Corrigendum: Identification of the first congenital ichthyosis case caused by a homozygous deletion in the ALOX12B gene due to chromosome 17 mixed uniparental disomy
 Lei Zhang1†
Lei Zhang1† Yanqiu Hu1†Jingjing Lu2†Peiwei Zhao1Xiankai Zhang1Li Tan1Jun Li3Cuiping Xiao1*
Yanqiu Hu1†Jingjing Lu2†Peiwei Zhao1Xiankai Zhang1Li Tan1Jun Li3Cuiping Xiao1* Linkong Zeng4*
Linkong Zeng4* Xuelian He1*
Xuelian He1*- 1Precision Medical Center, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology, Wuhan, China
- 2Dermatology Department, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology, Wuhan, China
- 3Otolaryngology Department, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology, Wuhan, China
- 4Neonatology Department, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology, Wuhan, China
Uniparental disomy (UPD) is a rare genetic event caused by errors during gametogenesis and fertilization leading to two copies of a chromosome or chromosomal region inherited from one parent. MixUPD is one type of UPD that contains isodisomic and heterodisomic parts because of meiotic recombination. Using whole-exome sequencing (WES), we identified the first case of ichthyosis due to a maternal mixUPD on chromosome 17, which results in a homozygous deletion of partial intron 8 to exon 10 in ALOX12B, being predicted to lead to an internal protein deletion of 97 amino acids. We also performed a retrospective analysis of 198 patients with ALOX12B mutations. The results suggested that the exon 9 and 10 are located in the mutational hotspots of ALOX12B. In addition, our patient has microtia and congenital stenosis of the external auditory canals, which is very rare in patients with ALOX12B mutations. Our study reports the first case of autosomal recessive congenital ichthyosis (ARCI) due to a mixUPD of chromosome 17 and expands the spectrum of clinical manifestations of ARCI caused by mutations in the ALOX12B gene.
Introduction
Uniparental disomy (UPD), first reported by Engel in 1980, refers to the inheritance of two copies of chromosomes or their segments from one parent, different from the classical Mendelian inheritance (Engel, 1980). The two copies can be identical (uniparental isodisomy or iUPD), in which a single chromosome from one parent is duplicated (caused by meiosis II error), or not identical (uniparental heterodisomy or hUPD), in which a pair of nonidentical chromosomes are inherited from one parent (caused by a meiosis I error) (Liehr, 2010). If a UPD contains iUPD and hUPD, with either terminal region of homozygosity (ROH) (meiosis I error) or centromeric ROH (meiosis II error), it is referred to as a mixed UPD (mixUPD) due to meiotic recombination (Yauy et al., 2020). UPD can result in imprinting disorders when it involves chromosomes 6, 7, 11, 14, 15, or 20 (Scuffins et al., 2021), such as Prader-Willi/Angelman syndrome in 15q11-q13 (Sharp et al., 2010). In addition, UPD is a cause of autosomal recessive (AR) disorders through the inheritance of recessive disease alleles from a carrier parent, which only occurs in iUPD or mixUPD patients (Kotzot, 2001; Spiekerkoetter et al., 2002; King et al., 2014; Xiao et al., 2019).
Autosomal recessive congenital ichthyosis (ARCI) is a group of genetic diseases related to defective epidermal barriers; to date, 13 known genes have been causally associated with this disorder, namely, ALOX12B, ABCA12, ALOXE3, CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7, SLC27A4, SULT2B1, ST14, and TGM1 (Williams and Elias, 1985; Richard, 1993; Vahlquist et al., 2018; Simpson et al., 2020). The ALOX12B gene encodes the keratinocyte lipid transporter ATP-binding cassette subfamily A, and mutations in this gene, which lead to the disruption of lamellar granule lipid transport in the upper epidermal keratinocytes, cause nonbullous congenital ichthyosiform erythroderma (NCIE, MIM: #242100) or mild phenotypes such as self-improving collodion ichthyosis (SICI) (Jobard et al., 2002; Harting et al., 2008; Vahlquist et al., 2010). In addition to skin symptoms, overfolded ear and hearing loss have been presented in cases with ALOX12B mutations in a recent study (Simpson et al., 2020).
Here, we report the first case with SICI caused by homozygous deletion of the entire exon 9 and partial exon 10 in the ALOX12B gene, and this deletion has induced exon 9 and 10 skipping, likely resulting in a mutation protein with an internal absence of 97 amino acids. Further homozygosity mapping and B allele frequency (BAF) analysis have indicated that the homozygous deletion of ALOX12B was caused by a maternal mixUPD of chromosome 17. In addition to SICI, our patient had congenital auricular deformities, microtia, and stenosis of the external auditory canals.
Case presentation
Clinical report
The newborn boy was the second child of healthy non-consanguineous parents without skin disorders (Figure 1B). He was delivered by cesarean section at 39 weeks of gestation after an uneventful pregnancy. His birth weight was 3,060 g, and his height was 50 cm with an Apgar score of 9 at 1 minute and 9 at 5 minutes. At birth, he was covered by a collodion membrane with underlying erythroderma. His skin was covered with multiple fissures, together with partial desquamation and oozing through its cracks. He had bilateral ectropion and eyelid swelling. In addition, he had a significant malformation of the external ear and microtia, and further CT scanning of the inner ear showed that the cartilaginous part was narrow (left side) (Figure 1A). Left inguinal canal cryptorchidism was noted.
