
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 05 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.924573
This article is part of the Research TopicGenetic and Environmental Factors in the Occurrence of Paediatric Disorders – Volume IIView all 10 articles
 Jianlong Zhuang1†
Jianlong Zhuang1† Chunnuan Chen2†
Chunnuan Chen2† Yuanbai Wang1Shuhong Zeng1Yu’e Chen3Yuying Jiang1
Yuanbai Wang1Shuhong Zeng1Yu’e Chen3Yuying Jiang1 Yingjun Xie4,5*Gaoxiong Wang6*
Yingjun Xie4,5*Gaoxiong Wang6*Background: Pathogenic mutations in the KCNH2 gene were associated with long QT syndrome 2 (LQT2), which typically manifest in a prolonged QT interval and may lead to recurrent syncopes, seizure, or sudden death. Limited reports indicated that the KCNH2 mutations would result in LQT2 combined with tetralogy of fallot. Our goal was to present an additional case of LQT2 combined with the tetralogy of fallot in a fetus with a novel KCNH2 mutation.
Case presentation: Enrolled in this study was a 23-year-old pregnant woman from Quanzhou Fujian province, China. In her pregnancy, fetal ultrasound anomalies were identified, including tetralogy of fallot, coronary sinus enlargement, and persistent left superior vena cava. No chromosomal abnormality was detected by fetal karyotype analysis. However, 238.1-kb duplication in the 2q14.2 region containing the GLI2 gene was observed in the fetus by chromosomal array analysis, which was inherited from the mother with normal clinical features and interpreted as a variant of uncertain significance (VOUS). Furthermore, whole-exome sequencing (WES) detection identified a novel nonsense c.1907C > G (p.S636*) mutation in the KCNH2 gene in the fetus, and it was classified as a likely pathogenic variant, according to the ACMG guidelines. Parental verification analysis indicated that c.1907C > G (p.S636*) mutation was inherited from the mother.
Conclusion: In this study, we believe that 2q14.2 duplication may not be the reason for fetal heart defects; moreover, we described an additional case with KCNH2 gene mutation, which may lead to LQTS and be associated with congenital heart defects. In addition, our study further confirms the application value of the WES technology in prenatal genetic etiology diagnosis of fetuses with structural anomalies and unexplained structural variants.
Microdeletions or microduplications in the 2q14.2 region are extremely rare variants, with approximately 10 case reports being available in the literature. The 2q14.2 microscopic deletions usually manifest phenotypic variability and incomplete penetrance; the clinical phenotypes are typically characterized as holoprosencephaly, abnormal pituitary gland formation and/or function, craniofacial dysmorphisms, branchial arch anomalies, and polydactyly (Kevelam et al., 2012; Kordaß et al., 2015; Elizabeth et al., 2020). Isolated 2q14.2 duplications are rarer with no defined specific syndrome. To the best of our knowledge, only one report was referred to 2q14.2 duplication, and it demonstrated that it may be responsible for microphthalmia/microcornea and congenital cataracts in an affected family (Schilter et al., 2013).
Congenital long QT syndrome (LQTS) is a genetic disorder that typically manifests arrhythmia of a prolonged QT interval and a risk of recurrent syncopes, seizures, or sudden death (Crotti et al., 2008; Hedley et al., 2009; Christiansen et al., 2014). A previous study indicated that LQTS showed a high incidence of approximately 1/2,500 in healthy live births (Schwartz et al., 2009). Among them, KCNQ1, KCNH2 (HERG), and SCN5A are the three most frequently affected genes, which would lead to LQT1, LQT2, and LQT3, respectively. The main mutation types in LQT2 are nonsense mutation and missense mutation, with more than half of them being nonsense mutations and predict to result in haploinsufficiency through nonsense-mediated RNA decay (Ono et al., 2020). At present, no specific forms of congenital heart defects (CHD) have been reported to associate with LQT2, while several cases with KCNH2 mutations showed coexistence of CHD (Bhuiyan et al., 2014; Ebrahim et al., 2017; Song et al., 2018).
