 Rebecca Kingdom
Rebecca Kingdom Caroline F. Wright
Caroline F. Wright
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 25 July 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.920390
This article is part of the Research TopicDNA-based Population Screening for Precision Public HealthView all 28 articles
The same genetic variant found in different individuals can cause a range of diverse phenotypes, from no discernible clinical phenotype to severe disease, even among related individuals. Such variants can be said to display incomplete penetrance, a binary phenomenon where the genotype either causes the expected clinical phenotype or it does not, or they can be said to display variable expressivity, in which the same genotype can cause a wide range of clinical symptoms across a spectrum. Both incomplete penetrance and variable expressivity are thought to be caused by a range of factors, including common variants, variants in regulatory regions, epigenetics, environmental factors, and lifestyle. Many thousands of genetic variants have been identified as the cause of monogenic disorders, mostly determined through small clinical studies, and thus, the penetrance and expressivity of these variants may be overestimated when compared to their effect on the general population. With the wealth of population cohort data currently available, the penetrance and expressivity of such genetic variants can be investigated across a much wider contingent, potentially helping to reclassify variants that were previously thought to be completely penetrant. Research into the penetrance and expressivity of such genetic variants is important for clinical classification, both for determining causative mechanisms of disease in the affected population and for providing accurate risk information through genetic counseling. A genotype-based definition of the causes of rare diseases incorporating information from population cohorts and clinical studies is critical for our understanding of incomplete penetrance and variable expressivity. This review examines our current knowledge of the penetrance and expressivity of genetic variants in rare disease and across populations, as well as looking into the potential causes of the variation seen, including genetic modifiers, mosaicism, and polygenic factors, among others. We also considered the challenges that come with investigating penetrance and expressivity.
Approximately 72% (Nguengang Wakap et al., 2020) of all rare diseases are genetic in origin, and most of these are thought to be monogenic in nature (Haendel et al., 2020). Rare, deleterious variants are known to cause thousands of different genetic disorders in humans (Boycott et al., 2017; Rahit and Tarailo-Graovac, 2020), and while the molecular basis of over 6,000 monogenic diseases has been uncovered (OMIM, 2022), with more than 200,000 pathogenic variants described (QIAGEN, 2022; Stenson et al., 2017), the genetic basis of most rare disorders remains to be determined. With advances in next-generation sequencing (NGS) and the increasing availability of whole exome/genome sequencing (WES/WGS), the study of genotype–phenotype relationships has become more widespread as determining how the genotype causes a phenotype is a fundamental step toward understanding disease pathology (Stephanou et al., 2019). Protein-coding variants that are associated with disease phenotypes directly link DNA variation to altered protein function or dosage and to the phenotypic outcome, and so much of what we know about the genotype–phenotype relationship is based on the study of rare variants that cause monogenic disease (Chong et al., 2015). Monogenic genotypes can be highly predictive for specific individual disorders, but sometimes this relationship can be complicated, with some damaging dominant monogenic variants not following the expected Mendelian inheritance patterns (Schacherer, 2016). Individuals with the same genotype can display distinctly different clinical phenotypes (McDermott et al., 2017; Kumar et al., 2019; Crawford et al., 2021), including being clinically asymptomatic. Currently, there are gaps in translating how the individual genomic variation affects phenotypic presentation and how genetic variants exert their functional impact to cause disease.