FIGURE 1
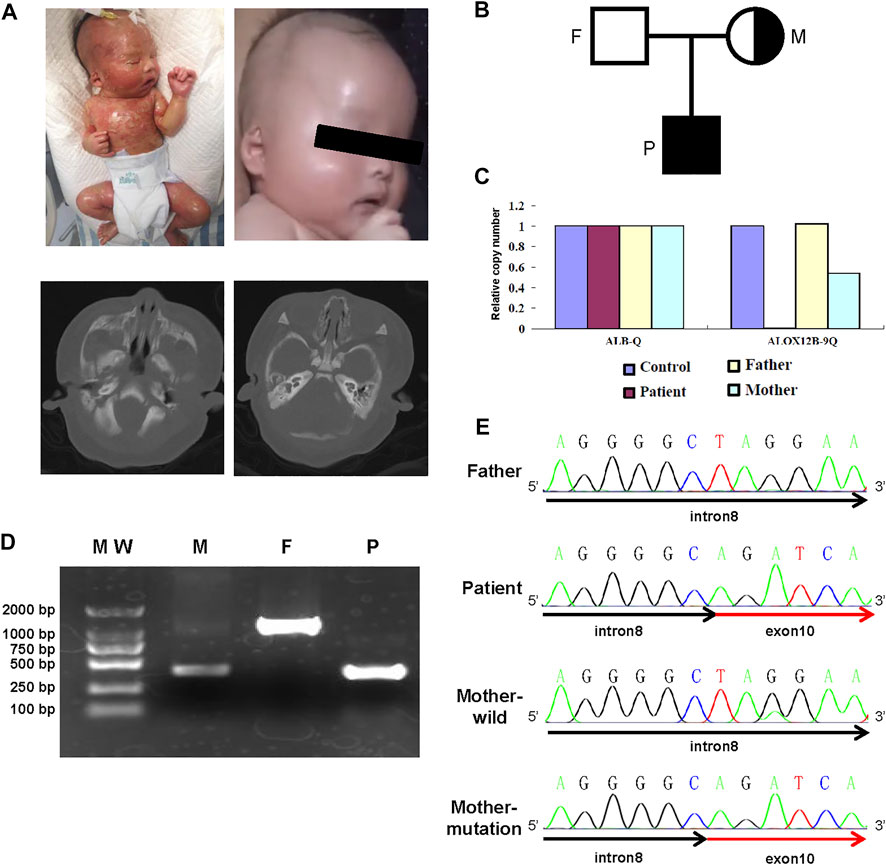
FIGURE 1. Clinical and genetic information of the family. (A) Photograph and inner ear CT of the case. Lateral view of the patient at the age of 1 day (upper left panel) and 3 months (upper right panel). CT plain scan of the inner ear (lower panel). (B) Pedigree of the reported family. (C) Quantitative PCR histogram of ALOX12B to ALB gene. (D) Agarose gel electrophoresis of PCR amplification for samples from the patient (P), heterozygous carrier mother (M), and wild-type father (F). (E) Sanger sequencing confirms the breakpoints of the deletion encompassing the 3′ end of intron 8 and 5′ end of exon 10.
The patient was immediately admitted to the intensive care unit and given liver oil ointment to the entire body to keep the affected area moist and mupirocin ointment to reduce secondary bacterial infection where the skin was broken. Erythromycin ophthalmic ointment was applied on the eyelid margins to reduce eyelid swelling. After 2 weeks of treatment, the membrane was shed, and the skin looked almost normal after a 3-month follow-up (Figure 1A). In addition, he had no significant hearing problems.
Methods
The study was approved by the Clinical Research Ethics Committee of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, and written informed consent was obtained from the patient’s parents.
Whole-exome sequencing and homozygosity mapping analysis
Genomic DNA was isolated from the peripheral blood, and trio-WES was conducted on the proband and his parents. The protocol used for WES was the same as previously described (Luo et al., 2021). WES data analysis showed a loss of heterozygosity (LOH) region of 15.33 Mb in the short arm of chromosome 17. To further confirm the LOH, the clean data were aligned to the human reference genome (hg19) using BWA-MEM (v.0.7.12), then sorted and indexed with SAMtools (version 1.3.1). Single-nucleotide polymorphism (SNP) calling and mapping were performed using the GATK (version 3.8) Haplotype Caller. After common CNV (>1% population frequency) regions were filtered out with the aid of PLINK(R), SNPs were selected based on the population databases (dbSNP153) with minor allele frequency values between 20 and 80%.
Genotypic status at each locus was assessed as BAF, the locus with a value of 0.5 is the heterozygous position and 1 or 0 is for the homozygous genotype. By estimating the proportion of homozygous/heterozygous genotypes for all candidate loci in each chromosome, a preliminary judgment was made on whether partial or whole chromosome UPD was present. For the sliding window homozygosity analysis, the window size was set to 50 SNPs, and the sliding stride was set to 5 SNPs with r2 > 0.5 in 2 SNPs. A fragment of homozygous region (ROH) was defined as follows: a segment ≥2.5 Mb in length contains over 50 homozygous SNPs distributed with a density of at least 1 SNP per 5,000 kb.