In this study, the whole-exome sequencing (WES) technology was first employed for further prenatal diagnosis in a fetus who harbored familial 2q14.2 duplication and exhibited fetal congenital heart defects including tetralogy of fallot. In addition, our study further strengthened the application value of the WES technology in genetic etiology of fetuses with structural anomalies and unexplained copy number variants.
A 23-year-old Chinese pregnant woman, gravida 1, para 0, from Quanzhou Fujian province, was enrolled in this study. Her husband was 29 years old, and they denied consanguinity marriage and had a family history of inherited disease. In this pregnancy, low progesterone was observed in the first trimester and was treated with didroxyprogesterone. No obvious abnormality was found by ultrasound examination during her first-trimester pregnancy, with a normal nuchal translucency (0.7 mm). In the second trimester, a low-risk screening result was detected by Down’s screening. However, at the gestational age of 23+2 weeks, three-level color Doppler ultrasound examination results revealed abnormal heart defects in the fetus, including tetralogy of fallot (ventricular septal defect, overriding aorta, and pulmonary artery stenosis), coronary sinus enlargement, and persistent left superior vena cava (Figure 1). Amniocentesis was performed at the gestational age of 25+5 weeks, after sufficient genetic consultation.
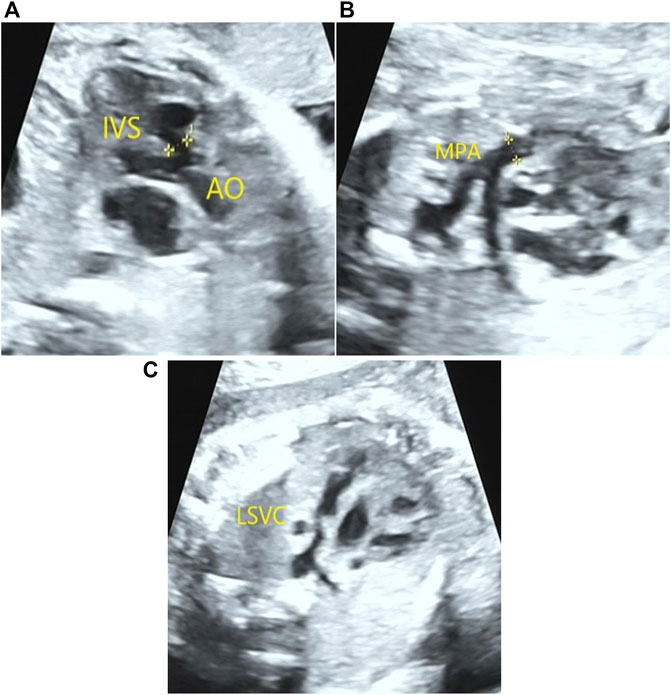
FIGURE 1. Congenital heart defects detected in the fetus by prenatal ultrasound examination. A 0.41-cm ventricular septal defect and overriding aorta (A) were detected in the fetus by prenatal ultrasound examination. In addition, pulmonary artery stenosis (B) and persistent left superior vena cava (C) were also observed.
No obvious chromosomal abnormality was detected in the fetus by karyotype analysis, and her parents showed normal karyotypes as well. However, chromosomal array analysis (CMA) results demonstrated a 238.1-kb duplication in the 2q14.2 region ([GRCh37]2q14.2 (121,477,769–121,715,896)x3) in the fetus containing the GLI2 gene (OMIM:165230). According to the ACMG guidelines, the 2q14.2 duplication was interpreted as variants of uncertain significance. Parental CMA verification demonstrated that the 2q14.2 duplication was inherited from the mother who exhibited normal clinical features.