The study of genetic disease has often been divided into rare monogenic forms of disease and more common polygenic complex disorders (Claussnitzer et al., 2020). Current evidence suggests that these groups may be more overlapping than previously thought as the genetic variation present across the genome highlights the complexity underlying the phenotypic presentation. There are both rare variants in individual genes that cause monogenic forms of complex disease (Vuckovic et al., 2020; Muse et al., 2021) and common variants that affect the severity of monogenic disease (Niemi et al., 2018; Goodrich et al., 2021). Such complexity makes investigating the genotype–phenotype relationships more complicated, which is only exacerbated by erroneous variant associations due to study design problems (Wright et al., 2019a). Human genetic diversity displays considerable variability, with individual genomes differing from the reference at 4.1–5 million sites (Auton et al., 2015). Although most variation is common and predicted to be functionally neutral (Ng et al., 2008), each individual has on average 85 heterozygous and 35 homozygous protein-truncating variants (PTVs) (Lek et al., 2016). Population cohort studies have shown that the average genome contains around 200 very rare variants per person (Gudmundsson et al., 2021) and 54 variants previously reported as disease-causing, including 7.6 rare non-synonymous coding variants in monogenic disease genes (Lek et al., 2016; Walsh et al., 2017). Variant interpretation is an ongoing challenge within diagnostic medicine, making understanding the phenotypic consequences of underlying genetic variation a key aim of genomics research.
A deleterious genotype should be no more prevalent in the population than the disease that it causes (Minikel et al., 2016). However, the same genetic variant can result in different disease presentations in different people, from clinically asymptomatic to severely affected, even among members of the same family (Mahat et al., 2021). The proportion of individuals who possess a particular genotype and exhibit the expected clinical symptoms is defined as the penetrance of that genotype (Cooper et al., 2013; Shawky, 2014). If everyone with the genotype presents with clinical symptoms by a particular age, then it is said to be fully penetrant, whereas if it falls below this, it is said to exhibit reduced or incomplete penetrance. Genotypes can also display variable expressivity, where the severity of the phenotype caused by the genotype can vary among affected individuals (Shawky, 2014) (Table 1); this differs from pleiotropy, where different variants in the same gene can cause different, potentially unrelated phenotypes that may even be categorized as different diseases (Ittisoponpisan et al., 2017) (Figure 1). Although penetrance, expressivity, and pleiotropy are three distinct concepts, biological reality means that their overall effects often overlap, especially in population cohorts where it is difficult to identify the cause of the phenotypic diversity. Multiple distinct phenotypes, in aggregate, could either be classified as a single more severe phenotype or different disease subtypes. As these three are likely to be caused by overlapping or similar mechanisms (Gruber and Bogunovic, 2020), especially in genetically heterogenous conditions, we will discuss them together in this review.
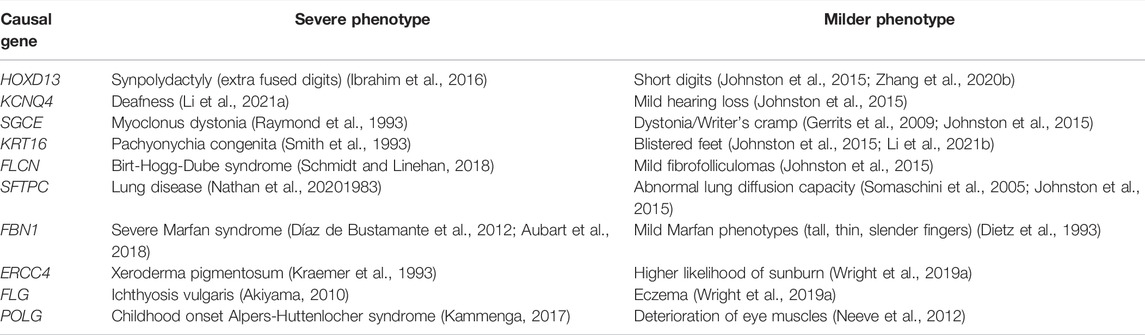
TABLE 1. Examples of variable expressivity in monogenic diseases. Deleterious variants in these genes are known to cause a spectrum of phenotypes, from severe disease to mild subclinical effects.
