 Jun Ren1,2,3
Jun Ren1,2,3 Cuiting Peng1,2,3
Cuiting Peng1,2,3 Fan Zhou1,2,3Yutong Li1,2,3Yuezhi Keqie1,2,3Han Chen1,2,3Hongmei Zhu1,2,3Xinlian Chen1,2,3*†
Fan Zhou1,2,3Yutong Li1,2,3Yuezhi Keqie1,2,3Han Chen1,2,3Hongmei Zhu1,2,3Xinlian Chen1,2,3*† Shanling Liu1,2,3*
Shanling Liu1,2,3*- 1Department of Medical Genetics, Center of Prenatal Diagnosis, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, China
- 3Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
Preimplantation genetic testing (PGT) has been increasingly used to prevent rare inherited diseases. In this study, we report a case where PGT was used to prevent the transmission of disease-caused variant in a SCID-X1 (OMIM:300400) family. SCID-X1 is an X-linked recessive inherited disease whose major clinical manifestation of immune deficiency is the significant reduction in the number of T-cells and natural killer cells. This family gave birth to a boy who was a hemizygous proband whose IL2RG gene was mutated (c.315T > A, p(Tyr105*), NM_000206.3, CM962677). In this case, Sanger sequencing for mutated allele and linkage analysis based on single-nucleotide polymorphism (SNP) haplotype via next-generation sequencing were performed simultaneously. After PGT for monogenic disorder, we detected the aneuploidy and copy number variation (CNV) for normal and female carrier embryos. Four embryos (E02, E09, E10, and E11) were confirmed without CNVs and inherited variants at the IL2RG gene. Embryo E02 (ranking 4BB) has been transferred after considering the embryo growth rate, morphology, and PGT results. Prenatal genetic diagnosis was used to detect amniotic fluid cells, showing that this fetus did not carry the variant of the IL2RG gene (c.315T > A). Ultimately, a healthy girl who had not carried disease-causing variants of SCID-X1 confirmed by prenatal diagnosis was born, further verifying our successful application of PGT in preventing mutated allele transmission for this SCID family.
Introduction
Severe combined immune deficiency (SCID, OMIM:300400) is a fatal genetic defect (Torii, 1996). Although SCID morbidity varies from country to country, there is no report on its exact incidence in China. According to research on newborn screening in the United States, SCID affects one in 58,000 infants (95% CI, 1/46000–1/80000) (Kwan et al., 2014). SCID is a prenatal disorder of T lymphocyte development (Cossu, 2010), in which the affected infant gradually develops a pediatric emergency after birth. In general, the affected infant presents severe opportunistic infections within 1 month of birth because of defects in humoral and cellular immunity (Mamcarz et al., 2019). Typical laboratory inspections show a lack of T cells, natural killer (NK) cells, and functional B cells. As a result, affected infants cannot usually live beyond their first year of life.
The most common cause of SCID is a variant of the IL2RG gene (OMIM: 300400), also called SCID-X1. The IL2RG gene, situated in Xq13.1, encodes the interleukin-2 receptor common gamma chain and is shared by several cytokine receptors necessary for the development and function of lymphocytes (Mamcarz et al., 2019). Allogeneic hematopoietic stem-cell transplantation or autologous gene therapy is considered the most effective treatment for SCID (Cavazzana-Calvo et al., 2000). Although hematopoietic stem cell transplantation (HSCT) from a matched sibling donor is effective, it can only be used in a minority of patients, as transplantation from an alternative donor is related to an increased risk of graft-versus-host disease and incomplete immune reconstitution (Mamcarz et al., 2019). Gene therapy has also shown potential in this regard, but the carcinogenicity of retroviral vectors remains to be solved. Leukemia was caused when gene therapy was first used to treat SCID-X1, owing to an insertional variant induced by enhancers of the adenovirus vectors (Fischer and Hacein-Bey-Abina, 2020). With the development of gene therapy vectors, such as lentivirus, increasingly exciting research has been carried out to prove their application value in SCID therapy (Blanco et al., 2020).