WES technology was carried out to look for additional variants in the fetus with unexplained structural variants of 2q14.2 duplication. A novel nonsense mutation of c.1907C > G (p.S636*) in the KCNH2 gene was identified in the fetus by WES technology, which was inherited from the mother and further confirmed by Sanger sequencing (Figure 2). No frequency was found in the databases of gnomAD, 1,000 genomes, dbSNP, and ExAC. In addition, several computer-aided analysis software applications predicted that this variation may affect the protein structure/function (MetaSVM_score: 0.717; GERP++_RS: 5.14; dbscSNV_ADA: 0; dbscSNV_RF: 0). According to the ACMG Guidelines (Richards et al., 2015), the nonsense mutation was interpreted as a likely pathogenic variant (PVS1, PM2_Supporting). At present, no obvious cardiac abnormality was observed in the pregnant woman by electrocardiogram and echocardiography. Finally, the family chose to terminate her pregnancy at a gestational age of 30 weeks.
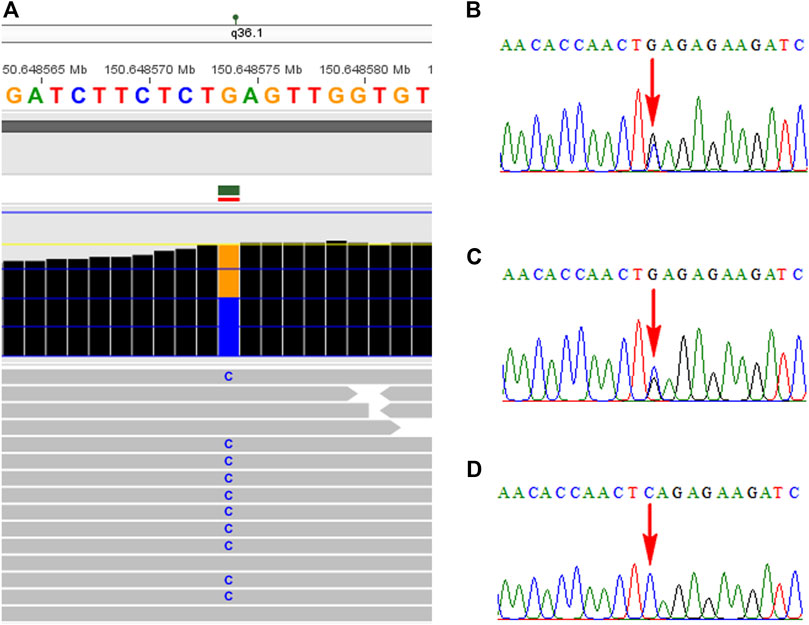
FIGURE 2. Identification of the KCNH2 mutation in the family using the WES technology and Sanger sequencing. (A) Novel nonsense mutation in the KCNH2 gene was identified in the fetus by WES. (B) Mutation of c.1907C > G was confirmed by Sanger sequencing. Parental Sanger sequencing revealed that her mother carried the same mutation (C), while the mutation was not found in her father (D).
With the advanced application of the CMA technology, an increasing rate of variants of uncertain significance (VOUS) and variants with phenotypical diversity may be followed, which will result in a big challenge for genetic consultation. The WES technology showed a great effectiveness in genetic etiology diagnosis in patients at the single-gene level, while few studies were available to further reveal the phenotypical diversity of unexplained structure variants using WES technology. Additional mutations besides the structural variants were identified by WES detection in patients with familial 1q21.1 microduplication/microdeletion, duplication of Xp22.31, and 16p11.2 duplication who exhibited phenotypical variability (Dastan et al., 2016; Qiao et al., 2017; Qiao et al., 2019). In addition, a recent study conducted by Granata et al. (2022) identified variants in CECR2, MTOR, RICTOR, and LRRK2 genes by WES detection in the affected patients who also had a 16p13.11 microdeletion and proposed that WES technology could be used as a fundamental tool to identify additional mutations in patients with a predisposing variant. In this study, the WES technology was employed to further investigate the genetic etiology in a fetus with familial 2q14.2 duplication who had more severe phenotypes.