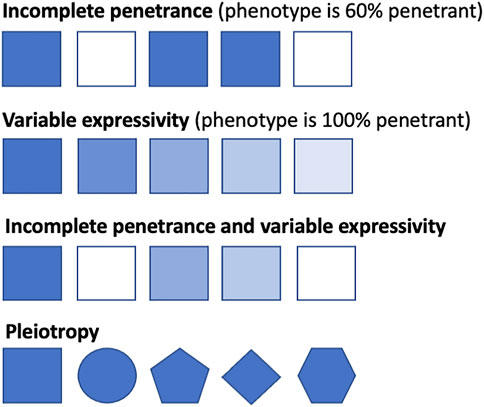
FIGURE 1. Conceptual representation of penetrance, expressivity, and pleiotropy. Squares represent individuals with the same genotype, with shaded squares indicating the individual displays the related phenotype and non-shaded squares indicating the individual does not display the related disease phenotype. Line one shows incomplete penetrance, where 60% of the individuals display the related phenotype. Line two shows that all individuals display the related phenotype, from severe manifestations to milder presentations. Line three shows incomplete penetrance and variable expressivity, where the genotype varies both in the severity of presentation and in penetrance across the population. Line four shows pleiotropy, whereby different phenotypes are caused by variants (represented by different shapes) in one gene.
Incomplete penetrance can be observed in both dominant and recessive conditions. However, the cause of variability in genotype–phenotype correlations can be difficult to elucidate; phenotypic variation has been observed in mice with identical environmental and genetic backgrounds, including variability in lethality for gene knockouts despite the introduction of identical variants (Dickinson et al., 2016). Establishing that a identified variant is the sole (or primary) cause of an individual’s clinical phenotype can be difficult (Shieh, 2019), which is an important concern when it comes to diagnosis and providing accurate genetic counseling, and such difficulties can lead to incorrect or delayed diagnosis (Maroilley and Tarailo-Graovac, 2019). The widespread presence of incomplete penetrance and variable expressivity through many overlapping mechanisms (Figure 2) can explain why apparently unaffected parents can pass on pathogenic variants to affected offspring (McDermott et al., 2017) and why seemingly healthy individuals’ genomes can contain a large number of putatively damaging variants and yet not suffer any obvious adverse effects (Xue et al., 2012).
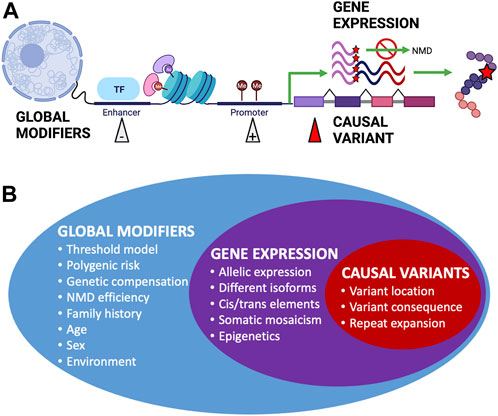
FIGURE 2. Factors affecting penetrance and expressivity. (A) Examples of different biological mechanisms that can affect the overall penetrance and expressivity of monogenic disease-causing genetic variants. Figure created using BioRender.com. (B) Summary of factors affecting penetrance and expressivity across the genome, from global modifiers that can have wide-ranging overall effects to expression of the gene containing causal variants and to specific causal variants that have more distinctive effects.
Although databases of clinically identified variants in affected individuals are useful for assessing pathogenicity (van Rooij et al., 2020), population-based datasets that include WES/WGS alongside phenotypic and medical information are increasingly important for investigating the penetrance and expressivity of these variants. Large population cohort studies have shown the occurrence of apparently pathogenic variants is much higher than previously estimated through small clinical or familial cohort studies (Wright et al., 2019a; Lacaze et al., 2020; van Rooij et al., 2020), and their frequency highlights either the incomplete penetrance, variable expressivity, or misclassification of such variants. The existence of PTVs in dosage-sensitive genes in healthy individuals also remains problematic when it comes to determining pathogenicity (Cummings et al., 2020). The potential for genomic technologies and WGS to detect individuals at risk of genetic disease is enormous, but incomplete penetrance and variable expressivity present a challenge for clinicians, especially when an incidental finding occurs without any prior clinical indication, leading to uncertainty over whether a clinical phenotype will develop, and if so, when. This problem is highlighted when testing unselected population cohorts, who may or may not have phenotypes of relevance to genomic findings at the point of testing. To understand how genetic disorders develop, we need to consider how deleterious variants interact with the rest of the variation in the genome and how variation can affect phenotypic presentation. This may also identify targets that help prevent disease progression (Downs et al., 2019). The presence of putatively pathogenic variants in asymptomatic adults also highlights the possibility that there are disease resistance mechanisms we can identify through the sequencing of general population cohorts.