However, for these couples with an inherited disease that can be clearly diagnosed genetically, adequate genetic counseling and prenatal or preimplantation genetic diagnostic techniques are important (De Rycke and Berckmoes, 2020, Group et al., 2020). With the development of assisted reproduction and molecular genetic technology, preimplantation genetic testing (PGT) has been used in birth defect prevention and control (Group et al., 2020). PGT for monogenic disorders (PGT-M) can select unaffected embryos to prevent the transmission of disease-causing variants. PGT technology avoids the adverse effects of repeated abortion on women’s physical and mental health.
Through this case, we report a PGT-M case based on MALBAC and next-generation sequencing (NGS)-based single-nucleotide polymorphism (SNP) haplotype for SCID. To further verify our PGT results, chromosome microarray (CMA) for copy number variation (CNV) analysis and Sanger sequencing for mutated alleles were conducted in amniotic cells for prenatal genetic diagnosis.
Patients and Methods
Patients
A 31-year-old couple had given birth to a boy who was the proband. The female carrying IL2RG mutated allele visited the Department of Medical Genetics, West China Second University Hospital, Sichuan University. The boy was diagnosed with X-linked SCID (OMIM:300400) in the West China Second University Hospital for repeated high fever, severe anemia, hepatosplenomegaly, immune system deficiency, coagulation dysfunction, hemangioma, and severe sepsis. The boy died at less than 1 year of age. We found that the boy had a variant in the IL2RG gene (c.315T > A, p(Tyr105*), NM_000206.3, CM962677). According to the standards and guidelines for sequence variant interpretation of the American College of Medical Genetics and Genomics (ACMG/AMP) (Richards et al., 2015), Clinical Interpretation of Sequence Variants (zhang et al., 2020) and ClinGen Sequence Variant Interpretation Recommendation for PM2 - Version 1.0, we evaluated this variant with PVS1 + PM2_Supporting, likely pathogenic variant. Parents received genetic counseling and signed an informed consent form. Sample collection, library preparation, NGS and data analysis were conducted at the Department of Medical Genetics, West China Second University Hospital. This study was approved by the Internal Ethical Committees of the West China Second University Hospital.
Assisted Reproductive Technology Procedure and Embryo Trophectoderm Biopsy
Controlled ovarian stimulation, intracytoplasmic sperm injection (ICSI), blastocyst culture, trophectoderm biopsy, and blastocyst transfer were conducted in the Reproductive Medicine Center of West China Second University Hospital, according to the standard protocol (McArthur et al., 2005; Schoolcraft et al., 2011). In this cycle, twelve embryos were finally developed into blastocysts, and the trophectoderm (TE) cells were biopsied on day 5 or 6 after insemination. A total of five to eight biopsied cells from the TE were transferred into 4.5 μL lysis buffer (Yikon Genomics) in 0.2 μL PCR tubes for whole-genome amplification (WGA) (MALBAC).
gDNA Extraction and WGA
Peripheral blood of the couple was collected for gDNA extraction by a DNeasy Blood and Tissue Kit (Qiagen). Although the proband (hemizygote) was deceased, the couple kept his gDNA sample. In the second trimester, amniotic cells were collected by transabdominal amniocentesis; gDNA extraction was also carried out using a DNeasy Blood and Tissue Kit (Qiagen). The buccal mucosa cells (BMCs) of the woman (variant carrier) were diluted by phosphate buffer saline; five to eight cells were selected for WGA. WGA involves multiple annealing and looping-based amplification cycles (MALBAC, Yikon genomics) applied for diluted mucosa and TE cells. Additionally, 5 μL WGA products were used for 1% agarose gel electrophoresis; a 300–2000 bp diffused band indicated successful amplification. Whole-genome products were purified using a DNA Clean-up Kit (CWBIO). All operations were performed according to manufacturers’ protocols.
Variant Site Detection
PCR amplification and Sanger sequencing were conducted to validate the mutated IL2RG allele. Primer was designed to amplify the segment containing c.315 of IL2RG. Forward primer (CTCCCAG GTACCCCACTGTT) and reverse primer (TCCAATGTCCCACAGTATCCC) were designed (Primer 5.0 software) and synthesized (TsingKe Biotechnology, Beijing). PCR was performed in a 25 μL system using 2× GoldStar Best Master Mix (CWBIO) on a 96-Well Thermal Cycler Veriti Dx (Life Technologies). The amplification system contained 2 μL of primer mix, 12.5 μL of enzyme mix, gDNA, or purified WGA products as templates. The reaction condition was as follows: 95°C for 5 min; 95°C for 30 s, 57°C for 30 s, 72°C for 40 s (35 cycles); 72°C for 5 min; held at 4°C. Sanger sequencing data were analyzed by ChromasPro software.
