
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 29 August 2022
Sec. Livestock Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.929889
This article is part of the Research TopicSex Control and Sex Differentiation in Aquatic AnimalsView all 6 articles
 Juan Sui1,2†
Juan Sui1,2† Sheng Luan1,2†Jiawang Cao1,2Ping Dai1,2Xianhong Meng1,2Kun Luo1,2Baolong Chen1,2Jian Tan1,2Qiang Fu1,2
Sheng Luan1,2†Jiawang Cao1,2Ping Dai1,2Xianhong Meng1,2Kun Luo1,2Baolong Chen1,2Jian Tan1,2Qiang Fu1,2 Jie Kong1,2*
Jie Kong1,2*Penaeus vannamei is the most important economic shrimp in the world. Many selective breeding programs are carried out to improve its production and performance traits. Although significant differences in the reproductive ability of female P. vannamei under artificial breeding conditions have been reported, the genome-wide adaption of the reproductive ability of domesticated female P. vannamei is less investigated. In this study, whole-genome analysis was performed along with pooled DNA sequencing on two fecundity separated bulks, high fecundity bulk (HB), and low fecundity bulk (LB). Each bulk contained 30 individuals from 3 commercial populations. A sequencing depth of >30× was achieved for each bulk, leading to the identification of 625,181 and 629,748 single nucleotide polymorphisms (SNPs) in HB and LB, respectively. Fixation index (Fst) combined with p ratio allowed for the identification of 145 selective sweep regions, with a sequence length of 14.5 Mb, accounting for 0.59% of the genome. Among the 145 selective sweep regions, a total of 64,046 SNPs were identified, and further verification was performed by genotyping 50 candidate SNPs on 60 samples from the offspring of the three populations. Furthermore, 121 genes were screened from the sweep regions. GO annotation and KEGG enrichment analyses showed that partial genes were essential for fecundity regulation. This study provides important information for in-depth investigation of genomic characteristics for long-term selective breeding on the fecundity of female P. vannamei and will also be important for genome-assisted breeding of P. vannamei in the future.
Pacific white shrimp (Penaeus vannamei) is the most important economic shrimp in the world, with a global aquaculture production of 5,446,216 tons and a value of 32,190,978 dollars in 2019 (FAO, 2022). The breeding of P. vannamei has lasted for 30 years and made great achievements in growth-related traits, survival, and specific disease resistance (Benzie, 2009; Gjedrem, 2012; Castillo-Juárez et al., 2015; Gjedrem and Rye, 2016).
The reproductive performance of parent shrimp is an essential trait for commercial larval production (Arcos et al., 2004; Caballero-Zamora et al., 2015). However, there are significant differences in the fecundity of female P. vannamei in seedling production. In a production cycle, some females can spawn up to 15 times, whereas some other females never spawn (Arcos et al., 2003; Tan et al., 2019). To improve larval production, selecting female shrimp with high fecundity is a vital strategy (Racotta et al., 2003; Arcos et al., 2004). However, knowledge about the genetic basis of such significant differences in the reproductive traits of female P. vannamei is very limited.
Both artificial breeding and directional selection would gradually affect the phenotype of domesticated animals, which will lead to the abundant phenotypic diversity and genetic adaptability of domesticated animals (Andersson, 2012 and 2013). The process of artificial breeding would leave detectable signatures on the genomes of domestic species (Utsunomiya et al., 2015; López et al., 2019). Analyzing these signatures is critical to study the genomic regions that changed due to domestication and genetic improvement.
Whole-genome resequencing has become a practical approach for the analysis of genome-wide variations and genomic signatures of domestication or artificial selection (Stratton, 2008; Rubin et al., 2010; Xia et al., 2015; Zhang et al., 2018). The method has been applied to many aquatic animals, such as Atlantic salmon (Bertolotti et al., 2020), Nile tilapia (Cadiz et al., 2019), channel catfish (Nam et al., 2020), large yellow croaker (Kon et al., 2021), and black tiger shrimp (Wong et al., 2020). Such analyses are usually costly because a large number of individuals need to be genotyped individually. In the population with a wide variation of target traits, selecting extreme or representative individuals can significantly reduce the scale and cost. The DNA pooling approach has been widely used to study genomic signatures of artificial selection in animals (Bertolini et al., 2016; Naval-Sanchez et al., 2020).
Fecundity is one of the noninvasive predictive criteria to identify females with high spawning frequency (Arcos et al., 2003). However, there are still no studies focusing on the identification of selection signatures in the reproductive traits of farmed Pacific white shrimp populations to date. In this study, we focused on the egg-laying frequency (ELF) and average egg amount of female parent P. vannamei in a production cycle. To increase the knowledge of selection effects on female reproduction in farmed populations of this species, selective sweeping analysis was performed on two fecundity separated bulks of P. vannamei using whole-genome resequencing with pooled DNA sequencing (DNA-seq). Single nucleotide polymorphism (SNPs) and genes associated with reproductive traits were identified. This work will contribute to the deep investigation of the molecular mechanisms of reproduction and the development of molecular breeding for high reproductive traits of P. vannamei.
In 2018, three commercial populations of P. vannamei were introduced to BLUP Aquabreed Co., Ltd (Weifang, Shandong Province, China). The three populations that contained 65 families were selected for years and labeled A, B, and C, respectively, among which A and B were characterized by strong resistance and C by fast growth. In 2019, the shrimps were cultivated as candidate parents. By the age of 10 months, 8–10 females and 8–9 males from each family were selected. Females subjected to unilateral eyestalk ablation to promote the synchronous development of ovaries and a number-coded ring were placed on the remaining eyestalk. At the same time, the parent shrimp started to undergo nutrition fortification for about 20 days. A total of 604 females and 500 males underwent nutrition fortification (Table 1). The diet consisted of two meals of squid and two meals of Nereis for females and three meals of squid and one meal of Nereis for males, which was accounted for a total daily supply of 20% of wet weight biomass, and was adjusted daily. Females were cultured in six 16-m2 tanks with a density of six to seven shrimp/m2. Males from the same family were placed in a 3-m2 tank, with a density of two to three shrimp/m2.
