). The molecular genetics of intrahepatic cholestasis of pregnancy. Obstet. Med. 1, 65–71. doi:10.1258/om.2008.080010
Eloranta, M. L., Heinonen, S., Mononen, T., and Saarikoski, S. (2001). Risk of obstetric cholestasis in sisters of index patients. Clin. Genet. 60, 42–45. doi:10.1034/j.1399-0004.2001.600106.x
Geenes, V., Chappell, L. C., Seed, P. T., Steer, P. J., Knight, M., Williamson, C., et al. (2014). Association of severe intrahepatic cholestasis of pregnancy with adverse pregnancy outcomes: A prospective population-based case-control study. Hepatology 59, 1482–1491. doi:10.1002/hep.26617
Genomes Project, C., Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., et al. (2012). An integrated map of genetic variation from 1, 092 human genomes. Nature 491, 56–65. doi:10.1038/nature11632
Glantz, A., Marschall, H. U., and Mattsson, L. A. (2004). Intrahepatic cholestasis of pregnancy: Relationships between bile acid levels and fetal complication rates. Hepatology 40, 467–474. doi:10.1002/hep.20336
Hayashi, H., Takada, T., Suzuki, H., Onuki, R., Hofmann, A. F., Sugiyama, Y., et al. (2005). Transport by vesicles of glycine- and taurine-conjugated bile salts and taurolithocholate 3-sulfate: A comparison of human BSEP with rat bsep. Biochim. Biophys. Acta 1738, 54–62. doi:10.1016/j.bbalip.2005.10.006
Huusko, J. M., Karjalainen, M. K., Graham, B. E., Zhang, G., Farrow, E. G., Miller, N. A., et al. (2018). Whole exome sequencing reveals HSPA1L as a genetic risk factor for spontaneous preterm birth. PLoS Genet. 14, e1007394. doi:10.1371/journal.pgen.1007394
Jacquemin, E., Cresteil, D., Manouvrier, S., Boute, O., and Hadchouel, M. (1999). Heterozygous non-sense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy. Lancet 353, 210–211. doi:10.1016/S0140-6736(05)77221-4
Kakizaki, S., Takizawa, D., Tojima, H., Horiguchi, N., Yamazaki, Y., Mori, M., et al. (2011). Nuclear receptors CAR and PXR; therapeutic targets for cholestatic liver disease. Front. Biosci. 16, 2988–3005. doi:10.2741/3893
Kamisako, T., Leier, I., Cui, Y., Konig, J., Buchholz, U., Hummel-Eisenbeiss, J., et al. (1999). Transport of monoglucuronosyl and bisglucuronosyl bilirubin by recombinant human and rat multidrug resistance protein 2. Hepatology 30, 485–490. doi:10.1002/hep.510300220
Kawakita, T., Parikh, L. I., Ramsey, P. S., Huang, C. C., Zeymo, A., Fernandez, M., et al. (2015). Predictors of adverse neonatal outcomes in intrahepatic cholestasis of pregnancy. Am. J. Obstet. Gynecol. 213, 570 e571–e8. doi:10.1016/j.ajog.2015.06.021
Lai, H., Liu, X., Xin, S., Zheng, J., Liu, H., Ouyang, Y., et al. (2022). Identification of two novel pathogenic variants of the NR1H4 gene in intrahepatic cholestasis of pregnancy patients. BMC Med. Genomics 15, 90. doi:10.1186/s12920-022-01240-w
Lefebvre, P., Cariou, B., Lien, F., Kuipers, F., and Staels, B. (2009). Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191. doi:10.1152/physrev.00010.2008
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Liu, X., Lai, H., Xin, S., Li, Z., Zeng, X., Nie, L., et al. (2021). Whole-exome sequencing identifies novel mutations in ABC transporter genes associated with intrahepatic cholestasis of pregnancy disease: A case-control study. BMC Pregnancy Childbirth 21, 110. doi:10.1186/s12884-021-03595-x