Before the scheme could be used for the TE cells, we used the wife’s BMCs to imitate biopsied trophectoderm (TE) cells because the proband had passed away. The purpose is to test the effectiveness of the primers and the allele drop-out (ADO) rate of the WGA (WGA) method. MALBAC was applied for five to eight BMCs. WGA production of BMCs was used to amplify the target segments and Sanger sequencing.
Library Preparation and NGS
Purified WGA products were used for the PCR and SNP-based haplotype via NGS. An NGS library preparation kit (Yikon Genomics) was used to prepare the SNP library. For the CNV library, unpurified WGA products were used via an NGS library preparation kit (Yikon Genomics). All operations followed the manufacturers’ protocols. Library sequencing was performed in the MiSeq Dx platform using a MiSeq Reagent Kit v3 (150-cycles) (Illumina). The raw data were automatically filtered, generating FASTQ files; the Q30 should be greater than 90%.
CNV Analysis and NGS-Based SNP Haplotype
For CNV analysis, FASTQ files were disposed of in the local analysis platform ChromGo (Yikon Genomics). More than 4 Mb deletions or duplications were reported. For CNV analysis, valid reads should be more than 1 Mb, CV (1000K_bin_size), and valid read GC contents should be in an acceptable range. To detect ADO and recombination, an NGS-based haplotype was conducted using SNP within the 2-million base pair (Mb) region, flanking the targeted gene. In this case, 60 SNPs were selected. For the SNP haplotype, bioinformatics analysis was conducted by Yikon Genomics, Ltd. The informative SNP sites are homozygous in the spouse and heterozygous in the variant carrier. Proband haplotypes were used for reference.
Embryo Transplantation and Prenatal Genetic Diagnosis
For PGT-M, embryo selection involves comprehensive considerations (Munné et al., 2019), including quality, the developmental stage of the embryo, and the results of PGT-A&M. Results were confirmed by the prenatal genetic diagnosis of amniotic fluid cells at gestational weeks 18–22+6.
Results
Patients and Genetic Background
In this family, this variant is the cause of X-linked SCID, whose hereditary mode is X-linked recessive inheritance (Figure 1). The size of the IL2RG gene is about 4.23 kb, containing eight exons (Figure 1). The c.315T > A variant occurred in exon 3. According to the ACMG/AMP guidelines, we evaluated this variant with PVS1 + PM2_Supporting as likely pathogenic. Both the husband and wife had normal karyotypes (320 bands). The peripheral blood of the couple and gDNA of the proband were detected by amplifying the target segments and Sanger sequencing (Supplementary Figure S1). Results showed that the female and proband were the carrier and hemizygote, respectively. The male did not carry this variant in the IL2RG gene (c.315T > A). These tests were carried out on the WGA production of BMCs from the carrier. The same diagnostic result was obtained as that of the peripheral blood cell test. The results indicate that the primers and MALBAC method could detect pathogenic variants in WGA products. (Supplementary Figure S1).
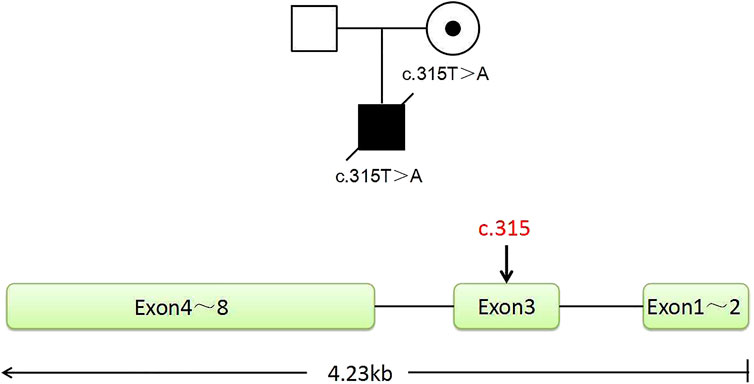
FIGURE 1. Pedigree of the family. The proband is a deceased hemizygous patient, and the woman is a disease-causing gene carrier, and the man is normal. The IL2RG gene with eight exons is located in the X chromosome Xq13.1. The graph shows the position of the mutant sites of c.315, located in exon 3.