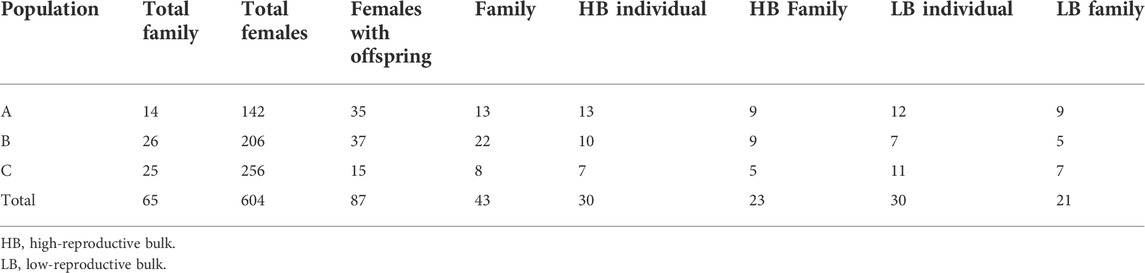
TABLE 1. The composition of high- and low-reproductive bulks.
The next generation was produced by mating selected male and female families at a ratio of 1:2 to 1:5, imposing certain restrictions for controlling inbreeding (e.g., avoiding full-sib, half-sib, and cousin mating). Sexually matured females were placed into the tank with males for natural mating at 9:00 a.m. and collected at 7:00 p.m. every day (recording the female shrimp number-coded ring). The mated females were put into the 120-L spawning tank (recording number-coded ring), whereas the unmated females were put back to the original tank. Male shrimp of one family mated with female shrimp from two different families at most at 1 day. The family construction period was from September 4 to 29 September 2019.
The ELF of 604 females during this period and the average egg number (AEN) from each spawn per female was recorded. Among these individuals, 179 had spawning records and 87 had offspring that successfully developed to juvenile stage. We only selected high reproductive individuals from the 87 females with offspring, because the vitality of offspring can also reflect the reproductive performance of females to some extent, although not absolutely. According to the ELF and AEN, we developed a fecundity index to measure the fecundity of female shrimp. The calculation formula was as follows:
where yi is the fecundity index of the ith individual of the 87 spawning individuals; aelfi and aaeni are the ELF and AEN of the ith individual, respectively; μelf and μaen are the average of the ELF and AEN of the 87 spawning individuals; and σelf and σaen are the standard error of the ELF and AEN of the 87 spawning individuals.
According to the reproductive index, a total of 30 females with high reproductive index and 30 individuals with no sexual maturity record were selected from the three populations to comprise a high reproductive bulk (HB) and a low-reproductive bulk (LB), respectively.
Muscle samples of 30 high- and 30 low-reproductive shrimp were collected and frozen in liquid nitrogen. For genotyping, the total genomic DNA of each individual was isolated using cetyltrimethylammonium bromide (CTAB). Equal DNA content quantified by Qbit 2.0 (Invitrogen, CA, United States) was pooled separately to generate a “high” pool and a “low” pool.
Paired-end sequencing libraries with an insert size of about 500 bp were constructed using the Paired-End DNA Sample Prep Kit (Illumina Inc., San Diego, CA, United States). The libraries were sequenced on an Illumina HiSeq 2500 system using standard protocol (Illumina, Inc.; San Diego, CA, United States). The sequence depth of two pools was 30× on average.
Raw reads were processed to obtain high-quality clean reads according to three stringent filtering standards: 1) removing reads with ≥10% unidentified nucleotides (N); 2) removing reads with >50% bases having Phred quality scores of ≤20; and 3) removing reads aligned to the barcode adapter.
To identify SNPs, the Burrows–Wheeler Aligner (BWA, Li and Durbin, 2009) was used to align the high-quality reads from each pool against the reference genome (GenBank accession numbers QCYY01000001-QCYY01004682; Zhang et al., 2019) with the settings “mem 4-k 32-M.” Variant calling was performed using the GATK’s UnifiedGenotyper. SNPs were filtered using GATK’s Variant Filtration with proper standards (-Window 4, -filter “QD < 2.0 || FS > 60.0 || MQ < 40.0,” -G_filter “GQ < 20”), and those exhibiting segregation distortion or sequencing errors were discarded. To determine the physical positions of each SNP, the software tool ANNOVAR (Wang et al., 2010) was used to align and annotate SNPs. Insertions/deletions (indels) were not considered in this study because the reliability of next generation sequencing to detect such variants was relatively low (Boland et al., 2013).
Selective sweep regions were selected according to the interception of two parameters, Fixation Index (Fst) (Hudson et al., 1992) and p ratio (ratio of nucleotide diversity between two populations, πA/πB) (Nei and Li, 1979). A 50-kb sliding window approach with 25-kb step size was applied to quantify these parameters with in-house PERL scripts. Regions under selection were identified using an empirical procedure (Li et al., 2018). In brief, genomic regions with significantly biased p ratios (p < 0.01) and significantly high Fst values (p < 0.01) of the empirical distribution were regarded as regions with strong selection signals along the genome. All related graphs were drawn using R scripts (R Core Team, 2006).
Candidate SNPs and genes within the sweep regions were extracted for further analysis. To evaluate the reliability of SNP prediction using pooled DNA-seq data, 50 candidate SNPs were genotyped in 60 samples based on individual DNA-seq data using the MALDI-TOF method. The 60 samples were obtained from the offspring of the three populations with contrasting reproduction traits, among which 30 were of high fecundity and 30 were of low fecundity. The SNP index of each locus and the frequency difference distribution in the two bulks were calculated. The direction was as follows: Δ(index) = index (HB) − index (LB). Only significant SNPs were selected as candidates for validation. The locus with Δ index close to 1 or to −1 was used as the preferred sites for verification.
All candidate genes were mapped to GO terms in the Gene Ontology database (http://www.geneontology.org/). Gene numbers were calculated for every term, and significantly enriched GO terms were defined using hypergeometric test with p < 0.05. KEGG is a major public pathway–related database. The calculation formula is the same as that in GO analysis. Pathways with p < 0.05 were defined as significantly enriched pathways in genes.