Liu, X., Lai, H., Zeng, X., Xin, S., Nie, L., Liang, Z., et al. (2020). Whole-exome sequencing reveals ANO8 as a genetic risk factor for intrahepatic cholestasis of pregnancy. BMC Pregnancy Childbirth 20, 544. doi:10.1186/s12884-020-03240-z
Manna, L. B., and Williamson, C. (2021). Nuclear receptors, gestational metabolism and maternal metabolic disorders. Mol. Asp. Med. 78, 100941. doi:10.1016/j.mam.2021.100941
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Ontsouka, E., Epstein, A., Kallol, S., Zaugg, J., Baumann, M., Schneider, H., et al. (2021). Placental expression of bile acid transporters in intrahepatic cholestasis of pregnancy. Int. J. Mol. Sci. 22, 10434. doi:10.3390/ijms221910434
Ovadia, C., Seed, P. T., Sklavounos, A., Geenes, V., Di Ilio, C., Chambers, J., et al. (2019). Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: Results of aggregate and individual patient data meta-analyses. Lancet 393, 899–909. doi:10.1016/S0140-6736(18)31877-4
Ovadia, C., and Williamson, C. (2016). Intrahepatic cholestasis of pregnancy: Recent advances. Clin. Dermatol. 34, 327–334. doi:10.1016/j.clindermatol.2016.02.004
Papacleovoulou, G., Abu-Hayyeh, S., Nikolopoulou, E., Briz, O., Owen, B. M., Nikolova, V., et al. (2013). Maternal cholestasis during pregnancy programs metabolic disease in offspring. J. Clin. Invest. 123, 3172–3181. doi:10.1172/JCI68927
Piatek, K., Kurzawinska, G., Magielda, J., Drews, K., Barlik, M., Malewski, Z., et al. (2018). The role of ABC transporters' gene polymorphism in the etiology of intrahepatic cholestasis of pregnancy. Ginekol. Pol. 89, 393–397. doi:10.5603/GP.a2018.0067
Puljic, A., Kim, E., Page, J., Esakoff, T., Shaffer, B., LaCoursiere, D. Y., et al. (2015). The risk of infant and fetal death by each additional week of expectant management in intrahepatic cholestasis of pregnancy by gestational age. Am. J. Obstet. Gynecol. 212, 667 e661–e5. doi:10.1016/j.ajog.2015.02.012
Reyes, H., Ribalta, J., and Gonzalez-Ceron, M. (1976). Idiopathic cholestasis of pregnancy in a large kindred. Gut 17, 709–713. doi:10.1136/gut.17.9.709
Reyes, H., Taboada, G., and Ribalta, J. (1979). Prevalence of intrahepatic cholestasis of pregnancy in La Paz, Bolivia. J. Chronic Dis. 32, 499–504. doi:10.1016/0021-9681(79)90111-5
Rook, M., Vargas, J., Caughey, A., Bacchetti, P., Rosenthal, P., Bull, L., et al. (2012). Fetal outcomes in pregnancies complicated by intrahepatic cholestasis of pregnancy in a Northern California cohort. PLoS One 7, e28343. doi:10.1371/journal.pone.0028343
Saleh, M. M., and Abdo, K. R. (2007). Intrahepatic cholestasis of pregnancy: Review of the literature and evaluation of current evidence. J. Womens Health 16, 833–841. doi:10.1089/jwh.2007.0158
Sampson, J. A. (1927). Metastatic or embolic endometriosis, due to the menstrual dissemination of endometrial tissue into the venous circulation. Am. J. Pathol. 3, 93–110.43. doi:10.1016/s0002-9378(15)30003-x
Simjak, P., Parizek, A., Vitek, L., Cerny, A., Adamcova, K., Koucky, M., et al. (2015). Fetal complications due to intrahepatic cholestasis of pregnancy. J. Perinat. Med. 43, 133–139. doi:10.1515/jpm-2014-0089