TE Cell Biopsy and Detection of Pathogenic Variant Allele
In this PGT cycle, 12 embryos finally developed into blastocysts after ICSI (Table1). The TE biopsy was conducted by a mechanical method on day 5 or 6 post-insemination. A total of five to eight cells were collected and transferred into 0.2 μL PCR tubes, to which lysis buffer was added beforehand. The MALBAC two-step method was used for WGA. To detect the variant allele, parts of WGA products were purified to obtain 100 bp–10 kb fragments and eliminate primers, enzymes, and oligonucleotides of the previous reaction. Then, the purified products were detected by amplifying the target segments and Sanger sequencing (Figure 2). The data show that embryos E1, E3, E4, and E12 had a variant in the IL2RG gene c.315 (T > A). The remaining embryos either did not indicate variants or there was morbigenous ADO. Therefore, linkage analysis is necessary to determine if there is an ADO or chromosome recombination.

TABLE 1. Summary of detection results.
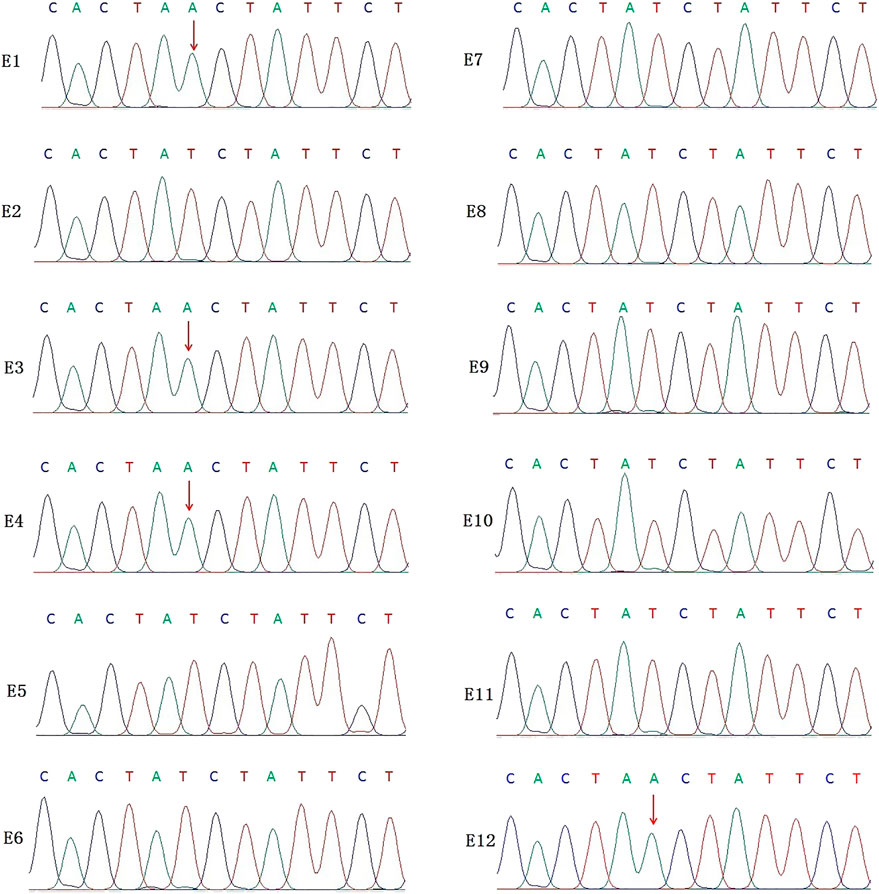
FIGURE 2. Results of the Sanger sequencing for biopsied TE cells. The figure shows that E1, E3, E4, and E12 are carriers of the IL2RG c.315 (T > A) variant; no variants were detected in the remaining embryo samples. For X-linked recessive diseases, this technique alone cannot distinguish between male patients and female carriers. At the same time, misdiagnosis lead by ADO cannot be ruled out.