Quantitative polymerase chain reaction of ALOX12B
The genomic DNA was adjusted to 20 ng/μL using the Qubit 2.0 fluorometer and Qubit dsDNA HS Assay kit. Quantitative PCR was performed using the quantitative TAKARA SYBR Green PCR kit and ABI StepOnePlus PCR instrument according to the following conditions: 95°C for 2 min, followed by 95°C for 15 s, and 60°C for 60 s (40 cycles). We quantified ALB gene (encoding albumin, 4q13.3) as the internal reference gene. The primers for these DNA fragments were ALB exon 13 (forward: 5′-AGT GCA CTT GTT GAG CTC GTG-3′, reverse: 5′-GCA AAG CAG GTC TCC TTA TCG-3′), ALOX12B exon 9 (forward: 5′-GCC ACC CCC TCT ACA AGG TAT-3′, reverse: 5′-TAT GCC AAG CCC TCT CTT GTG-3′).
Deletion breakpoint analysis of the ALOX12B gene
The WES analysis suggested that the proband carried a deletion of exon 9 in the ALOX12B gene. To determine the breakpoint, primers were designed from the breakpoint interval, with the upstream primer being complementary to sequences in intron 8, and the downstream primer being complementary to sequences in intron 10 (forward: 5′-GTC AGG TCC CTA CTG GAA ATAC-3′, reverse: 5′-ATG ATG ACC CAA AGG CAA A-3′). A long-range PCR was performed using FastPfu DNA Polymerase (TransGen Biotech), and the products were separated on 1.5% agarose gels and sequenced by the ABI 3500 DNA Sequencer (Applied Biosystems, United States).
Review of reported ALOX12B cases from literature
The databases such as PubMed, Scopus, Embase, and Web of Science (including MEDLINE) were systematically searched through 2022/02. The search terms included the combination of the following groups of keywords: 1) “ALOX12Bgene,” or “Arachidonate 12-Lipoxygenase”; 2) “variant” or “mutation”; and 3) “recessive congenital ichthyosis” or “self-improving collodion ichthyosis.” The initial electronic search identified 90 articles in PubMed, 46 in Scopus, 92 in Embase, and 77 in Web of Science. After removing the duplicates and screening the articles by title and abstract, we discarded 281 articles, and 24 remained (Jobard et al., 2002; Eckl et al., 2005; Ashoor et al., 2006; Lesueur et al., 2007; Harting et al., 2008; Eckl et al., 2009; Akiyama et al., 2010; Kurban et al., 2010; Vahlquist et al., 2010; Li et al., 2012; Chaitidis et al., 2013; Israeli et al., 2013; Osorio et al., 2013; Numata et al., 2015; Lolas et al., 2016; Bastaki et al., 2017; Alavi et al., 2020; Fioretti et al., 2020; Anker et al., 2021; Frommherz et al., 2021; Hotz et al., 2021; Mohamad et al., 2021; Srinivas et al., 2021). Mutations in the ALOX12B gene were described according to the Human Genome Variation Society, using the NCBI reference sequence NM_001139.3. After completing the statistical analysis, a lollipop diagram was created by the trackViewer (version 1.28.1) in R software (version 3.6.3). Density plots were generated with ggplot2, and “geom_density” function was used to analyze the possible mutation spectrum and hotspots.
Results
Trio-whole-exome sequencing data analysis reveals a reduction in homozygosity for a maternal ALOX12B deletion mutation due to maternal mixUPD
Trio-WES and homozygosity mapping detected the LOH region of 15.33 Mb in the short arm of chromosome 17 (17p), which included the ALOX12B gene. To further investigate the LOH occurrence on chromosome 17, BAF was performed by checking Mendelian inheritance errors in the trio-WES data. A total of 1,286 SNPs, ranging 80 Mb on chromosome 17, were analyzed, and 1,119 (87.0%) SNPs, which included homozygous and heterozygous SNPs, were identified as being maternally inherited, suggesting a maternal mixUPD in the patient (Figure 2A). Thus, the homozygous deletion of ALOX12B exon 9 was caused by a maternal isodisomy. Figure 2B explains the mechanisms of chromosome 17 mixUPD caused by errors in stages I and II of meiosis.