Traditionally, rare pathogenic variants were identified in small phenotypically enriched clinical cohorts of individuals and families with similar monogenic disease. Population cohorts allow us to utilize the information from small clinical studies to investigate the penetrance of variants in the general “healthy” population, where such severe monogenic phenotypes are likely to be depleted, and the potential to identify the causes of clinical heterogeneity. Ascertainment bias can occur with any study design, with volunteer population cohorts tending to be healthier than the average individual (Fry et al., 2017) and clinical cohorts tending to have more severe phenotypes. Estimates of the maximum and minimum variant effect sizes across different ascertainment contexts are needed to avoid falsely predicting that a significant proportion of the healthy population is at risk for a monogenic condition (Flannick et al., 2013). The proportion of individuals affected and the average age of onset (i.e., age-dependent penetrance) can vary depending on the ascertainment context (Figure 3). For example, individuals with putatively pathogenic variants in HNF1A and HNF4A, known for causing maturity-onset diabetes of the young (MODY), develop diabetes significantly later or not at all when tested outside of the context of clinical referrals for the suspected MODY (Mirshahi et al., 2021).
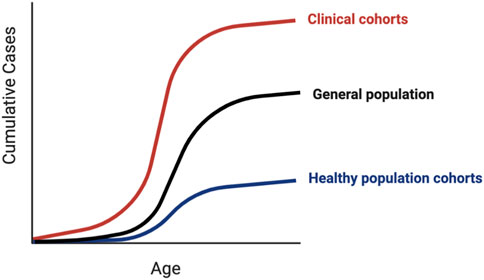
FIGURE 3. Penetrance in clinical versus population cohorts. Penetrance of genetic variants identified in clinical cohorts tends to be higher than the same variants identified in population cohorts, which can manifest as earlier disease onset, less severe disease, or a larger proportion of affected individuals. Due to inherent ascertainment biases in both types of cohorts, the penetrance of variants in the general unselected population is likely to lie somewhere in-between.
For almost all human genetic diseases, individual variability in the phenotype is influenced by background variation in the genome. As genetic testing has become more widely available, both through healthcare systems, direct-to-consumer testing (Stoeklé et al., 2016), our understanding of how genomic variation affects disease progression and prevalence has become significantly more important, both for clinical utility (Shieh, 2019) and for our functional understanding of the disease (Tarailo-Graovac et al., 2017). Variation in the genome can predispose individuals to disease through traditional monogenic variants that disrupt physiological pathways and exert a large effect on the phenotype, or through the accumulation of polygenic effects that involve many variants of small effect sizes in different pathways (Fahed et al., 2020), or as is increasingly becoming clear, through their combined effect.
Within population cohorts, penetrance estimates for monogenic variant carriers average 60% or lower for most conditions (Goodrich et al., 2021), illustrating that many individuals have highly penetrant, pathogenic variants in known monogenic disease-causing genes who never develop the corresponding phenotype (Chen et al., 2016). Generally, 70% of the “Wellderly” healthy aging cohort, all of whom reached 80 without any chronic diseases, had one heterozygous deleterious variant in genes listed in the American College of Medical Genetics and Genomics (ACMG) secondary findings (Erikson et al., 2016). Similarly, one in 75 (1.3%) of healthy elderly individuals in the APSREE trial carried a previously identified pathogenic variant, including in Lynch syndrome and familial hypercholesterolemia genes, without having the associated phenotype (Lacaze et al., 2020). These cases demonstrate that carrying such pathogenic variants does not always cause the associated disease and that other mechanisms may contribute to the protection of human health, including genetic modifiers that ‘rescue’ individuals from a disease phenotype.