SNP Haplotype
ADO may lead to misdiagnosis. To minimize its interference with the diagnostic results, SNP markers within the 2 Mb region flanking the target gene were used for linkage analysis. The peripheral blood samples of this couple, gDNA of the proband, and WGA products of embryos were applied to the SNP haplotype (Figure 3). For the IL2RG gene, 60 SNPs were chosen for linkage analysis. In this case, within 1 Mb upstream and downstream of the variant site, there were 7 and 13 SNPs, respectively. The selection of informative SNP was based on the ESHRE PGT Consortium good practice recommendations for detecting monogenic disorders (Group et al., 2020). Combined with the pedigree haplotype, the high-risk maternal chromosome was indicated by a red bar (Figure 3); the dark blue and dark orange represent paternal and low-risk maternal chromosomes, respectively. Results show that E02, E06, E07, E08, E09, E10, and E11 did not carry the high-risk maternal chromosome. E05 is not listed in the graph because its linkage analysis cannot be determined, as it only had the paternal X chromosome; therefore, as we think it might be X monosomy, we will confirm our conjecture through PGT-A soon. The results of pathogenic variant site detection and linkage analysis were considered, and we can conclude that E02, E06, E07, E08, E09, E10, and E11 do not carry maternal c.315 (T > A) in the IL2RG gene.
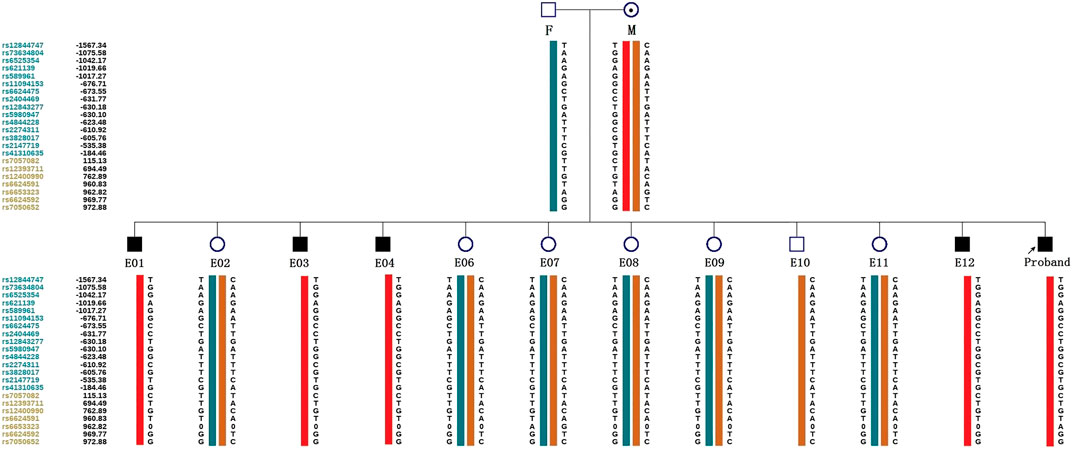
FIGURE 3. Schematic diagram representing the SNP-based haplotype of this family members and embryos. F, M, and P represent the father, mother, and proband, respectively. The reference SNP cluster ID numbers were listed on the left side. The ID numbers highlighted in dark blue and orange refer to the upstream and downstream informative SNPs, respectively. The red bar refer to the high-risk haplotypes, the dark blue bar represent the normal or low-risk haplotype of the father, and the dark orange bar represent the normal or low-risk haplotype of the mother. Results show that the E01, E03, E04, and E12 are male patient embryos; the E02, E06, E07, E08, E09, and E11 are normal female embryos; and the E10 is a normal male embryo. E05 was not included because of anomalies found in the linkage analysis.
Preimplantation Genetic Testing for Aneuploidy (PGT-A)
The above seven embryos that did not carry the maternal variant were detected for aneuploidy. NGS-based CNV-Seq (CNV sequencing) was performed in the Illumina MiseqDx platform, indicating large than 10 Mb CNV and 30–70% mosaicism (large than 30 Mb). The CNV results are summarized in Table 1. No CNV larger than 10 M and aneuploidy were found in E02, E09, E10, and E11. E05, which could be determined for linkage analysis, indicated X monosomy (Supplementary Figure S2). E06 was aneuploidy, and E07 and E08 had fragment deletion or mosaicism (Supplementary Figure S2).
