 Yuhao Wu
Yuhao Wu Long Wen
Long Wen Peiru Wang
Peiru Wang Xiuli Wang
Xiuli Wang Guolong Zhang
Guolong Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 25 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.926451
This article is part of the Research Topic The Genetics of Human Mendelian Skin Disorders View all 11 articles
Congenital poikiloderma is an extremely rare autosomal dominant genetic syndrome, characterized by a combination of early onset poikiloderma, telangiectasia, and epidermal atrophy. FAM111B gene with multiple mutations has been identified as a potential causative gene for congenital poikiloderma. In this report, we described a boy with congenital poikiloderma confirmed by clinical manifestations. Next-generation sequencing based on a gene probe panel consisting of 541 genetic loci of genodermatoses, was used to screen mutations of the proband and his parents. Results showed that a missense mutation in the FAM111B gene c.1883G>A (rs587777238) was identified in the proband, but absent in his parents, indicating the mutation is de novo. In conclusion, a new case of congenital poikiloderma in China was reported, and the hotspot mutations in codon 628 of FAM111B gene was reviewed, as well as authenticating the uncertain association between genotypes and phenotypes in this rare disease.
Congenital poikiloderma (hereditary fibrosing poikiloderma) is an extremely rare syndromic form of the autosomal dominant disease. It is characterized by a combination of early onset poikiloderma, telangiectasia, epidermal atrophy, tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP), accompanied with the deficiency of eccrine sweat glands (also called hypohidrosis), sparse scalp hair and absent body hair, including eyebrows and eyelashes (Rayinda et al., 2021). In 2013, family with sequence similarity 111 member B (FAM111B) mutations were reported to be responsible for congenital poikiloderma (Mercier et al., 2013). Its mode of inheritance and primary clinical features were first described in two generations of a multiplex South African family. FAM111B has also been reported to be associated with inherited exocrine pancreatic dysfunction and prostate cancer (Akamatsu et al., 2012; Seo et al., 2016). In addition, FAM111B has been confirmed as a direct target of p53 and identified as an oncogene for lung adenocarcinoma (Sun et al., 2019). However, the underlying pathogenic mechanism concerning FAM111B mutations is still unclear.
Herein, we reported a 5-year-old boy with mottled pigmentation, telangiectasia, epidermal atrophy, and a missense mutation (c.1883G>A) of FAM111B gene was identified. Furthermore, the mutations in codon 628 of FAM111B gene were reviewed and the uncertain association between genotypes and phenotypes in this rare disease was also authenticated.
The current study conformed to the tenets of the Helsinki declaration and was approved by Ethical Committee of Shanghai skin disease hospital. The proband, his parents and 120 ethnically matched control individuals were informed regarding the purpose of the study and written consent was provided prior to recruitment and sampling.
A 5-year-old boy was admitted to the Shanghai Skin Disease Hospital outpatient department for developed blisters on the scalp, that were present 1 month after his birth, which gradually spread to the whole body and turned into poikiloderma after 3 months (Figure 1). The lesion was predominantly located on the face and in the other sun-exposed areas, which were typical manifestations for the diagnosis of congenital poikiloderma (Figures 1A,B). He had hypohidrosis and also eczematous lesions on the trunk and legs (Figures 1C–F). No lymphoedema of the upper or lower extremities was observed. Since the onset of the disease, the rash has occurred repeatedly and aggravated in winter. His scalp hair was sparse, with eyelashes and eyebrows absent. His nails and teeth were normal. In addition, from the first year of life, elevated liver transaminase levels were observed on repeat blood samples, including aspartate aminotransferase (316 U/L; normal range, 15–40 U/L), alanine transferase (354 U/L; normal range, 9–50 U/L), γ-glutamyl transferase (334 U/L; normal range, 10–60 U/L), alkaline phosphatase (532 U/L; normal range, 0–500 U/L) and lactate dehydrogenase (362 U/L; normal range, 120–230 U/L). Vasodilation and hyperemia were also observed. The results were consistent with the manifestation in congenital poikiloderma.