In the present study, a 238.1-kb duplication in the 2q14.2 region was identified in the fetus, which was inherited from her mother with a normal phenotype. As shown in the DECIPHER database and listed in Table 1, all small fragments of 2q14.2 duplication were interpreted as VOUS without specific syndrome. In addition, as elicited in the ClinGen database, the dosage sensitivity of GLI2 gene indicated sufficient evidence of haploinsufficiency (3) but without triplosensitivity evidence (0). In addition, a patient harbored both 2q14.1q14.2 duplication and 2q37.3 deletion and exhibited autism, while the authors believed that the clinical phenotype may ascribe to 2q37.3 deletion (Devillard et al., 2010). In this study, we believe that the 2q14.2 duplication may not contribute to the ultrasonic abnormalities in the fetus. However, a previous study indicated that 2q14.2 duplication was co-segregated with microphthalmia/microcornea and congenital cataracts in an affected family and suggested that the 2q14.2 duplication may be the reason for the clinical features (Schilter et al., 2013). Nevertheless, no obvious ocular abnormalities were observed in our study, and the congenital heart defects including tetralogy of fallot in the fetus were hard to be explained by 2q14.2 duplication.
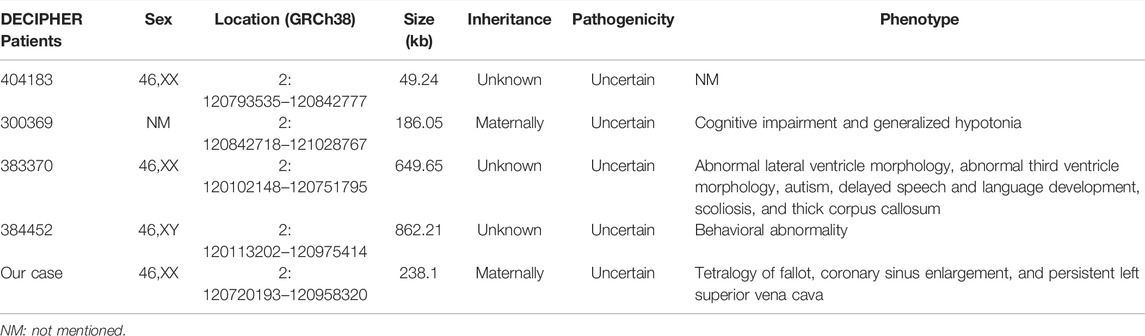
TABLE 1. Cases with less than 1.0-Mb duplications in the 2q14.2 region are presented in the DECIPHER database.
Further WES detection revealed a novel nonsense mutation of c.1907C > G (p.S636*) in the KCNH2 gene in the fetus, which was interpreted as a likely pathogenic variant. In addition, no pathogenic or uncertain variants in the known genes that referred to CHD features were identified in the fetus. KCNH2 encodes the pore-forming subunit of a rapidly activating delayed rectifier potassium channel; loss-of-function mutations in the KCHN2 gene would lead to LQT2 (Gianulis and Trudeau, 2011). As delineated by previous studies, several patients diagnosed with long QT syndrome are also accompanied by congenital heart defects (Massin et al., 2010; Hsiao et al., 2007; Wu et al., 1999). The study conducted by Murugan et al. (2005) presented a family with LQTS and coexisting persistent patency of the arterial duct, and they proposed that it may not be a coincidence, and hypothesis about a possible genetic mechanism may exist. In addition, a new form of LQTS was indicated in three patients who also had LQTS and associated with structural heart disease and syndactyly (Marks et al., 1995). Moreover, a previous study (Ebrahim et al., 2017) presented 11 patients who harbored single-gene mutations that resulted in long QT syndrome, combined with congenital heart defects. Among them, four patients carried KCNQ1 gene mutation, and one patient had a KCNQ1 mutation associated with KCNH2 mutation. Most of them (six cases) had KCNH2 mutations, of which two cases had tetralogy of fallot (TOF) (Ebrahim et al., 2017). In addition, patients with KCNH2 mutations who had LQTS combined with tetralogy of fallot (TOF) are summarized in Table 2. In the present study, the fetus had a novel KCNH2 mutation and manifested congenital heart defects including TOF, which further enhanced the genotype–phenotype correlation.