FIGURE 2
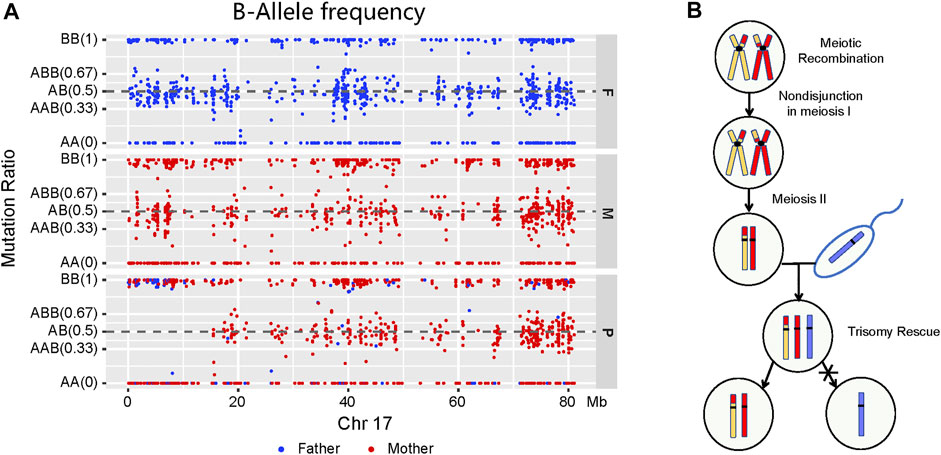
FIGURE 2. Molecular characterization and a model for the origin of mixUPD. (A) A loss of LOH region in the short arm of chromosome 17 described by B-allele frequency plot. Paternal and maternal sites are highlighted in blue and red, respectively. A point is classified as AA—if the BAF value is <0.1, as AB/AAB/ABB—if it is between 0.1 and 0.9, and as BB if it is >0.9. LOH takes place on the short arm of chromosome 17 (p11.2–p13.3) in the patient, and most points are generated maternally. (B) Schematic illustration of genetic events with mixUPD of chromosome 17.
Validation of the partial ALOX12B gene deletion and breakpoint analysis
The partial deletion of ALOX12B gene was validated by quantitative PCR analysis, and the results revealed that the ratios in exon 9 regions were 0, 0.5, and 1 in the proband, his mother, and his father, respectively (Figure 1C). PCR using primers complementary to sequences in introns 8 and 10 revealed that the proband was homozygous for deletion, whereas his mother was heterozygous and his father was normal (Figure 1D). Sanger sequencing was used to detect the breakpoint, and alignment to hg19 revealed the breakpoints located within IVS8 at the g.8077544_8077545 position and the distal one within exon10 at the g.8076713_8076714 position, resulting in 831 bp deletion, which included the full length of exon 9 and intron 9 and partial exon 10 (Figure 1E).
Review of ALOX12B mutations in self-improving collodion ichthyosis
We reviewed and listed 134 different pathologic mutation sites in the ALOX12B gene reported in 198 ARCI patients (Figure 3A). Among these, missense and frameshift mutations were the most common types, accounting for more than 80% of the cases. The high-frequency mutation sites were more concentrated in exons 9,10,12, and 14, which are located in the lipoxygenase domain (Figure 3B). The homozygous mutation in our case is in the hotspot region and presents with SICI.
FIGURE 3
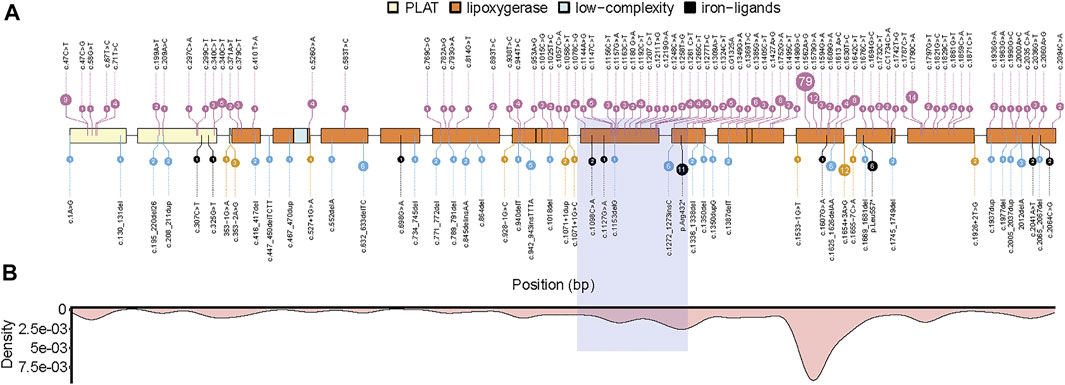
FIGURE 3. Summary of pathogenic mutations in ALOX12B. (A) ALOX12B domains, exons, and site of variants (missense mutations in pink, frameshift mutations in blue, nonsense mutations in black, and splicing mutations in yellow). (B) The density plot of all mutations is shown below, and the light purple box represents the deletion in this case. PLAT: polycystin-1, lipoxygenase, α-toxin domain.
Discussion
We report the first case of ARCI resulting from a homozygous deletion of the 3’ end of intron 8, exon 9, intron 9, and part of exon 10 (831 bp) in the ALOX12B gene due to a mixUPD of maternal chromosome 17. In addition to ichthyosis, our patient had microtia and congenital stenosis of the external auditory canals, which is exceedingly rare in patients with gene mutations.