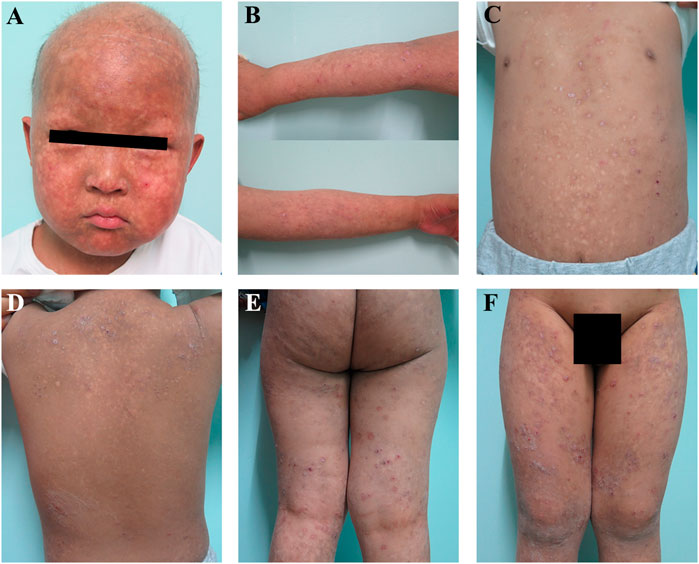
FIGURE 1. Clinical features of the patient (A,B) Congenital poikiloderma, including mottled pigmentation, telangiectasia, epidermal atrophy, sparse scalp hair, as well as absent eyelashes and eyebrows (C–F) The patient also had eczematous lesions on the trunk and legs.
To investigate the underlying mutation of congenital poikiloderma, next-generation sequencing based on a multi-gene probe panel consisting of 541 genes of monogenic hereditary diseases was used to screen mutations of the proband and his parents. In detail, genomic DNA was extracted from the peripheral blood using the Wizard Genomic DNA purification kit (Promega Corporation). A total of 120 unrelated population-matched control samples were also used to exclude the possibility that these were polymorphisms. Total DNA was isolated from peripheral blood using QIAamp DNA Mini kit (Qiagen, Inc.) according to the manufacturer’s instructions. DNA was concentrated and quality controled using a Qubit 3.0 Fluorometer instrument (Invitrogen; Thermo Fisher Scientific, Inc.) to ensure the concentration was higher than 40 ng/μL. The Illumina Hiseq X Ten sequencing platform (Illumina, Inc.) was used, with an average sequencing depth >200× and Q30 > 90%. To verify the accuracy of the identified mutation, direct Sanger sequencing was performed to confirm whether the variants co-segregated with the disease phenotype in the proband and his parents using an ABI PRISM 3730XL automated sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.). The sequencing reactions were all performed in forward and reverse directions. The American College of Medical Genetics (ACMG) classification of the variant was performed using the online tool Varsome (https://varsome.com/) (Kopanos et al., 2019).
A heterozygous point mutation, c.1883G>A (rs587777238) in FAM111B was detected, leading to an amino acid alternation from serine to asparagine (p.628S > N) (Figure 2). This mutation was absent from his unaffected parents, which indicates that it is a de novo event. According to the ACMG variant classification guideline, this variant was categorized as a pathogenic variant. Moreover, it was not found in any of the healthy controls also showing that it is a novel pathogenic mutation, not a common polymorphism. This mutation causes protein structural and functional changes, which induces the occurrence of this disease.
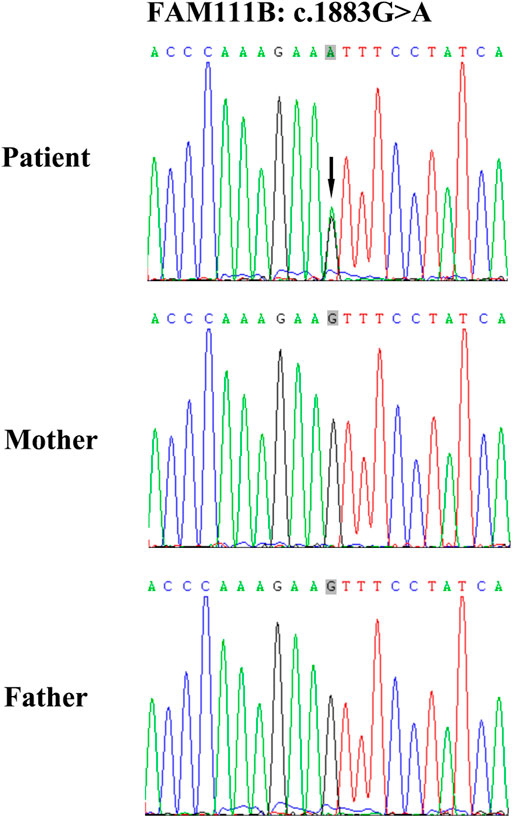
FIGURE 2. Genomic DNA of the patient was analyzed using a gene probe consisting of 541 genetic loci of Geno dermatoses. Sequences were aligned to GRCh38. The c.1883G>A mutation in exon four exhibited a heterozygous point mutation in the patient, indicated by the black arrow, which was absent in his unaffected parents.