For genetically heterogenous monogenic diseases, the penetrance and expressivity can vary between different genes or variants, with the same phenotype potentially caused by numerous different variants across multiple genes (Wright et al., 2018). Even within the same gene, some deleterious variants in known monogenic disease genes may exhibit complete penetrance, while others show incomplete or low penetrance. Variation can be due to functional redundancy of genes, or the location and type of variant, with missense and PTVs in the same gene often causing different phenotypes. For example, hereditary angioedema can show great phenotypic diversity, even among members of the same family, and individuals with missense variants in SERPING1 typically display a milder and later onset of disease than patients with PTVs (Speletas et al., 2015). In contrast, missense variants in BMPR2 cause earlier and more severe pulmonary hypertension than PTVs in the same gene (Austin et al., 2009).
Pathogenic PTVs typically cause disease through loss of function (LoF) due to degradation of the RNA by nonsense-mediated decay (NMD) (Lu and Krebber, 2021). NMD is an mRNA surveillance pathway that recognizes and degrades damaged mRNA transcripts that would produce misfolded or shortened proteins that can accumulate in the cell and initiate the endoplasmic reticulum (ER) stress response (Haeri and Knox, 2012). However, the production of a variant protein can either exacerbate disease severity through the accumulation of toxic proteins in the cell (Nguyen et al., 2014) or alleviate it through providing a residual function that protects against haploinsufficiency-mediated disease in the heterozygous state (van Leeuwen et al., 2017; Coban-Akdemir et al., 2018; Kennedy et al., 2019), meaning the occurrence of NMD can affect phenotypic severity depending on the mechanism of disease. PTVs may also cause LoF through aberrant splicing (Cummings et al., 2020), which is also regulated by NMD (Lareau and Brenner, 2015). In some cases, the location of NMD boundaries at the 5′ and 3′ ends of genes containing causal variants can explain phenotypic variation between individuals with different PTVs in the same gene (Nagy and Maquat, 1998; Lindeboom et al., 2016). For example, PTVs located outside of the region that triggers NMD in SOX10 escape NMD and produce proteins that have dominant-negative activity, causing the severe complex neurological disorder PCWH, whereas PTVs located within the NMD region produce transcripts that are recognized by NMD and removed, causing the relatively milder WS4 syndrome via haploinsufficiency (Inoue et al., 2004; Miller and Pearce, 2014). This variability in penetrance or expressivity could potentially be classed as distinct subtypes of disease, with different variants causing disease through different mechanisms and producing distinct syndromes. Pathogenic variants in KAT6B show a similar disease manifestation, with two distinct syndromes depending on whether NMD is triggered or not (Zhang et al., 2020a). Variants in KAT6A cause severe intellectual disability (ID) and neurodevelopmental disorders (NDD), with late PTVs more likely to cause a severe phenotype, compared to 60% of early PTVs which conferred a mild phenotype (Kennedy et al., 2019), potentially due to whether NMD is activated or not. The position of the PTV within the gene has also been seen to modulate the severity of clinical phenotypes in Marfan syndrome (Taniguchi et al., 2021) and Charcot-Marie-Tooth disease (Pipis et al., 2022). Disease due to SFTPB variants typically presents in neonates as respiratory distress syndrome, resulting in death within the first few months; variants that allow partial production of the SP-B protein confer longer survival times and later onset of disease, whereas the variants that cause complete deficiency of SP-B due to NMD cause fatal neonatal respiratory distress syndrome (van Moorsel et al., 2021).