UPD is an underestimated cause of AR disorders. Previously, the most cited prevalence of UPD was 1/3,500 live births (Robinson, 2000). However, recent studies have estimated that UPD occurs 1 in 2,000 births, and maternal UPD is twice as prevalent as paternal UPD (Nakka et al., 2019; Scuffins et al., 2021). Chromosome 17 UPD is uncommon, and the first case of maternal UPD involving chromosome 17 was reported in a child with a normal phenotype in 1999 (Genuardi et al., 1999). Although UPD of chromosome 17 is not associated with imprinting disease, it has the potential to unmask recessive mutations. To date, only a few cases with genetic conditions caused by UPD of chromosome 17 have been reported (Lebre et al., 2009; Natsuga et al., 2010; Labrijn-Marks et al., 2019).
ARCI is a rare dermatological condition, and to date, mutations in 13 genes have been identified to cause it (Williams and Elias, 1985; Richard, 1993; Vahlquist et al., 2018). However, there are only five cases of ichthyosis caused by UPD involving CERS3 (2), ABCA12 (2), and SPINK5 (1) (Castiglia et al., 2009; Numata et al., 2014; Shibata et al., 2015; Polubothu et al., 2018; Muthusamy et al., 2020). To our knowledge, no case involving UPD of chromosome 17 has been reported to have ichthyosis, and our patient is the first case with ARCI caused by UPD.
Interestingly, we reported that the baby showed congenital auricular deformities. The phenomenon was not reported in a large cohort until nearly 2 years ago and the total number does not exceed 10 (Simpson et al., 2020; Hotz et al., 2021). However, the location and types of mutations on ALOX12B do not seem to affect the severity of dysplastic ears (Simpson et al., 2020), and the mechanism by which ALOX12B mutations cause dysplastic ears is definitively unknown. Furthermore, in this case, the proband presented with cryptorchidism, which has not been reported yet to be associated with the ALOX12B gene.
By literature review, we found 134 different pathologic mutations reported in the ALOX12B gene among 198 ARCI patients, with missense mutation being the most frequent type, followed by frameshift mutations. The hotspot regions are exons 9, 10, 12, and 14, which encode the lipoxygenase domain (Figures 3A,B). The deletion in our case was due to UPD and was in the hotspot region, exon 9, and part of exon 10. To date, neither homozygous deletion nor mutation due to UPD has been reported. In this study, a homozygous deletion of the 3’ end of intron 8, exon 9, and part of exon 10 was predicted to cause a shortened ALOX12B production (in-frame transcripts) or degradation of aberrant ALOX12B mRNA induced by the nonsense-mediated decay, both of which would cause the loss of function of ALOX12B. It has been previously reported that a 35-year-old male with homozygous deletion of ALOX12B exon 3–15 had a mild body erythema in his childhood and only very mild face erythema with palmar hyperlinearity in adulthood (Fioretti et al., 2020). It is unlikely that the mutant ALOX12B genes with deletion of exon 3–15 can produce a protein with residual enzyme activity, thus we speculate that ALOX12B deficiency alone causes a mild, self-improving, ARCI phenotype, and that more severe ALOX12B-related phenotypes could depend on additional modifiers, genetic or epigenetic ones. Our patient with the homozygous deletion of partial intron 8 to exon 10 in ALOX12B also displayed relative mild skin phenotypes, and the deletion is predicted to cause the in-frame skipping of exon 9 and 10, with exon 8 joined to exon 11, resulting in an internal protein deletion of 97 amino acids (from Leu358 to Lys454), suggesting that this region is not critical for enzyme activity.
Lastly, our study supports that the WES data can identify distinct types of genomic lesions, such as SNVs, LOH, and CNVs (Xiao et al., 2019). Detecting UPD events in the trio-WES data does not incur additional costs but increases the diagnostic rate, especially in cases with non-Mendelian inheritance (Xiao et al., 2019; Yauy et al., 2020).
Conclusion
Here, we report the first case of ARCI due to homozygous deletion of exon 9 and partial exon 10 in the ALOX12B gene due to mixUPD of maternal chromosome 17. Our study extends the clinical phenotype spectrum and further confirms that congenital auricular deformity is one of the phenotypes in patients with ALOX12B gene mutations. Furthermore, our work supports the value that trio-WES can have as an auxiliary diagnostic method for testing UPD.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Clinical Research Ethics Committee of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
Study concepts: XH, CX, and LZ. Study design: LZ, YH, and XH. Literature research: LZ, YH, and Yufeng Huang. Clinical information collection: JLu, PZ, and JLi. Data acquisition: LZ, YH, PZ, and XZ. Data analysis/interpretation: LZ, YH, PZ, and XZ. Manuscript preparation: XH, PZ, and CX. Manuscript editing: XH and LT. Manuscript final version approval: CX, LZ, and XH.