Embryo Transplantation and Prenatal Genetic Diagnosis
For the embryo transplant decision, parents had discussions with a reproductive doctor, embryologist and medical genetic doctor in the Reproductive Medicine-Medical Genetic MDT (Multi-disciplinary Treatment, MDT) of the West China Second University Hospital. The decision was based on the developmental stage and grade of the embryo, according to PGT-M and PGT-A results (Table1). Finally, E02 was used for the transplantation, and the pregnancy was successful. At the 20th week of gestation, the pregnant woman underwent amniocentesis for prenatal genetic diagnosis. Amniotic cells were applied for CMA (cytoscan 750K, Affymetrix) (Lwvy and Wapner, 2018), amplification of the target segments, and Sanger sequencing (data not shown). The prenatal diagnosis was consistent with the preimplantation genetic testing. The woman eventually gave birth to a healthy girl who did not carry the variant.
Discussion
SCID is an X-linked recessive monogenic hereditary disease characterized by defects in humoral and cellular immunity (Verbsky et al., 2012). Affected children often do not survive their first year of life due to recurrent and severe infections. PGT-M is an effective technique for blocking the transmission of this disease-causing gene. Until now, allogenic HSCT (Haddad and Hoenig, 2019) and gene therapy (Ferrua and Aiuti, 2017) are promising treatments for SCID (Suk See De Ravin, 2016). However, these two technologies still have limitations in clinical applications (Fischer and Hacein-Bey-Abina, 2020). For some inherited immunodeficiency like SCID, the PGT has become a practical technique for these at-risk couples to avoid affected pregnancies and have a healthy progeny free from genetic and chromosomal disorders (Rechitsky et al., 2018). At present, there is no mature gene therapy program for clinical application for children born with SCID, HSCT from a matched sibling donor is the most effective way. HLA matched stem cell transplantation improves significantly the outcome of transplantation treatment (Rechitsky et al., 2018). If children born with SCID are treated and protected early by pediatric ICU, they may be able to wait until PGT-M with HLA typing technique helps their family have a sibling who can provide a suitable transplantation donor.
We report a case blocking offspring with a rare inherited disease SCID by PGT-M. MALBAC is applied for WGA. An SNP-based haplotype and NGS were used for PGT-M&A by Illumina MiseqDx. Recently, MALBAC has been gradually recognized for its comparable single nucleotide variations detection efficiency, false-positive ratio, ADO ratio with MDA (Hou et al., 2015). Moreover, MALBAC is suited for the detection of CNVs because of its lower long-range variability in read mapping and weaker inherent bias in amplification than MDA (Charles F. A. de Bourcy, 2014).
Before the test protocol is applied to biopsied TE cells, we first detected gDNA of peripheral blood cells and WGA products of BMCs to evaluate the effectiveness and ADO rate of this method. Then we performed a PGT-M test on biopsied samples. For X-linked recessive inherited SCID, female carrier and normal embryo proceed to the next step of PGT-A. In linkage analysis, it was found that embryo 05 carried only the male normal X chromosome haplotype. In the Sanger sequencing of the pathogenic site, embryo 05 showed no variant. We hold the opinion that embryo 05 may be the X-monosomy inherited from the father; subsequent PGT-A results confirmed our conjecture. Therefore, to use PGT-M for X-linked genetic diseases, it is necessary to design a pair of primers to amplify the partial loci of the SRY gene or apply with PGT-A for embryo sex identification. For autosomal genetic diseases, PGT-A is also recommended after PGT-M. This helps to prevent us from misjudging the embryo in these cases. After PGT-M and PGT-A, E02, E09, E10, and E11 did not carry pathogenic variants, and no clear chromosomal abnormalities were found. E02 was eventually used for transplantation after considering the PGT results and morphological scores.
After a successful pregnancy by the PGT technique, prenatal diagnosis by amniocentesis during the second trimester is indispensable. Due to the possibility of mosaicism, biopsied trophectoderm cells represent only the genetic makeup of the placenta; there may be a normal placenta and abnormal fetal development. Therefore CMA and amplification of the target segments for Sanger sequencing were performed in amniocytes. This ensures that healthy babies are born through this double-check method.