Missense variants can also result in LoF due to substantially reduced protein function or stability (Høie et al., 2022). Although many missense variants have little or no effect, they can result in conformational changes, increased protein misfolding, and aberrant protein trafficking, which can lead to intracellular retention or accumulation, increased ER stress, activation of the unfolded protein response, or increased pro-apoptotic signaling and apoptosis (van Moorsel et al., 2021). Some missense variants, small insertions/deletions, and gene duplications can also result in gain of function (GoF) effects due to increased activity (Niday and Tzingounis, 2018), increased protein production (Stefl et al., 2013), or via protein products that gain a new damaging function (Li and Babu, 2018). Some GoF variants can exhibit a more severe phenotype than LoF variants in the same gene; for example, GoF variants in KCNA2 were associated with more severe epilepsy phenotypes than LoF variants (Syrbe et al., 2015). Where in a gene a variant is located can affect the mechanism of disease, as well as penetrance and expressivity through molecular subregional effects (Platzer et al., 2017); the impact of a variant depends on whether it is located at sites that undergo post-translational modification, within sites that are critical for tertiary and quaternary structure, at protein–protein interaction interfaces or ligand binding sites, or inside versus outside of functional domains (Faure et al., 2022). For example, missense variants in GRIN2A located in transmembrane or linker domains were more frequently associated with severe developmental phenotypes than those located elsewhere, such as within amino-terminal or ligand-binding domains (Liu et al., 2021), with a wide range of phenotypes observed from normal to mild epilepsy, to severe developmental phenotypes and epileptic encephalopathy (Strehlow et al., 2019); similarly, GoF variants in highly conserved regions of the potassium channel of KCNA2 were associated with more severe epileptic encephalopathy than variants located elsewhere (Masnada et al., 2017). An improved understanding of the protein structure and the functionality of interacting domains will help elucidate specific variant effects on the resulting phenotypic presentation (Ittisoponpisan et al., 2021).
Finally, there are a small but increasing number of pathogenic non-coding variants that have been identified as causes of monogenic diseases. These variants can operate either through LoF or GoF mechanisms by altering the gene or isoform expression (Ellingford et al., 2021). For example, biallelic variants in the PTF1A enhancer are a well-established cause of recessive pancreatic agenesis through tissue-specific LoF (Weedon et al., 2014); de novo LoF variants in the 5′ untranslated region (UTR) of MEF2C have been shown to account for around a quarter of developmental disorder diagnoses in this gene (Wright et al., 2021); and a single GoF variant that creates a novel promoter has been shown to cause α-thalassemia (Bozhilov et al., 2021). However, establishing the pathogenicity of non-coding variants is often much more challenging than coding variants, and thus, studies of penetrance and expressivity of these variants are likely to lag behind.
Repeat expansion disorders are caused by genomic expansions of short tandem repeat (STR) sequences that either affect the gene expression or protein sequence (Paulson, 2018), with the penetrance and expressivity affected by the number of repeats (Table 2). Anticipation is often observed in families due to molecular instability around the repeats; in each generation, the repeat length can increase, resulting in the earlier onset of disease and increased severity. For example, Fragile X syndrome is caused by the expansion of over 200 repeats in the CGG motif in the 5′UTR of FMR1 on the X chromosome, resulting in hypermethylation of the promoter, silencing the gene (Hagerman et al., 2017). Fragile X exhibits incomplete penetrance and reduced expressivity, with 100% of males and 60% of females presenting with ID and 50–60% of males and 20% of females diagnosed with autism spectrum disorder (ASD) (Payán-Gómez et al., 2021). Wild type (WT) alleles contain <44 CGG repeats, while full mutations in affected individuals typically have >200 repeats. Those with premutation alleles of 55–200 repeats have milder phenotypes than full mutation carriers, although they have an increased risk of Fragile X-associated tremor/ataxia syndrome (Cabal-Herrera et al., 2020) and primary ovarian insufficiency prior to age 40 (Fink et al., 2018) compared to WT. Monotonic dystrophy shows a similar mechanism, with unaffected individuals having 5–37 CTG repeats in the 3′UTR of DMPK and fully affected individuals having >80 repeats (although repeats of >1,000 have been seen in congenitally affected children (Morales et al., 2016)), with an number of repeats correlating with the earlier age of onset.