Funding
This work was supported by the grants of Wuhan Municipal Health Commission (NO. WX19C19, WX14A06); the Youth Program of National Natural Science Foundation of China (NO.81700302); and the Natural Science Foundation of Hubei Province (2017CFB322).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akiyama, M., SaKai, K., Yanagi, T., TabataN., , YaMadaM., , and SHimizu, H. (2010). Partially disturbed lamellar granule secretion in mild congenital ichthyosiform erythroderma with ALOX12B mutations. Br. J. Dermatol. 163 (1), 201–204. doi:10.1111/j.1365-2133.2010.09745.x
Alavi, A., Darki, F., Bidgoli, M. M. R., Zare-Abdollahi, D., Moini, A., Shahshahani, M. M., et al. (2020). Mutation in ALOX12B likely cause of POI and also ichthyosis in a large Iranian pedigree. Mol. Genet. Genomics 295 (4), 1039–1053. doi:10.1007/s00438-020-01663-z
Anker, P., Kiss, N., Kocsis, I., Czemmel, E., Becker, K., Zakarias, S., et al. (2021). Report of a novel ALOX12B mutation in self-improving collodion ichthyosis with an overview of the genetic background of the collodion baby phenotype. Life (Basel) 11 (7), 624. doi:10.3390/life11070624
Ashoor, G., Masse, M., Garcia Luciano, L. M., Sheffer, R., Martinez-Mir, A., Christiano, A. M., et al. (2006). A novel mutation in the 12(R)-lipoxygenase (ALOX12B) gene underlies nonbullous congenital ichthyosiform erythroderma. Br. J. Dermatol. 155 (1), 198–200. doi:10.1111/j.1365-2133.2006.07188.x
Bastaki, F., Mohamed, M., Nair, P., Saif, F., Mustafa, E. M., Bizzari, S., et al. (2017). Summary of mutations underlying autosomal recessive congenital ichthyoses (ARCI) in Arabs with four novel mutations in ARCI-related genes from the United Arab Emirates. Int. J. Dermatol. 56 (5), 514–523. doi:10.1111/ijd.13568
Castiglia, D., CastoriM., , PisanEschi, E., SoMMiM., , CovaCiu, C., Zambruno, G., et al. (2009). Trisomic rescue causing reduction to homozygosity for a novel ABCA12 mutation in harlequin ichthyosis. Clin. Genet. 76 (4), 392–397. doi:10.1111/j.1399-0004.2009.01198.x
Chaitidis, P., Adel, S., Anton, M., Heydeck, D., Kuhn, H., and Horn, T. (2013). Lipoxygenase pathways in Homo neanderthalensis: Functional comparison with Homo sapiens isoforms. J. Lipid Res. 54 (5), 1397–1409. doi:10.1194/jlr.M035626
Eckl, K. M., Krieg, P., Kuster, W., Traupe, H., Andre, F., Wittstruck, N., et al. (2005). Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum. Mutat. 26 (4), 351–361. doi:10.1002/humu.20236
Eckl, K. M., de Juanes, S., Kurtenbach, J., Natebus, M., Lugassy, J., Oji, V., et al. (2009). Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: Evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J. Invest. Dermatol. 129 (6), 1421–1428. doi:10.1038/jid.2008.409
Engel, E. (1980). A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 6 (2), 137–143. doi:10.1002/ajmg.1320060207
Fioretti, T., Auricchio, L., Piccirillo, A., Vitiello, G., Ambrosio, A., Cattaneo, F., et al. (2020). Multi-gene next-generation sequencing for molecular diagnosis of autosomal recessive congenital ichthyosis: a genotype-phenotype study of four Italian patients. Diagn. (Basel) 10 (12), E995. doi:10.3390/diagnostics10120995
Frommherz, L., Krause, A., Kopp, J., Hotz, A., Hubner, S., Reimer-Taschenbrecker, A., et al. (2021). High rate of self-improving phenotypes in children with non-syndromic congenital ichthyosis: case series from south-western Germany. J. Eur. Acad. Dermatol. Venereol. 35 (11), 2293–2299. doi:10.1111/jdv.17524
Genuardi, M., Tozzi, C., Pomponi, M. G., Stagni, M. L., Della Monica, M., Scarano, G., et al. (1999). Mosaic trisomy 17 in amniocytes: phenotypic outcome, tissue distribution, and uniparental disomy studies. Eur. J. Hum. Genet. 7 (4), 421–426. doi:10.1038/sj.ejhg.5200333
Harting, M., Brunetti-Pierri, N., Chan, C. S., Kirby, J., Dishop, M. K., Richard, G., et al. (2008). Self-healing collodion membrane and mild nonbullous congenital ichthyosiform erythroderma due to 2 novel mutations in the ALOX12B gene. Arch. Dermatol. 144 (3), 351–356. doi:10.1001/archderm.144.3.351
Hotz, A., Kopp, J., Bourrat, E., Oji, V., Komlosi, K., Giehl, K., et al. (2021). Meta-analysis of mutations in ALOX12B or ALOXE3 identified in a large cohort of 224 patients. Genes (Basel) 12 (1), 80. doi:10.3390/genes12010080
Israeli, S., Goldberg, I., Fuchs-Telem, D., BeRgman, R., Indel Man, M., Bitterman-Deutsch, O., et al. (2013). Non-syndromic autosomal recessive congenital ichthyosis in the Israeli population. Clin. Exp. Dermatol. 38 (8), 911–916. doi:10.1111/ced.12148
Jobard, F., Lefevre, C., Karaduman, A., Blanchet-Bardon, C., Emre, S., Weissenbach, J., et al. (2002). Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum. Mol. Genet. 11 (1), 107–113. doi:10.1093/hmg/11.1.107