In conclusion, PGT-M has important clinical application value in the prevention and control of birth defects. It can avoid the psychological and physical harm caused by induced labor after prenatal diagnosis. In this case, PGT (monogenic disorder and aneuploidy) combined with follow-up prenatal diagnosis helped a family with IL2RG gene variant have a healthy infant. We believe double detection at the blastocyst stage and second trimester, as well as the rational choice of multiple technologies can guarantee the birth of healthy children. At the same time, it also minimizes the risk of misdiagnosis caused by chromosome recombination, mosaicism, and ADO.
Data Availability Statement
The data analyzed in this study is subject to the following licenses/restrictions: The raw datasets analysed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources and patient anonymity, but are available from the corresponding author on reasonable request. Requests to access these datasets should be directed to Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Second Hospital, Sichuan University. Written informed consent to participate in this study was provided by the participants’legal guardian/next of kin.
Author Contributions
Conceptualization: JR and SL; Methodology: CP, YL, YK, HC, and HZ; Formal analysis and investigation: FZ and XC; Writing—original draft preparation: JR; Writing—review and editing: CL; Funding acquisition: SL; Resources: JR; Supervision: XC and SL.
Funding
This study was supported by the National Key Research and Development Program of China (2021YFC1005303) and the Technology Research and Development Program of the Science and Technology, Sichuan Province, China (2021YFS0078).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the family for their cooperation and participation, to the embryology team in the Center of Reproductive Medicine for the help with sample preparation and to Zhen Liu from Yikon Genomics for the technical help for data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.926060/full#supplementary-material
References
Blanco, E., Izotova, N., Booth, C., and Thrasher, A. J. (2020). Immune Reconstitution After Gene Therapy Approaches in Patients with X-Linked Severe Combined Immunodeficiency Disease. Front. Immunol. 11, 608653. doi:10.3389/fimmu.2020.608653
Carvalho, F., Moutou, C., Dimitriadou, E., Dreesen, J., Gimenez, C., et al. ESHRE PGT-M Working Group (2020). ESHRE PGT Consortium Good Practice Recommendations for the Detection of Monogenic Disorders. Hum. Reprod. Open 2020 (3), hoaa018. doi:10.1093/hropen/hoaa018
Cavazzana-Calvo, M., Hacein-Bey, S., Basile, G. d. S., Gross, F., Yvon, E., Nusbaum, P., et al. (2000). Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 288, 669–672. doi:10.1126/science.288.5466.669
de Bourcy, C. F. A., De Vlaminck, I., Kanbar, J. N., Wang, J., Gawad, C., and Quake, S. R. (2014). A Quantitative Comparison of Single-Cell Whole Genome Amplification Methods. Plos One 9 (8), e105585. doi:10.1371/journal.pone.0105585
De Ravin, S. S., Wu, X., Moir, S., Anaya-O'Brien, S., Kwatemaa, N., Littel, P., et al. (2016). Lentiviral Hematopoietic Stem Cell Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 8 (335), 335ra57. doi:10.1126/scitranslmed.aad8856
De Rycke, M., and Berckmoes, V. (2020). Preimplantation Genetic Testing for Monogenic Disorders. Genes 11 (8), 871. doi:10.3390/genes11080871
Ferrua, F., and Aiuti, A. (2017). Twenty-Five Years of Gene Therapy for ADA-SCID: FromBubble Babiesto an Approved Drug. Hum. Gene Ther. 28 (11), 972–981. doi:10.1089/hum.2017.175
Fischer, A., and Hacein-Bey-Abina, S. (2020). Gene Therapy for Severe Combined Immunodeficiencies and beyond. J. Exp. Med. 217 (2), e20190607. doi:10.1084/jem.20190607