Stulic, M., Culafic, D., Boricic, I., Stojkovic Lalosevic, M., Pejic, N., Jankovic, G., et al. (2019). Intrahepatic cholestasis of pregnancy: A case study of the rare onset in the first trimester. Med. Kaunas. 55, 454. doi:10.3390/medicina55080454
Sun, Y., Li, Y., Chen, M., Luo, Y., Qian, Y., Yang, Y., et al. (2019). A novel silent mutation in the L1CAM gene causing fetal hydrocephalus detected by whole-exome sequencing. Front. Genet. 10, 817. doi:10.3389/fgene.2019.00817
Turro, E., Astle, W. J., Megy, K., Graf, S., Greene, D., Shamardina, O., et al. (2020). Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102. doi:10.1038/s41586-020-2434-2
Van Mil, S. W., Milona, A., Dixon, P. H., Mullenbach, R., Geenes, V. L., Chambers, J., et al. (2007). Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology 133, 507–516. doi:10.1053/j.gastro.2007.05.015
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wang, Q., Dhindsa, R. S., Carss, K., Harper, A. R., Nag, A., Tachmazidou, I., et al. (2021). Rare variant contribution to human disease in 281, 104 UK Biobank exomes. Nature 597, 527–532. doi:10.1038/s41586-021-03855-y
Wikstrom Shemer, E. A., Stephansson, O., Thuresson, M., Thorsell, M., Ludvigsson, J. F., Marschall, H. U., et al. (2015). Intrahepatic cholestasis of pregnancy and cancer, immune-mediated and cardiovascular diseases: A population-based cohort study. J. Hepatol. 63, 456–461. doi:10.1016/j.jhep.2015.03.010
Williamson, C., and Geenes, V. (2014). Intrahepatic cholestasis of pregnancy. Obstet. Gynecol. 124, 120–133. doi:10.1097/AOG.0000000000000346
Wolski, H., Kurzawinska, G., Ozarowski, M., Drews, K., Barlik, M., Piatek, K., et al. (2020). FokI vitamin D receptor polymorphism as a protective factor in intrahepatic cholestasis of pregnancy. Ginekol. Pol. 91, 719–725. doi:10.5603/GP.a2020.0135
Xiao, J., Li, Z., Song, Y., Sun, Y., Shi, H., Chen, D., et al. (2021). Molecular pathogenesis of intrahepatic cholestasis of pregnancy. Can. J. Gastroenterol. Hepatol. 2021, 6679322. doi:10.1155/2021/6679322
Yan, Z., Li, E., He, L., Wang, J., Zhu, X., Wang, H., et al. (2015). Role of OATP1B3 in the transport of bile acids assessed using first-trimester trophoblasts. J. Obstet. Gynaecol. Res. 41, 392–401. doi:10.1111/jog.12549
Keywords: ICP, WES, OTAPs, ABCs, receptors, novel mutations
Citation: Liu X, Zheng J, Xin S, Zeng Y, Wu X, Zeng X, Lai H and Zou Y (2022) Whole-exome sequencing expands the roles of novel mutations of organic anion transporting polypeptide, ATP-binding cassette transporter, and receptor genes in intrahepatic cholestasis of pregnancy. Front. Genet. 13:941027. doi: 10.3389/fgene.2022.941027
Received: 11 May 2022; Accepted: 01 July 2022;
Published: 15 August 2022.
Edited by:
Fengxiao Bu, Sichuan University, ChinaReviewed by:
Evgeny Suspitsin, Saint Petersburg State Pediatric Medical University, RussiaXiaodong Wang, Sichuan University, China
Copyright © 2022 Liu, Zheng, Xin, Zeng, Wu, Zeng, Lai and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoming Zeng, MTgwNzAwMzg2NzVAMTYzLmNvbQ==; Hua Lai, MTkzMzM2ODQxOEBxcS5jb20=; Yang Zou, em91eWFuZzgxQDE2My5jb20=