King, J. E., Dexter, A., Gadi, I., Zvereff, V., Martin, M., Bloom, M., et al. (2014). Maternal uniparental isodisomy causing autosomal recessive GM1 gangliosidosis: a clinical report. J. Genet. Couns. 23 (5), 734–741. doi:10.1007/s10897-014-9720-9
Kotzot, D. (2001). Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J. Med. Genet. 38 (8), 497–507. doi:10.1136/jmg.38.8.497
Kurban, M., Shimomura, Y., Bahhady, R., Ghosn, S., Kibbi, A. G., and Christiano, A. M. (2010). Nonsense mutation in the ALOX12B gene leads to autosomal recessive congenital ichthyosis in a Lebanese family. J. Eur. Acad. Dermatol. Venereol. 24 (2), 232–234. doi:10.1111/j.1468-3083.2009.03381.x
Labrijn-Marks, I., Somers-Bolman, G. M., In 't Groen, S. L. M., Hoogeveen-Westerveld, M., Kroos, M. A., Ala-Mello, S., et al. (2019). Segmental and total uniparental isodisomy (UPiD) as a disease mechanism in autosomal recessive lysosomal disorders: evidence from SNP arrays. Eur. J. Hum. Genet. 27 (6), 919–927. doi:10.1038/s41431-019-0348-y
Lebre, A. S., Moriniere, V., Dunand, O., Bensman, A., Morichon-Delvallez, N., and Antignac, C. (2009). Maternal uniparental heterodisomy of chromosome 17 in a patient with nephropathic cystinosis. Eur. J. Hum. Genet. 17 (8), 1019–1023. doi:10.1038/ejhg.2009.13
Lesueur, F., Bouadjar, B., Lefevre, C., Jobard, F., Audebert, S., Lakhdar, H., et al. (2007). Novel mutations in ALOX12B in patients with autosomal recessive congenital ichthyosis and evidence for genetic heterogeneity on chromosome 17p13. J. Invest. Dermatol. 127 (4), 829–834. doi:10.1038/sj.jid.5700640
Li, H., Lorie, E. P., Fischer, J., Vahlquist, A., and Torma, H. (2012). The expression of epidermal lipoxygenases and transglutaminase-1 is perturbed by NIPAL4 mutations: indications of a common metabolic pathway essential for skin barrier homeostasis. J. Invest. Dermatol. 132 (10), 2368–2375. doi:10.1038/jid.2012.160
Liehr, T. (2010). Cytogenetic contribution to uniparental disomy (UPD). Mol. Cytogenet. 3, 8. doi:10.1186/1755-8166-3-8
Lolas, I. B., Sommerlund, M., Okkels, H., Ramsing, M., and Petersen, M. B. (2016). A novel deletion mutation in the ALOX12B gene in a Kurdish family with autosomal recessive congenital ichthyosis. J. Eur. Acad. Dermatol. Venereol. 30 (11), e144–e145. doi:10.1111/jdv.13457
Luo, S., Chen, L., Wei, W., Tan, L., Zhang, M., Duan, Z., et al. (2021). Prenatal genetic diagnosis in three fetuses with left heart hypoplasia (LHH) from three unrelated families. Front. Cardiovasc. Med. 8, 631374. doi:10.3389/fcvm.2021.631374
Mohamad, J., Samuelov, L., Malchin, N., Rabinowitz, T., Assaf, S., Malki, L., et al. (2021). Molecular epidemiology of non-syndromic autosomal recessive congenital ichthyosis in a Middle-Eastern population. Exp. Dermatol. 30 (9), 1290–1297. doi:10.1111/exd.14345
Muthusamy, K., Macke, E. L., Klee, E. W., Tebben, P. J., Hand, J. L., Hasadsri, L., et al. (2020). Congenital ichthyosis in Prader-Willi syndrome associated with maternal chromosome 15 uniparental disomy: Case report and review of autosomal recessive conditions unmasked by UPD. Am. J. Med. Genet. A 182 (10), 2442–2449. doi:10.1002/ajmg.a.61792
Nakka, P., Pattillo Smith, S., O'Donnell-Luria, A. H., McManus, K. F., Mountain, J. L., Ramachandran, S., et al. (2019). Characterization of prevalence and Health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105 (5), 921–932. doi:10.1016/j.ajhg.2019.09.016
Natsuga, K., Nishie, W., Arita, K., Shinkuma, S., Nakamura, H., Kubota, S., et al. (2010). Complete paternal isodisomy of chromosome 17 in junctional epidermolysis bullosa with pyloric atresia. J. Invest. Dermatol. 130 (11), 2671–2674. doi:10.1038/jid.2010.182
Numata, S., Hamada, T., Teye, K., Matsuda, M., Ishii, N., Karashima, T., et al. (2014). Complete maternal isodisomy of chromosome 5 in a Japanese patient with Netherton syndrome. J. Invest. Dermatol. 134 (3), 849–852. doi:10.1038/jid.2013.398
Numata, S., Teye, K., Krol, R. P., Karashima, T., Fukuda, S., Matsuda, M., et al. (2015). Mutation study for 9 genes in 23 unrelated patients with autosomal recessive congenital ichthyosis in Japan and Malaysia. J. Dermatol. Sci. 78 (1), 82–85. doi:10.1016/j.jdermsci.2015.02.006
Osorio, F., Leao, M., Azevedo, F., and Magina, S. (2013). Lamellar ichthyosis due to ALOX12B mutation. Actas Dermosifiliogr. 104 (5), 443–444. doi:10.1016/j.ad.2012.07.011
Polubothu, S., Glover, M., Holder, S. E., and Kinsler, V. A. (2018). Uniparental disomy as a mechanism for CERS3-mutated autosomal recessive congenital ichthyosis. Br. J. Dermatol. 179 (5), 1214–1215. doi:10.1111/bjd.16999
Richard, G. (1993). “Autosomal recessive congenital ichthyosis,” in GeneReviews®. Editor M. P. Adam (Seattle, WA).