The fecundity indices of 87 females with offspring ranged from −1.59 to 1.52 for population A, −1.44 to 1.65 for population B, and −0.99 to 2.23 for population C (Figures 1A–C). In the QQ plots, most points were close to the fitting line, which showed that the fecundity indices of those females displayed normal distribution patterns in the three populations after the Shapiro–Wilk tests (p > 0.05, Figures 1D–F). In populations A, B, and C, 13, 10, and 7 individuals with the highest fecundity index were selected into HB, respectively. The average value of the fecundity indices of HB were 0.80, 0.91, and 0.57, respectively. In populations A, B, and C, 12, 7, and 11 individuals with no sexual maturity record were selected into LB, respectively (Table 1; Figures 1A–C). There were nine mutual families between HB and LB.
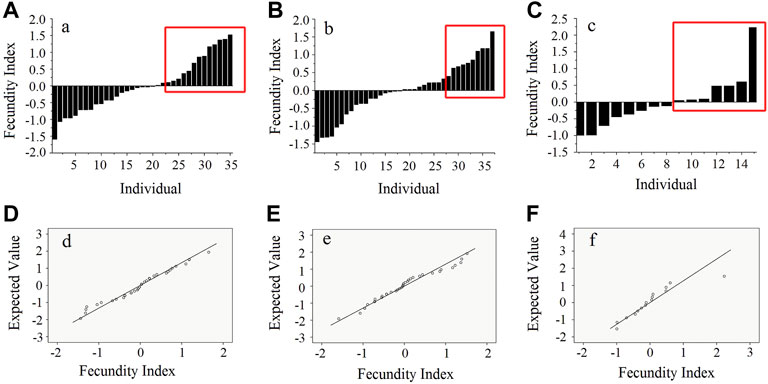
FIGURE 1. Distribution of fecundity indices. (A–C) Fecundity indices of 87 females with offspring in population. (A–C) (D–F) The QQ plot shows a normal distribution of females with offspring in three populations.
Using Illumina resequencing, a total of 1,192 million reads were obtained with about 548.87 million reads from HB and 643.51 million reads from LB (Table 2). Raw read data of HB and LB were deposited in the NCBI Sequence Read Archive under the accession number PRJNA610662. After quality control, 1,106 million reads were used for further analysis, giving rise to total clean data of 137.89 Gb with an average read length of 150 bp.
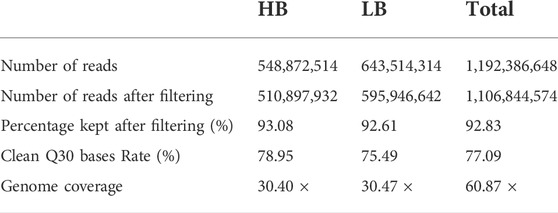
TABLE 2. Summary of Illumina sequencing of the high fecundity bulk (HB) and low-fecundity population (LB).
The Fst and p ratio were used to detect the selective sweep signals between HB and LB (Figure 2). The thresholds of each index determined by p < 0.01 were as follows: 0.1619 for Fst and 1.2977 for p ratio. The intersection part was considered as the reliable candidate interval. The significant interval and gene information in the interval were obtained according to the above threshold. The candidate region was generally the chromosome region with a high degree of genetic differentiation between HB and LB.
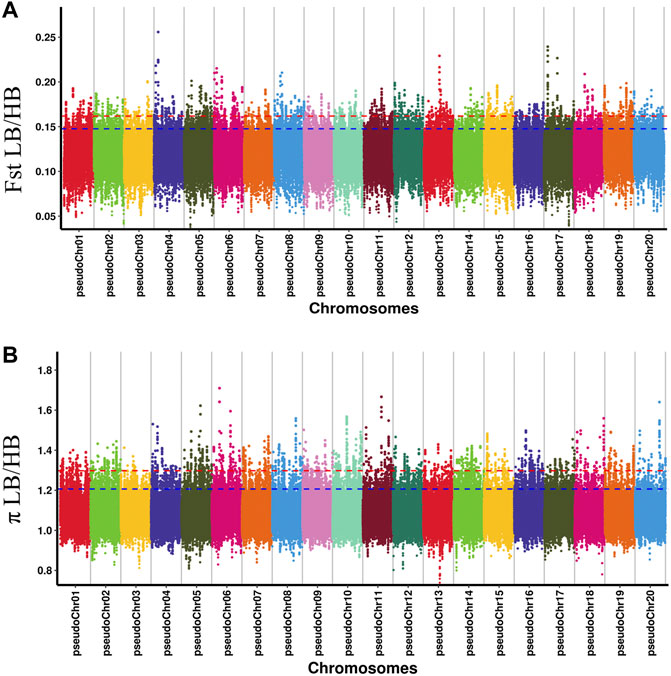
FIGURE 2. Manhattan distribution of fixation index (Fst) (A) and p ratio (B) of each pseudochromosome.
The intersection of selected parts of the Fst and p ratio is presented in Figure 3. The upper and bottom panels are the Fst and p ratio, respectively, for 100-kb windows compared between HB and LB. A total of 145 selected regions were screened, with a sequence length of 14.5 Mb, accounting for 0.59% of the genome.

FIGURE 3. Schematic diagram of selection signal.
All significant SNPs were assigned to 20 tentative chromosomes based on the P. vannamei genome sequence (GenBank accession number: PRJNA508983). A total of 625,181 SNPs were shared by HB and LB based on Fst, and 629,748 SNPs were shared based on the p ratio (p < 0.01). Among them, 377,714 and 376,872 were transitions (Ts) and 247,467 and 252,876 were transversions (Tv), with Ts/Tv ratios of 1.53 and 1.49, respectively (Figure 4). After the intersection of the limited SNP ranges of the two parameters, 64,046 significant SNPs were found, among which 1,136 were located in the coding regions, 12,048 in the intronic regions, 1,651 in the upstream or downstream regions, 49,187 in the intergenic regions, and 24 in the splicing regions.