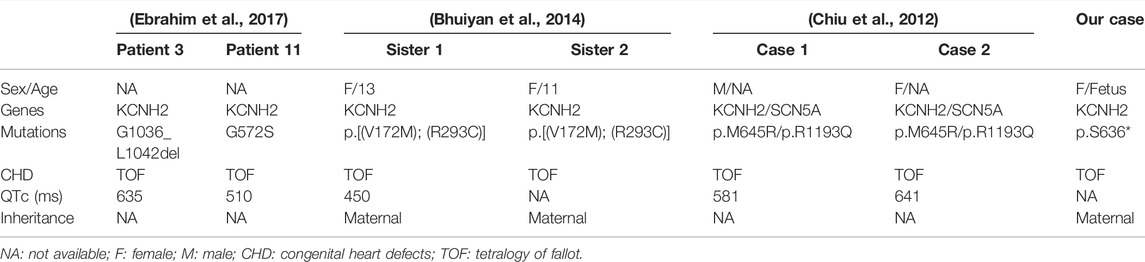
TABLE 2. KCNH2 mutations in patients who had LQTS combined with tetralogy of fallot in the literature.
In addition, the KCNH2 mutation in the fetus was inherited from her mother, who had no obvious cardiac phenotype. Similar to a previous study (Bhuiyan et al., 2014), two sisters in a close relative married family were found to carry the KCNH2 mutation, which was also inherited from their normal mother, which suggests incomplete penetrance of KCNH2, although it cannot be ruled out that the phenotype may appear in her mother in the future. Therefore, our study indicated that the additional variant in the KCNH2 gene identified in the fetus may be responsible for fetal ultrasound anomalies, rather than the 2q14.2 duplication. The fetus is likely to harbor a LQT2; unfortunately, a fetal electrocardiogram in utero was not available in this study, and whether the fetus has a prolonged QT interval is unknown. Moreover, the nonsense mutation of the KCNH2 gene identified in the fetus is classified as a secondary finding, according to the ACMG secondary finding v3.0 list; no sufficient evidence was available to reveal the causal relationship between KCNH2 mutations and CHD so far. Thus, more work needs to be conducted to clarify the existence of a genetic mechanism or if it was just a coincidence between KCNH2 mutations and CHD.
In conclusion, our study described an additional case of LQT2 combined with tetralogy of fallot in a fetus with a KCNH2 mutation. In addition, our study enriched the mutation spectrum of the KCNH2 gene and further indicated the application value of WES in prenatal diagnosis in fetuses with familial uncertain copy number variants.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee, and the approval was obtained from the Institutional Ethics Committee of Quanzhou women’s and children’s hospital to the commencement of the study (2020No.31). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JZ designed the article; JZ and CC wrote the article; YC and YJ recruited the participants and performed clinical consultation; YW and SZ performed the karyotype analysis and analyzed the data; GW, and YX revised and published the article. All authors approved the final article.
This research was supported by the Fujian Provincial Health Commission Youth Science and Technology Program (2020QNB045) and the Quanzhou City Science and Technology Program (2020C026R).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RL declared a shared parent affiliation with the author YX to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We wish to express our appreciation to the Fujian Provincial Health Commission and Quanzhou Science and Technology Bureau for funding this work. We also express our appreciation to the patients who participated in this study.