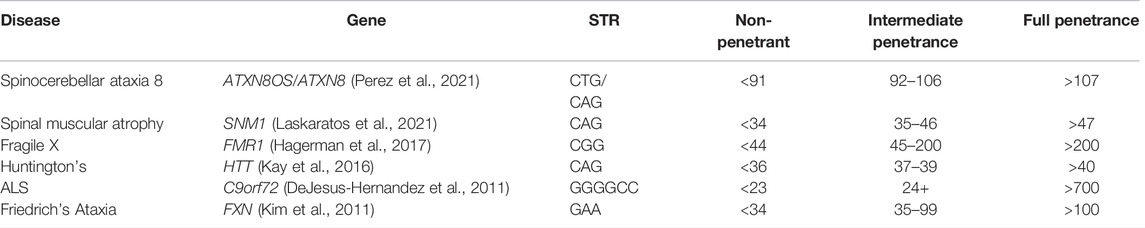
TABLE 2. Trinucleotide repeat disorders with varying penetrance depending on the number of repeats present.
Although the number of repeats accounts for a large proportion of variable expressivity, there are still missing genetic factors accounting for differences in the age of onset. For example, in Huntington’s disease, a lower number of N-terminal CAG repeats in HTT is associated with reduction in penetrance and later onset of clinical symptoms (Kay et al., 2016), but while the number of repeats is inversely correlated with the age of onset of motor symptoms, they only account for 70% of the variability (Holmans et al., 2017). The remaining unexplained variance displays a high degree of heritability, suggesting further genetic modifiers (Arning, 2016). Additional genetic variants in the DNA mismatch repair pathway have been linked with anticipation and overall severity of disease, and functional studies showing the knockout of base-excision repair or transcription-coupled repair pathways in animal and cellular models of nucleotide repeat disorders can inhibit the expansion and reduce the phenotypic severity (Goula and Merienne, 2013; Massey and Jones, 2018). Variants in the DNA repair gene MSH3 have also been linked with differences in disease severity through somatic instability (Flower et al., 2019). As non-penetrant individuals will not necessarily come to clinical attention and large triplet repeats are hard to genotype accurately using NGS (Bahlo et al., 2018), it is suspected that individuals with fewer than 41 CAG repeats in HTT may exist at a higher frequency than previously expected in the general asymptomatic population (Kay et al., 2016).