The following terms were combined in the search strategy [FAM111B (Title/Abstract)] AND [poikiloderma (Title/Abstract)] from PubMed database. Then, the retrieved literatures were analyzed in full text. Mutations in condon 628 of FAM111B identified in congenital poikiloderma were summarized.
Previous studies suggested that codon 628 of FAM111B could be a mutation hotspot. A total of 8 cases with mutations in codon 628 were retrieved from PubMed and results from the present case report were also illustrated for comparison (Supplementary Table S1). Poikiloderma and hypohidrosis were found in every patient carrying FAM111B mutations in codon 628. In contrast to the other patients, the proband in our report showed more severe liver damage, while muscle weakness was not found. Patients mostly present with hypotrichosis, but patients in one pedigree reported by Goussot et al. showed no signs of the symptom (Goussot et al., 2017).
Congenital poikiloderma is primarily characterized by early onset poikiloderma, combining with several symptoms, such as telangiectasia and epidermal atrophy, which occurs in neonates and infants. The susceptible gene, FAM111B, was identified by Mercier et al. (Chen et al., 2019) in 2013, which is the second member of the two-gene “family with sequence similarity 111” gene family. FAM111B contains four exons and encodes 734 amino acids, which is likely to contain a trypsin-like cysteine/serine peptidase domain. The identification of mutations in FAM111B provided definitive evidence for POIKTMP and distinguishes it from other types of hereditary poikiloderma, such as Rothmund-Thomson syndrome (RTS), hereditary sclerosing poikiloderma of Weary, Kindler syndrome and Clericuzio-type poikiloderma with neutropaenia (Arnold et al., 2010; Küry et al., 2016; Gatinois et al., 2020). In approximately 50% of affected individuals, FAM111B pathogenic variant is de novo (Mercier et al., 1993), which is the same as the present study.
In the present study, a rare case of congenital poikiloderma with a missense mutation (c.1883G>A) in FAM111B was reported. This mutation was within the putative protease domain and predicted to be pathogenic by Varsome database (https://varsome.com/), revealing that the mutation would promote the development of this disease. Recent studies found that disease-associated FAM111B mutants forms a complex with Family with sequence similarity 111 member A (FAM111A), hyperactivating the intrinsic protease activity of FAM111A via a common gain‐of‐function mechanism, which may become the cause of the hereditary fibrosing poikiloderma syndrome (Hoffmann et al., 2020).
Inter-familial phenotypic variability has been observed in congenital poikiloderma, indicating that it may be a multisystem disorder. The same mutation could lead to different phenotypes and an association between genotypes and phenotypes was not established (Mercier et al., 2013; Mercier et al., 2015; Goussot et al., 2017), suggesting that other factors, such as racial factor and environmental variables, might influence the clinical characteristics of this disease. To date, including our case in the present study, a total of 37 patients with this rare disorders have been reported globally (Arowolo et al., 2022a). For patients with congenital poikiloderma, the predominant manifestation is early onset poikiloderma, telangiectasia, and epidermal atrophy. However, patients display a wide spectrum of disease phenotypes. With respect to the genotype-phenotype association, the mutations in the FAM111B gene can be classified into two categories according to their positions (Table 1). The codons 621, 625, 627, and 628 are located within the putative protease domain of FAM111B, which may be associate with more severe clinical symptoms in skin, muscle and internal organs, and worse prognosis (Arowolo et al., 2022b). The clinical manifestations in affected individuals with mutations located outside the domain, such as codons 416 and 430, may be characterized by sclerosis, lymphoedema, bullous lesions, and pancreatic cancer (Takeichi et al., 2017; Arowolo et al., 2022b). In our reported case, the patient showed no symptoms or had mild symptoms such as tendon contractures and myopathy, which might be a result of the young age. Longer-term clinical follow-up is required.

TABLE 1. A comparison of clinical features of different mutation spots of FAM111B.
In conclusion, we reported a new case of congenital poikiloderma with FAM111B mutation c.1883G>A in China. Diverse phenotypes of congenital poikiloderma associated with FAM111B mutations in codon 628 were observed. Our results will expand the current knowledge and also verify the incomplete association between genotypes and phenotypes of this extremely rare disorder.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
YW performed the gene analyses and data interpretation. YW and LW wrote the manuscript. PW contributed to the acquisition of clinical data. XW and GZ contributed to the conception and design of the study, critically revised the manuscript and provided final approval of the version to be published.