Bhuiyan, Z. A., Alswaid, A., Belfiore, M., Al-Ghamdi, S. S., Liang, J., and Schlaepffer, J. (2014). Not all Pathogenic Mutations Are Pathogenic: KCNH2 Mutations in Two Sisters with Tetralogy of Fallot. Int. J. Cardiol. 172 (1), 276–277. doi:10.1016/j.ijcard.2013.12.242
Chiu, S.-N., Wu, M.-H., Su, M.-J., Wang, J.-K., Lin, M.-T., Chang, C.-C., et al. (2012). Coexisting Mutations/polymorphisms of the Long QT Syndrome Genes in Patients with Repaired Tetralogy of Fallot Are Associated with the Risks of Life-Threatening Events. Hum. Genet. 131 (8), 1295–1304. doi:10.1007/s00439-012-1156-4
Christiansen, M., Hedley, P. L., Theilade, J., Stoevring, B., Leren, T. P., Eschen, O., et al. (2014). Mutations in Danish Patients with Long QT Syndrome and the Identification of a Large Founder Family with p.F29L in KCNH2. BMC Med. Genet. 15, 31. doi:10.1186/1471-2350-15-31
Crotti, L., Celano, G., Dagradi, F., and Schwartz, P. J. (2008). Congenital Long QT Syndrome. Orphanet J. Rare Dis. 3, 18. doi:10.1186/1750-1172-3-18
Dastan, J., Chijiwa, C., Tang, F., Martell, S., Qiao, Y., Rajcan-Separovic, E., et al. (2016). Exome Sequencing Identifies Pathogenic Variants of VPS13B in a Patient with Familial 16p11.2 Duplication. BMC Med. Genet. 17 (1), 78. doi:10.1186/s12881-016-0340-0
Devillard, F., Guinchat, V., Moreno-De-Luca, D., Tabet, A.-C., Gruchy, N., Guillem, P., et al. (2010). Paracentric Inversion of Chromosome 2 Associated with Cryptic Duplication of 2q14 and Deletion of 2q37 in a Patient with Autism. Am. J. Med. Genet. 152A (9), 2346–2354. doi:10.1002/ajmg.a.33601
Ebrahim, M. A., Williams, M. R., Shepard, S., and Perry, J. C. (2017). Genotype Positive Long QT Syndrome in Patients with Coexisting Congenital Heart Disease. Am. J. Cardiol. 120 (2), 256–261. doi:10.1016/j.amjcard.2017.04.018
Elizabeth, M. S. M., Verkerk, A. J. M. H., Hokken-Koelega, A. C. S., Verlouw, J. A. M., Argente, J., Pfaeffle, R., et al. (2020). Unique Near-Complete Deletion of GLI2 in a Patient with Combined Pituitary Hormone Deficiency and Post-axial Polydactyly. Growth Hormone IGF Res. 50, 35–41. doi:10.1016/j.ghir.2019.10.002
Gianulis, E. C., and Trudeau, M. C. (2011). Rescue of Aberrant Gating by a Genetically Encoded PAS (Per-Arnt-Sim) Domain in Several Long QT Syndrome Mutant Human Ether-Á-Go-Go-Related Gene Potassium Channels. J. Biol. Chem. 286 (25), 22160–22169. doi:10.1074/jbc.m110.205948
Granata, P., Cocciadiferro, D., Zito, A., Pessina, C., Bassani, A., Zambonin, F., et al. (2022). Whole Exome Sequencing in 16p13.11 Microdeletion Patients Reveals New Variants through Deductive and Systems Medicine Approaches. Front. Genet. 13, 798607. doi:10.3389/fgene.2022.798607
Hedley, P. L., Jørgensen, P., Schlamowitz, S., Wangari, R., Moolman-Smook, J., Brink, P. A., et al. (2009). The Genetic Basis of Long QT and Short QT Syndromes: a Mutation Update. Hum. Mutat. 30 (11), 1486–1511. doi:10.1002/humu.21106
Hsiao, S.-M., Wu, M.-H., Jou, H.-J., Lee, C.-N., Shyu, M.-K., Shih, J.-C., et al. (2007). Outcome for Fetuses with Prenatally Detected Congenital Heart Disease and Cardiac Arrhythmias in Taiwan. J. Formos. Med. Assoc. 106 (6), 423–431. doi:10.1016/s0929-6646(09)60291-6
Kevelam, S. H. G., van Harssel, J. J. T., van der Zwaag, B., Smeets, H. J. M., Paulussen, A. D. C., and Lichtenbelt, K. D. (2012). A Patient with a Mild Holoprosencephaly Spectrum Phenotype and Heterotaxy and a 1.3 Mb Deletion Encompassing GLI2. Am. J. Med. Genet. 158A (1), 166–173. doi:10.1002/ajmg.a.34350