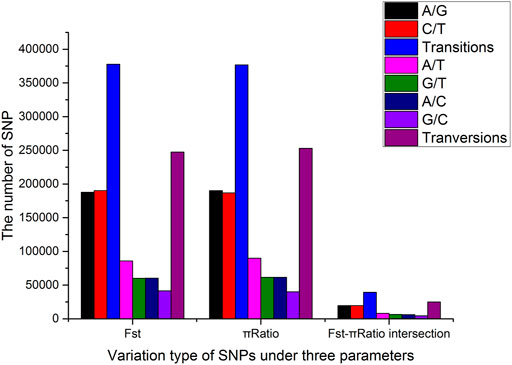
FIGURE 4. Type distribution of single nucleotide polymorphisms (SNPs) shared by the high fecundity bulk (HB) and low-fecundity population (LB) under two parameters.
To validate the reliability of SNP prediction using selected sweep analysis, 50 significant SNPs were genotyped in 60 independent samples based on their respective DNA. After removing the loci with a genotyping completion rate of less than 80%, there were 44 effective loci left. All the 44 candidate SNPs were polymorphic in HB, and 42 candidate SNPs were polymorphic in LB. The allele frequency distributions of these SNPs in HB and LB are presented in Supplementary Table S2. Six loci showed significant allelic imbalance between HB and LB at the level of p < 0.05, among which two loci were at the level of p < 0.01 (Table 3).
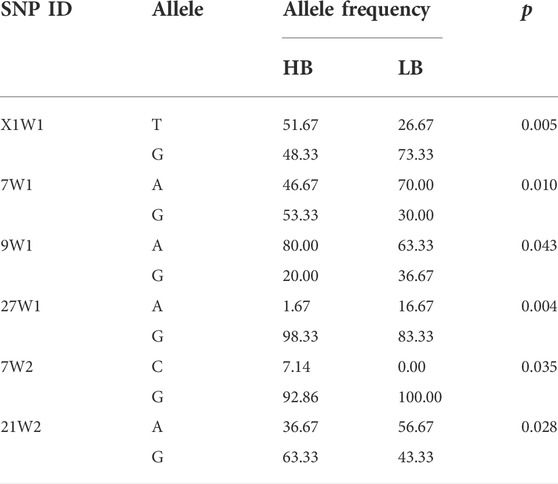
TABLE 3. Comparison of allele frequency distributions of 44 candidate single nucleotide polymorphisms (SNPs) between the HB and LB using Pearson’s chi-squared test.
Among the 145 selective sweep regions, 121 genes were identified (Figure 5, Supplementary Table S1). These genes provided genomic evidence of selection signatures and could be used as important loci for targeted genotyping in shrimp breeding. Thus, these genes are interestingly functional candidates for reproduction-related traits under selection on these populations. To further analyze the functions of these genes, we carried out GO annotation and KEGG enrichment analyses. As presented in Figure 6, the abscissa represented the three ontologies of GO: molecular function, including cellular process, localization, metabolic process, and single-organism process; cellular component, including cell, cell part, membrane, membrane part, and organelle; and biological process, including binding, catalytic activity, and transporter activity. Most of the highly enriched GO terms were associated with biological process and molecular function (Supplementary Table S3). The genes related to molecular function were included in the cyclohydrolase (GO: 0019238), acetylglucosaminyltransferase (GO: 0008375), and hydroxymethyl-, formyl-, and related transferase (GO: 0016742) activities. The genes related to biological process were included in calcium ion transport (GO: 0006816), divalent metal ion transport (GO: 0070838), divalent inorganic cation transport (GO: 0072511), and purine nucleotide metabolic process (GO: 0006163). Many genes were categorized into cellular and metabolic processes. Both categories had cell growth or anabolic/catabolic process resulting in cell growth, suggesting that these genes could be associated with fecundity in shrimp.
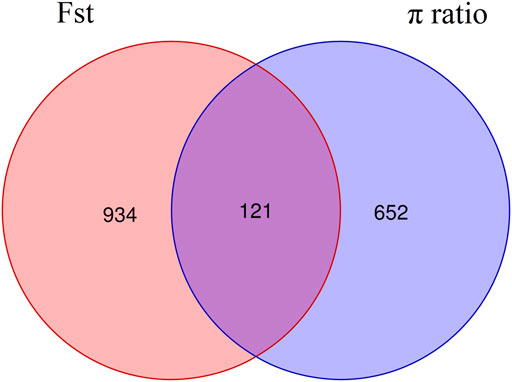
FIGURE 5. Venn diagram of genes in the selected regions screened by Fst and p ratio.
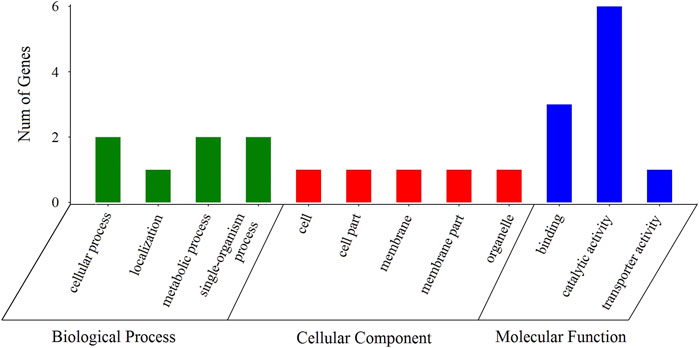
FIGURE 6. Distributions of candidate genes in different GO categories.
KEGG analysis showed that the 121 genes were enriched into 73 pathways, among which 19 pathways with p < 0.05 and 15 pathways with Q < 0.05 (adjusted p-value) were identified as significantly enriched pathways (Table 4, Supplementary Table S4). Most of the highly enriched pathways included cardiac muscle contraction (ko04260), adrenergic signaling in cardiomyocytes (ko04261), and cGMP–PKG signaling pathway (ko04022). The KEGG analysis showed that the pathways of some genes have been proven to be closely related to ovarian development, such as serotonergic synapse (ko04726) (Jayasankar et al., 2020). Some of the 121 candidate genes related to fecundity are presented in Table 5.

TABLE 4. Detailed information of significantly enriched KEGG pathways of the candidate genes.