Haddad, E., and Hoenig, M. (2019). Hematopoietic Stem Cell Transplantation for Severe Combined Immunodeficiency (SCID). Front. Pediatr. 7, 481. eCollection 2019. doi:10.3389/fped.2019.00481
Hou, Y., Ww, K., Shi, X., Li, F., Song, L., Wu, H., et al. (2015). Comparison of Variations Detection between Whole-Genome Amplification Methods Used in Single-Cell Resequencing. Gigascience 4, 37. doi:10.1186/s13742-015-0068-3
Kwan, A., Abraham, R. S., Currier, R., Brower, A., Andruszewski, K., Abbott, J. K., et al. (2014). Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 312 (7), 729–738. doi:10.1001/jama.2014.9132
Lwvy, B., and Wapner, R. (2018). Prenatal Diagnosis by Chromosomal Microarray Analysis. Fertil. Steril. 109, 201–212. doi:10.1016/j.fertnstert.2018.01.005
Mamcarz, E., Zhou, S., Lockey, T., Abdelsamed, H., Cross, S. J., Kang, G., et al. (2019). Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 380 (16), 1525–1534. doi:10.1056/NEJMoa1815408
Mcarthur, S. J., Leigh, D., Marshall, J. T., Dd Boer, K. A., and Jansen, R. P. (2005). Pregnancies and Live Births after Trophectoderm Biopsy and Preimplantation Genetic Testing of Human Blastocysts. Fertil. Steril. 84 (6), 1628–1636. doi:10.1016/j.fertnstert.2005.05.063
Munn, S., Kaplan, B., Frattarelli, J. L., Child, T., Nakhuda, G., Shamma, F. N., et al. (2019). Preimplantation Genetic Testing for Aneuploidy versus Morphology as Selection Criteria for Single Frozen-Thawed Embryo Transfer in Good-Prognosis Patients: a Multicenter Randomized Clinical Trial. Fertil. Steril. 112 (6), 1071–1079. e7. doi:10.1016/j.fertnstert.2019.07.1346
Rechitsky, S., Pakhalchuk, T., Prokhorovich, M., Ramos, G. S., Verlinsky, O., and Kuliev, A. (2018). Preimplantation Genetic Testing for Inherited Immunodeficiency. Hematol. Transfus. Int. J. 6 (6), 218–220. doi:10.15406/htij.2018.06.00187
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Mutations: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schoolcraft, W. B., Treff, N. R., Stevens, J. M., Ferry, K., Katz-Jaffe, M., and Scott, R. T. (2011). Live Birth Outcome with Trophectoderm Biopsy, Blastocyst Vitrification, and Single-Nucleotide Polymorphism Microarray–Based Comprehensive Chromosome Screening in Infertile Patients. Fertil. Steril. 96 (3), 638–640. doi:10.1016/j.fertnstert.2011.06.049
Tsuge, I., Matsuoka, H., Abe, T., Kamachi, Y., and Torii, S. (1996). Interleukin-2 Receptor Y-Chain Mutations in Severe Combined Immunodeficiency with B-Lymphocytes. Eur. J. Pediatr. 155 (12), 1018–1024. doi:10.1007/BF02532522
Verbsky, J. W., Baker, M. W., Grossman, W. J., Hintermeyer, M., Dasu, T., Bonacci, B., et al. (2012). Newborn Screening for Severe Combined Immunodeficiency; the Wisconsin Experience (2008-2011). J. Clin. Immunol. 32 (1), 82–88. doi:10.1007/s10875-011-9609-4
Keywords: preimplantation genetic testing, severe combined immune deficiency, IR2RG, haplotype, rare genetic disease, next generation sequencing
Citation: Ren J, Peng C, Zhou F, Li Y, Keqie Y, Chen H, Zhu H, Chen X and Liu S (2022) Case Report: Preimplantation Genetic Testing for X-Linked Severe Combined Immune Deficiency Caused by IL2RG Gene Variant. Front. Genet. 13:926060. doi: 10.3389/fgene.2022.926060
Received: 22 April 2022; Accepted: 16 May 2022;
Published: 01 June 2022.
Edited by:
Xi Wang, Nanjing Medical University, ChinaReviewed by:
Junyu Zhang, Shanghai Jiao Tong University School of Medicine, ChinaAndrea Grioni, Novartis Institutes for BioMedical Research, Switzerland
Copyright © 2022 Ren, Peng, Zhou, Li, Keqie, Chen, Zhu, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinlian Chen, Y2hlbnhpbmxpYW4xMTIxQDE2My5jb20=; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work and share last authorship