Robinson, W. P. (2000). Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22 (5), 452–459. doi:10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K
Scuffins, J., Keller-Ramey, J., Dyer, L., Douglas, G., Torene, R., Gainullin, V., et al. (2021). Uniparental disomy in a population of 32, 067 clinical exome trios. Genet. Med. 23 (6), 1101–1107. doi:10.1038/s41436-020-01092-8
Sharp, A. J., Migliavacca, E., Dupre, Y., Stathaki, E., Sailani, M. R., Baumer, A., et al. (2010). Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15. Genome Res. 20 (9), 1271–1278. doi:10.1101/gr.108597.110
Shibata, A., Sugiura, K., Suzuki, A., Ichiki, T., and Akiyama, M. (2015). Apparent homozygosity due to compound heterozygosity of one point mutation and an overlapping exon deletion mutation in ABCA12: a genetic diagnostic pitfall. J. Dermatol. Sci. 80 (3), 196–202. doi:10.1016/j.jdermsci.2015.10.003
Simpson, J. K., Martinez-Queipo, M., Onoufriadis, A., Tso, S., Glass, E., Liu, L., et al. (2020). Genotype-phenotype correlation in a large English cohort of patients with autosomal recessive ichthyosis. Br. J. Dermatol. 182 (3), 729–737. doi:10.1111/bjd.18211
Spiekerkoetter, U., Eeds, A., Yue, Z., Haines, J., Strauss, A. W., and Summar, M. (2002). Uniparental disomy of chromosome 2 resulting in lethal trifunctional protein deficiency due to homozygous alpha-subunit mutations. Hum. Mutat. 20 (6), 447–451. doi:10.1002/humu.10142
Srinivas, S. M., Dhar, S., and Gowdra, A. (2021). Pseudodominant inheritance of self-improving collodion ichthyosis with homozygous mutation in the ALOX12B gene. Indian J. Dermatol. Venereol. Leprol. 87 (5), 714–717. doi:10.25259/IJDVL_1376_20
Vahlquist, A., Bygum, A., Ganemo, A., Virtanen, M., Hellstrom-Pigg, M., Strauss, G., et al. (2010). Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in scandinavian patients. J. Invest. Dermatol. 130 (2), 438–443. doi:10.1038/jid.2009.346
Vahlquist, A., Fischer, J., and Torma, H. (2018). Inherited nonsyndromic ichthyoses: An update on pathophysiology, diagnosis and treatment. Am. J. Clin. Dermatol. 19 (1), 51–66. doi:10.1007/s40257-017-0313-x
Williams, M. L., and Elias, P. M. (1985). Heterogeneity in autosomal recessive ichthyosis. Clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch. Dermatol. 121 (4), 477–488. doi:10.1001/archderm.121.4.477
Xiao, B., Wang, L., Liu, H., Fan, Y., Xu, Y., Sun, Y., et al. (2019). Uniparental isodisomy caused autosomal recessive diseases: NGS-based analysis allows the concurrent detection of homogenous variants and copy-neutral loss of heterozygosity. Mol. Genet. Genomic Med. 7 (10), e00945. doi:10.1002/mgg3.945
Keywords: ARCI, ALOX12B, whole-exome sequencing, mixed UPD (mixUPD), microtia
Citation: Zhang L, Hu Y, Lu J, Zhao P, Zhang X, Tan L, Li J, Xiao C, Zeng L and He X (2022) Identification of the first congenital ichthyosis case caused by a homozygous deletion in the ALOX12B gene due to chromosome 17 mixed uniparental disomy. Front. Genet. 13:931833. doi: 10.3389/fgene.2022.931833
Received: 29 April 2022; Accepted: 27 June 2022;
Published: 08 August 2022.
Edited by:
Lidia Larizza, Research Lab of Medical Cytogenetics and Molecular Genetics, Italian Auxological Institute (IRCCS), ItalyReviewed by:
Daniele Castiglia, Fondazione Luigi Maria Monti, ItalyGabriella Esposito, University of Naples Federico II, Italy
Copyright © 2022 Zhang, Hu, Lu, Zhao, Zhang, Tan, Li, Xiao, Zeng and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuelian He, aGV4dWVsaWFuMjAxM0Bob3RtYWlsLmNvbQ==; Cuiping Xiao, NDU5NTE4MjQwQHFxLmNvbQ==; Linkong Zeng, ZnJlZW1hbjMxNUAxNjMuY29t
†These authors have contributed equally to this work