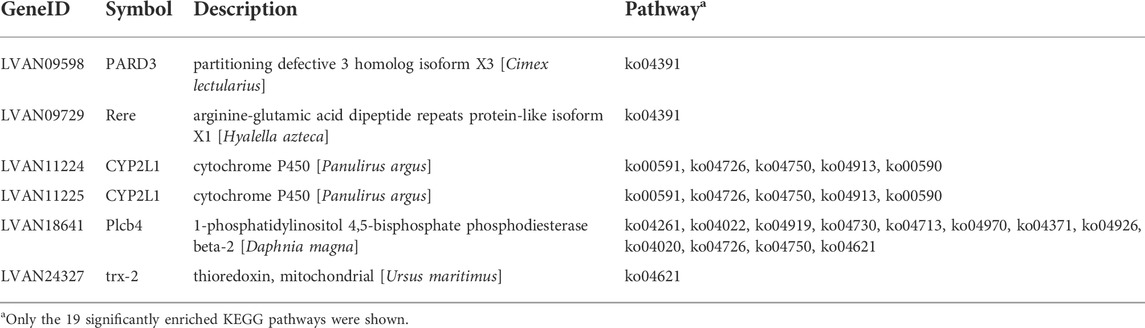
TABLE 5. Some of the 121 candidate genes related to fecundity.
Over the past 30 years, a variety of P. vannamei strains with specific phenotypic and genotypic characteristics have been cultivated in different artificial selection projects. Studies on Atlantic salmon have shown that even selecting the same trait may leave different marks in the genomes of different populations (Gutierrez et al., 2015a; Gutierrez et al., 2015b; López et al., 2019). The possible reason was that selection may act on different genes (Amaral et al., 2011), and similar phenotypes may form different genetic pathways among different populations (Vasemägi et al., 2012; Perrier et al., 2013; Elmer et al., 2014; Mäkinen et al., 2014; Pujolar et al., 2017). In this study, three populations were used, among which two were characterized by strong resistance and one by fast growth. To the best of our knowledge, this is the first study of genomic signatures of artificial selection in fecundity of female shrimp with pooled DNA-seq. The genomic signatures in fecundity obtained from populations with different characteristics may be less but more representative.
The combination of DNA-seq and the DNA pooling approach can associate specific regions of the genomic signatures with target traits. Using this strategy, Bertolini et al. (2016) identified genome-wide polymorphisms in two cultured European sea bass (Dicentrarchus labrax). Naval-Sanchez et al. (2020) analyzed the domestication of aquaculture species independently, which was the first analysis on the domestication of aquaculture species. In this study, more than 600,000 SNPs were mined for each bulk. Fst and p ratio were used to determine the selective sweep region. Fst was calculated using the Bayesian method, which might result in a high degree of false positives (Lotterhos and Whitlock, 2015). After the intersection of Fst and p ratio, only 64,046 SNPs were shared by both bulks, showing that the setting threshold was strict (p < 0.01). In the selective sweep regions, the shared sites exhibited a Ts/Tv ratio of 1.49 to 1.53, which is lower than another ratio reported in P. vannamei (2.0, Yu et al., 2014). In fish, the reported Ts/Tv ratio ranged from 0.95 to 1.49 in several Oncorhynchus species (Smith et al., 2005), 1.69 in Takifugu rubripes (Cui et al., 2014), and 1.37 in Salmo salar (Hayes et al., 2007). If transition and transversion randomly occurred, the Ts/Tv ratio should be 0.5, because there are two possibilities of transitions and four possibilities of transversion (Kraus et al., 2011). However, transition is usually more frequent than transversion in the genome (Rosenberg et al., 2003), which may be related to cytosine methylation (Shen et al., 1994). Different targeted regions may affect the Ts/Tv ratio, like whole-genome, exon, specific genes, which is also greatly affected by the CpG islands and GC content of the region (Dohm et al., 2008). The Ts/Tv ratio can reflect the characteristics of a targeted region of a specific genome to a certain extent.
As SNP calling was based on bioinformatics analysis of pooled DNA-seq data, it was necessary to validate the polymorphisms of SNPs at an individual level. Even though the most rigorous parameter setting was used to determine the selective sweep regions, in the subsequent SNP verification, only 6 of the 44 loci displayed significant differences in populations with much different fecundities. The SNPs had a high false-positive rate even in the offspring population, which showed genomic signatures might have strong population specificity in P. vannamei. If more samples were used for genotyping, the proportion would increase. The SNPs detected in this study could be used to calculate the genomic relationship matrix (GRM) and predict the genomic estimated breeding value (GEBVs) of candidate populations in the future GS programs (Luo et al., 2021), which have higher prediction accuracy than the GBLUP method.
Among the 121 identified genes in the selective sweep region, LvPlcb4 existed in 45 of the 73 enriched KEGG pathways. It belonged to the phospholipase C (PLC) family and was a membrane-related enzyme. The bioactive substances produced by the PLC family participate in a variety of cellular processes through intercellular and extracellular signal transduction pathways (Kim et al., 2015). PLC generated the second messengers inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) by catalyzing the hydrolysis of PIP2 to stimulate the release of Ca2+ in cells. Granulosa cells indirectly regulate the development and maturation of oocytes by transmitting signals of IP3, Ca2+, and other second messengers to oocytes (Buccione et al., 1990; Nance, 2005). The homeostasis of Ca2+ is closely related to the genesis of oocytes (Zhang et al., 2010). Lvpard3 is a homologue of partitioning defective 3 (PARD3) gene, which is a scaffold protein and plays an important role in regulating cell polarity, proliferation, migration, and tight junction (Assémat et al., 2008). Khadar found that PARD3 could promote cell proliferation and growth (Khadar and Kuo, 2018). Dong et al. observed in mice that luteinizing hormone can enhance the polarity of ovarian granulosa cells and promote cell proliferation and growth by stimulating the expression of pard3 in ovarian granulosa cells (Dong et al., 2021). Lvtrx2 is another important gene annotated in NOD-like receptor signaling pathway in this study. TRX2 is a member of thioredoxin (TRX) super protein family, which may be involved in the optimal growth and maturation of follicles (Damdimopoulos et al., 2002). Lvcyp2L1 is a member of the cytochrome P450 enzyme system, is widely distributed in the endoplasmic reticulum and mitochondria in the cell membrane system, and is one of the key enzymes for steroid transformation before oocyte maturation (Boyle et al., 1998). Sakai showed that the P450c17 gene was highly expressed in the prophase of rainbow trout ovary and oviposition (Sakai et al., 1992). In channel catfish, Kumar detected that the P450c17 gene was at a high level in the middle stage of oocyte development (Kumar et al., 2000). It is important to note that the use of pooled samples for DNA-seq does not inherently enable the evaluation of within-group variation in expression profiles, such as inter-individual or inter-family variation. Therefore, the genes identified in this study should be considered with caution until further validation.