Kordaß, U., Schröder, C., Elbracht, M., Soellner, L., and Eggermann, T. (2015). A familialGLI2deletion (2q14.2) Not Associated with the Holoprosencephaly Syndrome Phenotype. Am. J. Med. Genet. 167 (5), 1121–1124. doi:10.1002/ajmg.a.36972
Marks, M. L., Whisler, S. L., Clericuzio, C., and Keating, M. (1995). A New Form of Long QT Syndrome Associated with Syndactyly. J. Am. Coll. Cardiol. 25 (1), 59–64. doi:10.1016/0735-1097(94)00318-k
Massin, M. M., Dessy, H., Malekzadeh-Milani, S. G., Van Aerschot, I., and Verbeet, T. (2010). Prevalence of Preoperative Arrhythmias in Children with Delayed Treatment of Severe Congenital Heart Disease. Acta Cardiol. 65 (1), 37–42. doi:10.2143/ac.65.1.2045887
Murugan, S. J., Parsons, J. M., and Bennett, C. (2005). A Case of Long QT Syndrome Associated with Familial Occurrence of Persistent Patency of the Arterial Duct. Cardiol. Young 15 (3), 309–311. doi:10.1017/s1047951105000648
Ono, M., Burgess, D. E., Schroder, E. A., Elayi, C. S., Anderson, C. L., January, C. T., et al. (2020). Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules 10 (8), 1144. doi:10.3390/biom10081144
Qiao, Y., Badduke, C., Tang, F., Cowieson, D., Martell, S., Lewis, S. M. E., et al. (2017). Whole Exome Sequencing of Families with 1q21.1 Microdeletion or Microduplication. Am. J. Med. Genet. 173 (7), 1782–1791. doi:10.1002/ajmg.a.38247
Qiao, Y., Bagheri, H., Tang, F., Badduke, C., Martell, S., Lewis, S. M. E., et al. (2019). Exome Sequencing Identified a De Novo Mutation of PURA Gene in a Patient with Familial Xp22.31 Microduplication. Eur. J. Med. Genet. 62 (2), 103–108. doi:10.1016/j.ejmg.2018.06.010
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schilter, K., Reis, L., Schneider, A., Bardakjian, T., Abdul-Rahman, O., Kozel, B., et al. (2013). Whole-genome Copy Number Variation Analysis in Anophthalmia and Microphthalmia. Clin. Genet. 84 (5), 473–481. doi:10.1111/cge.12202
Schwartz, P. J., Stramba-Badiale, M., Crotti, L., Pedrazzini, M., Besana, A., Bosi, G., et al. (2009). Prevalence of the Congenital Long-QT Syndrome. Circulation 120 (18), 1761–1767. doi:10.1161/circulationaha.109.863209
Song, M. K., Bae, E. J., Kim, G. B., An, H. S., Ahn, K. J., Seong, M.-W., et al. (2018). Patients Diagnosed with Long QT Syndrome after Repair of Congenital Heart Disease. Pacing Clin. Electrophysiol. 41 (11), 1435–1440. doi:10.1111/pace.13512
Keywords: chromosomal array analysis, KCNH2, tetralogy of fallot, LQTS, whole-exome sequencing
Citation: Zhuang J, Chen C, Wang Y, Zeng S, Chen Y, Jiang Y, Xie Y and Wang G (2022) Case Report: Prenatal Whole-Exome Sequencing Identified a Novel Nonsense Mutation of the KCNH2 Gene in a Fetus With Familial 2q14.2 Duplication. Front. Genet. 13:924573. doi: 10.3389/fgene.2022.924573
Received: 20 April 2022; Accepted: 24 May 2022;
Published: 05 July 2022.
Edited by:
Oscar Campuzano, University of Girona, SpainReviewed by:
Ru Li, Guangzhou Medical University, ChinaCopyright © 2022 Zhuang, Chen, Wang, Zeng, Chen, Jiang, Xie and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaoxiong Wang, d2FuZ2dhb3hpb25nMjAxM0AxNjMuY29t; Yingjun Xie, eGlleWp1bkBtYWlsMi5zeXN1LmVkdS5jbg==
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.