The present study was supported by the grants from the National Natural Science Foundation of China (82073016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.926451/full#supplementary-material
Akamatsu, S., Takata, R., Haiman, C. A., Takahashi, A., Inoue, T., Kubo, M., et al. (2012). Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat. Genet. 44, 426–429. doi:10.1038/ng.1104
Arnold, A. W., Itin, P. H., Pigors, M., Kohlhase, J., Bruckner Tuderman, L., Has, C., et al. (2010). Poikiloderma with neutropenia: A novel C16orf57 mutation and clinical diagnostic criteria. Br. J. Dermatol. 163, 866–869. doi:10.1111/j.1365-2133.2010.09929.x
Arowolo, A., Rhoda, C., and Khumalo, N. (2022). Mutations within the putative protease domain of the human FAM111B gene may predict disease severity and poor prognosis: A review of POIKTMP cases. Exp. Dermatol. 31, 648–654. doi:10.1111/exd.14537
Arowolo, A., Rhoda, C., and Khumalo, N. (2022). Mutations within the putative protease domain of the human FAM111B gene may predict disease severity and poor prognosis: A review of POIKTMP cases. Exp. Dermatol. 31, 648–654. doi:10.1111/exd.14537
Chen, F., Zheng, L., Li, Y., Li, H., Yao, Z., Li, M., et al. (2019). Mutation in FAM111B causes hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Acta Derm. Venereol. 99, 695–696. doi:10.2340/00015555-3186
Gatinois, V., Desprat, R., Pichard, L., Becker, F., Goldenberg, A., Balguerie, X., et al. (2020). IPSC reprogramming of fibroblasts from a patient with a Rothmund-Thomson syndrome RTS. Stem Cell Res. 45, 101807. doi:10.1016/j.scr.2020.101807
Goussot, R., Prasad, M., Stoetzel, C., Lenormand, C., Dollfus, H., Lipsker, D., et al. (2017). Expanding phenotype of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis caused by FAM111B mutations: Report of an additional family raising the question of cancer predisposition and a short review of early-onset poikiloderma. JAAD Case Rep. 3, 143–150. doi:10.1016/j.jdcr.2017.01.002
Hoffmann, S., Pentakota, S., Mund, A., Haahr, P., Coscia, F., Gallo, M., et al. (2020). FAM111 protease activity undermines cellular fitness and is amplified by gain-of-function mutations in human disease. EMBO Rep. 21, e50662. doi:10.15252/embr.202050662
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980. doi:10.1093/bioinformatics/bty897
Küry, S., Mercier, S., Shaboodien, G., Besnard, T., Barbarot, S., Khumalo, N. P., et al. (2016). CUGC for hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP). Eur. J. Hum. Genet. 24, 779. doi:10.1038/ejhg.2015.205
Mercier, S., Küry, S., Salort-Campana, E., Magot, A., Agbim, U., Besnard, T., et al. (2015). Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J. Rare Dis. 10, 135. doi:10.1186/s13023-015-0352-4
Mercier, S., Küry, S., Shaboodien, G., Houniet, D. T., Khumalo, N. P., Bou-Hanna, C., et al. (2013). Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am. J. Hum. Genet. 93, 1100–1107. doi:10.1016/j.ajhg.2013.10.013
Mercier, S., Küry, S., and Barbarot, S. (1993). “Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis,” in GeneReviews(®). Editors M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.). Copyright © 1993–2022.
Rayinda, T., Steensel, M., and Danarti, R. (2021). Inherited skin disorders presenting with poikiloderma. Int. J. Dermatol. 60, 1343–1353. doi:10.1111/ijd.15498
Seo, A., Walsh, T., Lee, M. K., Ho, P. A., Hsu, E. K., Sidbury, R., et al. (2016). FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas 45, 858–862. doi:10.1097/MPA.0000000000000529
Sun, H., Liu, K., Huang, J., Sun, Q., Shao, C., Luo, J., et al. (2019). FAM111B, a direct target of p53, promotes the malignant process of lung adenocarcinoma. Onco. Targets. Ther. 12, 2829–2842. doi:10.2147/OTT.S190934
Keywords: congenital poikiloderma, FAM111B, mutation, case report, literature review
Citation: Wu Y, Wen L, Wang P, Wang X and Zhang G (2022) Case Report: Diverse phenotypes of congenital poikiloderma associated with FAM111B mutations in codon 628: A case report and literature review. Front. Genet. 13:926451. doi: 10.3389/fgene.2022.926451
Received: 22 April 2022; Accepted: 12 July 2022;
Published: 25 August 2022.
Edited by:
Ming Li, Shanghai Jiao Tong University, ChinaReviewed by:
Nancy Monroy-Jaramillo, National Institute of Neurology and Neurosurgery, MexicoCopyright © 2022 Wu, Wen, Wang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guolong Zhang, Z2x6aGFuZ3RqQHRvbmdqaS5lZHUuY24=; Xiuli Wang, d2FuZ3hpdWxpXzE0MDAwMjNAdG9uZ2ppLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.