In conclusion, in two fecundity separated populations of P. vannamei, 145 selective sweep regions in the genome were identified combining two parameters, namely, Fst and p ratio. A total of 64,046 SNPs and 121 genes were screened from the sweep regions, which need further verification. GO annotation and KEGG enrichment analyses showed that partial genes were essential for fecundity regulation. These results would provide important clues for the differences in fecundity of female Pacific white shrimp at the genomic level.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JS and SL analyzed the data; SL and JK acquired fund; JC, KL, BC, and JT cultivated the experimental materials; XM and JK administered the project; JS wrote the original draft; PD and QF performed review and editing.
This work was financially supported by the National Key R&D Program of China (2018YFD0901303), Shandong Province Agriculture Major Applied Technology Innovation Project (SD2019YY001), China Agriculture Research System (CARS-48), Science and Technology Innovation Team project of Chinese Academy of Fishery Sciences (No. 2020TD26), Talent Team bidding Project of Marine Equipment and Marine Biology in Zhanjiang (2021E05032), Scientific Research Project from Hainan Academician Innovation platform (SQ2021PTZ0014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.929889/full#supplementary-material
Amaral, A. J., Ferretti, L., Megens, H.-J., Crooijmans, R. P. M. A., Nie, H., Ramos-Onsins, S. E., et al. (2011). Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PLOS ONE 6 (4), e14782. doi:10.1371/journal.pone.0014782
Andersson, L. (2012). How selective sweeps in domestic animals provide new insight into biological mechanisms. J. Intern. Med. 271 (1), 1–14. doi:10.1111/j.1365-2796.2011.02450.x
Andersson, L. (2013). Molecular consequences of animal breeding. Curr. Opin. Genet. Dev. 23 (3), 295–301. doi:10.1016/j.gde.2013.02.014
Arcos, F. G., Ibarra, A. M., Vazquez-Boucard, C., Palacios, E., and Racotta, I. S. (2003). Feasible predictive criteria for reproductive performance of white shrimp Litopenaeus vannamei: Egg quality and female physiological condition. Aquaculture 228, 335–349. doi:10.1016/s0044-8486(03)00313-2
Arcos, F. G., Racotta, I. S., and Ibarra, A. M. (2004). Genetic parameter estimates for reproductive traits and egg composition in Pacific with shrimp Penaeus (Litopenaeus). Aquaculture 236, 151–165. doi:10.1016/j.aquaculture.2004.03.003
Assémat, E., Bazellières, E., Pallesi-Pocachard, E., Le Bivic, A., and Massey-Harroche, D. (2008). Polarity complex proteins. Biochim. Biophys. Acta 1778 (3), 614–630. doi:10.1016/j.bbamem.2007.08.029
Benzie, J. A. H. (2009). Use and exchange of genetic resources of penaeid shrimps for food and aquaculture. Rev. Aquac. 1, 232–250. doi:10.1111/j.1753-5131.2009.01018.x
Bertolini, F., Geraci, C., Schiavo, G., Sardina, M. T., Chiofalo, V., Fontanesi, L., et al. (2016). Whole genome semiconductor based sequencing of farmed European sea bass (Dicentrarchus labrax) Mediterranean genetic stocks using a DNA pooling approach. Mar. Genomics 28, 63–70. doi:10.1016/j.margen.2016.03.007
Bertolotti, A. C., Layer, R. M., Gundappa, M. K., Gallagher, M. D., Pehlivanoglu, E., Nome, T., et al. (2020). The structural variation landscape in 492 Atlantic salmon genomes. Nat. Commun. 11 (1), 5176. doi:10.1038/s41467-020-18972-x
Boland, J. F., Chung, C. C., Roberson, D., Mitchell, J., Zhang, X., Im, K. M., et al. (2013). The new sequencer on the block: Comparison of life technology's proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum. Genet. 132, 1153–1163. doi:10.1007/s00439-013-1321-4
Boyle, S. M., Greenberg, R. M., and Jame, M. O. (1998). Isolation of CYP2L2 and Two other cytochrome P450 sequences from a spiny lobster, Panzdirus argus, hepatopancreas cDNA library. Mar. Environ. Res. 46 (5), 21–24. doi:10.1016/S0141-1136(97)00103-7
Buccione, R., Schroeder, A. C., and Eppig, J. J. (1990). Minireview: Interactions between somatic cells and germ cells throughout mammalian oogenesis. Biol. Reprod. 43 (4), 543–547. doi:10.1095/biolreprod43.4.543
Caballero-Zamora, A., Cienfuegos-Rivas, E. G., Montaldo, H. H., Campos-Montes, G. R., Martínez-Ortega, A., and Castillo-Juárez, H. (2015). Genetic parameters for spawning and growth traits in the Pacific white shrimp (Penaeus (Litopenaeus) vannamei). Aquac. Res. 46, 833–839. doi:10.1111/are.12235
Cadiz, M. I., López, M. E., Díaz-Domínguez, D., Cáceres, G., Yoshida, G. M., Gomez-Uchida, D., et al. (2019). Whole genome re-sequencing reveals recent signatures of selection in three strains of farmed Nile tilapia (Oreochromis niloticus). Sci. Rep. 10, 11514. doi:10.1038/s41598-020-68064-5
Castillo-Juárez, H., Campos-Montes, G. R., Caballero-Zamora, A., and Montaldo, H. H. (2015). Genetic improvement of pacific white shrimp [Penaeus (Litopenaeus) vannamei]: Perspectives for genomic selection. Front. Genet. 6, 93. doi:10.3389/fgene.2015.00093
Cui, J., Wang, H. D., Liu, S., Qiu, X., Jiang, Z., and Wang, X. (2014). k., Qiu, X. M., Jiang, Z. Q., and Wang, X. LTranscriptome analysis of the gill of Takifugu rubripes using Illumina sequencing for discovery of SNPs. Comp. Biochem. Physiol. Part D. Genomics Proteomics 10, 44–51. doi:10.1016/j.cbd.2014.03.001
Damdimopoulos, A. E., Miranda-Vizuete1, A., Pelto-Huikko, M., Gustafsson, J., and Spyrou, G. (2002). Å., and Spyrou, GHuman mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 277 (36), 33249–33257. doi:10.1074/jbc.M203036200
Dohm, J. C., Lottaz, C., Borodina, T., and Himmelbauer, H. (2008). Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36, e105. doi:10.1093/nar/gkn425
Dong, Y. T., Chen, T. R., Ye, X. F., Hei, C. C., Cai, Y. F., Kong, B., et al. (2021). Proteomic analysis of the effects of luteinizing hormone on rat primary ovarian granulosa cells. J. Ningxia Med. Univ. 43 (06), 569–576. doi:10.16050/j.cnki.issn1674-6309.2021.06.004
Elmer, K. R., Fan, S., Kusche, H., Spreitzer, M. L., Kautt, A. F., Franchini, P., et al. (2014). Parallel evolution of Nicaraguan crater lake cichlid fishes via non-parallel routes. Nat. Commun. 5, 5168. doi:10.1038/ncomms6168
FAO (2022). FishStat database. AvaliableAt: www.fao.org/fishery/statistics/software/fishstatj/en.
Gjedrem, T. (2012). Genetic improvement for the development of efficient global aquaculture: A personal opinion review. Aquaculture 344–349, 12–22. doi:10.1016/j.aquaculture.2012.03.003
Gjedrem, T., and Rye, M. (2016). Selection response in fish and shellfish: A review. Rev. Aquac. 10 (1), 168–179. doi:10.1111/raq.12154
Gutierrez, A. P., Yáñez, J. M., and Davidson, W. S. (2015a). Evidence of recent signatures of selection during domestication in an Atlantic salmon population. Mar. Genomics 26, 41–50. doi:10.1016/j.margen.2015.12.007
Gutierrez, A. P., Yáñez, J. M., Fukui, S., Swift, B., and Davidson, W. S. (2015b). Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS One 10, e0119730. doi:10.1371/journal.pone.0119730
Hayes, B., Laerdahl, J. K., Lien, S., Moen, T., Berg, P., Hindar, K., et al. (2007). An extensive resource of single nucleotide polymorphism markers associated with Atlantic salmon (Salmo salar) expressed sequences. Aquaculture 265, 82–90. doi:10.1016/j.aquaculture.2007.01.037
Hudson, Richard R., Slatkin, M., and Maddison, W. P. (1992). Estimation of levels of gene flow from DNA sequence data. Genetics 132 (2, 1), 583–589. doi:10.1093/genetics/132.2.583
Jayasankar, V., Tomy, S., and Wilder, M. N. (2020). Insights on molecular mechanisms of ovarian development in decapod crustacea: Focus on vitellogenesis-stimulating factors and pathways. Front. Endocrinol. 11, 577925. doi:10.3389/fendo.2020.577925
Khadar, A., and Kuo, C. T. (2018). Laminating the mammalian cortex during development: Cell polarity protein function and Hippo signaling. Genes. Dev. 32 (11–12), 740–741. doi:10.1101/gad.316711.118
Kim, S. W., Cho, T., and Lee, S. (2015). Phospholipase C-β1 hypofunction in the pathogenesis of schizophrenia. Front. Psychiatry 6, 159. doi:10.3389/fpsyt.2015.00159
Kon, T., Pei, L., Ichikawa, R., Chen, C., Wang, P., Takemura, I., et al. (2021). Whole-genome resequencing of large yellow croaker (Larimichthys crocea) reveals the population structure and signatures of environmental adaptation. Sci. Rep-UK 11 (1), 1–11. doi:10.1038/s41598-021-90645-1(
Kraus, R. H. S., Kerstens, H. H. D., Van Hooft, P., Crooijmans, R. P. M. A., Van der Poel, J. J., Elmberg, J., et al. (2011). Genome wide SNP discovery, analysis and evaluation in mallard (Anas platyrhynchos). BMC Genomics 12 (150), 150. doi:10.1186/1471-2164-12-150
Kumar, R. S., Ijiri, S., and Trant, J. M. (2000). Changes in the expression of genes encoding steroidogenic enzymes in the channel catfish (Ictalurus punctatus) ovary throughout a reproductive cycle. Biol. Reprod. 63 (6), 1676–1682. doi:10.1095/biolreprod63.6.1676
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25 (14, 15), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, L., Li, A., Song, K., Meng, J., Guo, X., Li, S., et al. (2018). Divergence and plasticity shape adaptive potential of the Pacific oyster. Nat. Ecol. Evol. 2, 1751–1760. doi:10.1038/s41559-018-0668-2
López, M. E., Benestan, L., Moore, J. S., Perrier, C., Gilbey, J., Genova, A. D., et al. (2019). Comparing genomic signatures of domestication in two Atlantic salmon (Salmo salar L.) populations with different geographical origins. Evol. Appl. 12 (1), 137–156. doi:10.1111/eva.12689
Lotterhos, K. E., and Whitlock, M. C. (2015). The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 24 (5), 1031–1046. doi:10.1111/mec.13100
Luo, Z., Yu, Y., Xiang, J., and Li, F. (2021). Genomic selection using a subset of SNPs identified by genome-wide association analysis for disease resistance traits in aquaculture species. Aquaculture 539, 736620. doi:10.1016/j.aquaculture.2021.736620
Mäkinen, H., Vasemägi, A., McGinnity, P., Cross, T. F., and Primmer, C. R. (2014). Population genomic analyses of early-phase Atlantic Salmon (Salmo salar) domestication/captive breeding. Evol. Appl. 8 (1), 93–107. doi:10.1111/eva.12230
Nam, B.-H., Yoo, D. A., Kim, Y.-O., Park, J. Y., Shin, Y. G.-H., Park, C. I., et al. (2020). Author Correction: Whole genome sequencing reveals the impact of recent artificial selection on red sea bream reared in fish farms. Sci. Rep. 10 (1), 1625. doi:10.1038/s41598-020-58614-2
Nance, J. (2005). PAR proteins and the establishment of cell polarity during C. elegans development. Bioessays 27 (2), 126–135. doi:10.1002/bies.20175
Naval-Sanchez, M., McWilliam, S., Evans, B., Yáñez, J. M., Houston, R. D., Kijas, J. W., et al. (2020). Changed patterns of genomic variation following recent domestication: Selection sweeps in farmed atlantic salmon. Front. Genet. 11, 264. doi:10.3389/fgene.2020.00264
Nei, M., and Li, W.-H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76 (10), 5269–5273. doi:10.1073/pnas.76.10.5269
Perrier, C., Bourret, V., Kent, M. P., and Bernatchez, L. (2013). Parallel and nonparallel genome-wide divergence among replicate population pairs of freshwater and anadromous Atlantic salmon. Mol. Ecol. 22 (22), 5577–5593. doi:10.1111/mec.12500
Pujolar, J. M., Ferchaud, A. L., Bekkevold, D., and Hansen, M. M. (2017). Non-parallel divergence across freshwater and marine three-spined stickleback Gasterosteus aculeatus populations. J. Fish. Biol. 91 (1), 175–194. doi:10.1111/jfb.13336
R Core Team (2006). A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Racotta, I. S., Palacios, E., and Ibarra, A. M. (2003). Shrimp larval quality in relation to broodstock condition. Aquaculture 227, 107–130. doi:10.1016/s0044-8486(03)00498-8
Rosenberg, M. S., Subramanian, S., and Kumar, S. (2003). Patterns of transitional mutation biases within and among mammalian genomes. Mol. Biol. Evol. 20, 988–993. doi:10.1093/molbev/msg113
Rubin, C. J., Zody, M. C., Eriksson, J., Meadows, J. R. S., Sherwood, E., Webster, M. T., et al. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464 (7288), 587–591. doi:10.1038/nature08832
Sakai, N., Tanaka, M., Adachi, S., Miller, W. L., and Nagahama, Y. (1992). Rainbow trout cytochrome P-450c17 (17–hydroxylase/17, 20-lyase)-cDNA cloning, enzymatic properties and temporal pattern of ovarian P-450c 17 mRNA expression during oogenesis. FEBS Lett. 301, 60–64. doi:10.1016/0014-5793(92)80210-8
Shen, J. C., Rideout, W. M., and Jones, P. A. (1994). The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976. doi:10.1093/nar/22.6.972
Smith, C. T., Elfstrom, C. M., Seeb, L. W., and Seeb, J. E. (2005). Use of sequence data from rainbow trout and Atlantic salmon for SNP detection in Pacific salmon. Mol. Ecol. 14 (13), 4193–4203. doi:10.1111/j.1365-294X.2005.02731.x
Stratton, M. (2008). Genome resequencing and genetic variation. Nat. Biotechnol. 26 (1), 65–66. doi:10.1038/nbt0108-65
Tan, J., Luan, S., Cao, B. X., Luo, K., Meng, X. H., Kong, J., et al. (2019). Evaluation of genetic parameters for reproductive traits and growth rate in the Pacific white shrimp Litopenaeus vannamei reared in brackish water. Aquaculture 511, 734244. doi:10.1016/j.aquaculture.2019.734244
Utsunomiya, Y. T., Pérez, A. M., O’Brien, T. S., Sonstegard, J. S., and Garcia, J. F. (2015). Genomic data as the “hitchhiker’s guide” to cattle adaptation: Tracking the milestones of past selection in the bovine genome. Front. Genet. 6, 36. doi:10.3389/fgene.2015.00036
Vasemägi, A., Nilsson, J., McGinnity, P., Cross, T., O’Reilly, P., Glebe, B., et al. (2012). Screen for footprints of selection during domestication/captive breeding of Atlantic salmon. Comp. Funct. Genomics 2012 (1), 628204. doi:10.1155/2012/628204
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wong, L. L., Deris, Z. M., Igarashi, Y., Huang, S., Asakawa, S., Ayub, Q., et al. (2020). Skim-sequencing based genotyping reveals genetic divergence of the wild and domesticated population of black tiger shrimp (Penaeus monodon) in the Indo-Pacific Region. Biology 9 (9), 277. doi:10.3390/biology9090277
Xia, J. H., Bai, Z., Meng, Z., Zhang, Y., Wang, L. E., Liu, F., et al. (2015). Signatures of selection in tilapia revealed by whole genome resequencing. Sci. Rep. 5, 14168. doi:10.1038/srep14168
Yu, Y., Wei, J. K., Zhang, X. J., Liu, J. W., Liu, C. Z., Li, F. H., et al. (2014). SNP discovery in the transcriptome of white Pacific shrimp Litopenaeus vannamei by next generation sequencing. PLOS ONE 9 (1), e87218. doi:10.1371/journal.pone.0087218
Zhang, D. X., Li, X. P., Sun, S. C., Shen, X. H., Cui, X. S., Kim, N.-H., et al. (2010). Involvement of ER–calreticulin–Ca2+ signaling in the regulation of porcine oocyte meiotic maturation and maternal gene expression. Mol. Reprod. Dev. 77 (5), 462–471. doi:10.1002/mrd.21166
Zhang, X. J., Yuan, J. B., Sun, Y. M., Li, S. H., Gao, Y., and Yu, Y. (2019). Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat. Commun. 10, 356. doi:10.1038/s41467-018-08197-4
Keywords: fecundity, whole-genome analysis, single nucleotide polymorphisms (SNPs), Penaeus vannamei, selective sweep analysis
Citation: Sui J, Luan S, Cao J, Dai P, Meng X, Luo K, Chen B, Tan J, Fu Q and Kong J (2022) Genomic signatures of artificial selection in fecundity of Pacific white shrimp, Penaeus vannamei. Front. Genet. 13:929889. doi: 10.3389/fgene.2022.929889
Received: 27 April 2022; Accepted: 14 July 2022;
Published: 29 August 2022.
Edited by:
Xiangshan Ji, Shandong Agricultural University, ChinaCopyright © 2022 Sui, Luan, Cao, Dai, Meng, Luo, Chen, Tan, Fu and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Kong, YXF1YWdlbmVAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.