It has been hypothesized that the differential expression of alternative alleles in the gene containing causal variants could affect the presentation of phenotypic traits in individuals with identical genotypes. This mechanism has been proposed primarily for dominantly inherited conditions where haploinsufficiency is the cause of the disease (Ahluwalia et al., 2009; Jordan et al., 2019), including Lynch syndrome (Hesson et al., 2015) and hypertrophic cardiomyopathy (HCM) (Glazier et al., 2019), where an allelic imbalance could cause either higher expression of the WT allele, thus compensating for the haploinsufficiency and resulting in reduced penetrance, or lower expression of the WT allele, thus exacerbating the haploinsufficiency and resulting in higher penetrance. Significant allelic imbalance has been observed in up to 88% of genes in human tissues, potentially caused by genetic modifiers or stochastic factors (Aguet et al., 2017), and has been identified as both tissue-specific and genome-wide in mouse models (Pinter et al., 2015). Structural variants such as duplications that are in trans with a pathogenic LoF variant can alleviate the potential clinical phenotype when disease would be caused by haploinsufficiency, by providing an additional WT copy of a gene, thus resulting in a normal level of gene expression (Servetti et al., 2021), as has been observed in DiGeorge syndrome (Carelle-Calmels et al., 2009). Additional variants in the untranslated regions of mRNA can also affect the translational efficiency and gene expression can also vary widely across tissues, highlighting the importance of sequencing disease-relevant tissue in the interpretation of genetic variation (Cummings et al., 2017; Mignone et al., 2002). Compared to synonymous variants, rare missense variants show a significant reduction in allelic expression across many tissues in proportion to their predicted pathogenicity, suggesting deleterious variants are depleted from highly expressed haplotypes (Castel et al., 2018). Some highly differentially expressed genes have been shown to contain fewer disease-associated variants (Chen et al., 2008), which are less likely to accumulate on haplotypes that are highly expressed, or in high-penetrance combinations (Castel et al., 2018). For example, genetically heterogenous monogenic eye disorders display both incomplete penetrance and variable expressivity and also display significant variability in gene expression levels throughout the population (Green et al., 2020). The differential expression of alleles has also been shown to play a role in the variable expressivity of Marfan’s syndrome (Aubart et al., 2015).
The differential expression of alleles can also potentially cause recessive conditions to present in a dominant fashion. For example, Zellweger spectrum disorder (ZSD) is an autosomal recessive disorder caused by deleterious variants in any of 13 PEX genes, with the most common cause being variants in PEX1 or PEX6. Affected heterozygous carriers have been identified with ZSD despite lacking a second pathogenic allele, with all affected heterozygotes presenting with the allelic overexpression of the variant allele compared to WT, and a common polymorphism has been linked to this allelic overexpression (Falkenberg et al., 2017). In HCM, the proportion of sarcomeric proteins produced by variant alleles can vary with the allelic expression, and 30–80% of the sarcomere structure can be made up of proteins with reduced function (Marian and Braunwald, 2017; de Marvao et al., 2021), causing variation in overall phenotypic severity.
Stochastic variation within normal cellular and developmental processes can potentially be amplified by disease-causing variants and thus play a role in incomplete penetrance and variable expressivity (Binder et al., 2015). Random monoallelic expression (RME) is the transcription of only one allele from a homologous pair and can be constitutive, with all cells expressing the same allele throughout (as seen in imprinted genes), or somatic, with individual cells showing variation in expression levels (Eckersley-Maslin and Spector, 2014). Overall levels of RNA in cell populations tend to be stable, but dynamic allelic fluctuation through RME can present variability in the gene expression. Genes that show little RME are mostly housekeeping genes that have higher expression levels (Eckersley-Maslin and Spector, 2014). Although no variation in the disease trait has yet been definitively linked to somatic RME, conceptually it could explain the phenotypic variation either through alteration of gene dosage or the higher expression of a variant allele. RME during embryonic development has been tentatively linked with variation in developmental disorders such as Holt-Oram syndrome (Gui et al., 2017). Model organism research has suggested stochastic variation in the gene expression can affect the expressivity of variant genotypes, with 20% of genes causing variation in phenotypes in two different isolates with defined genetic backgrounds in C. elegans (Vu et al., 2015). Phenotypic variability has also been observed in inbred mice with a defined genetic background (Dickinson et al., 2016), as well as in monozygotic (MZ) twins (Baranzini et al., 2010), suggesting the influence of stochastic molecular events in variable expressivity.
Production of different transcripts of genes may also lead to the differential expression of traits and explain why potentially deleterious variants in haploinsufficient genes are found in population cohorts. Annotations based on transcription levels of different isoforms in haploinsufficient genes identified that 23% of LoF variants are in under-expressed exons and had similar effect sizes to synonymous variants (Cummings et al., 2020). In monogenic cardiomyopathies caused by LoF variants in the giant muscle protein titin, studies of TTN expression levels indicate that LoF variants found in unaffected population cohorts occur predominantly in exons that are absent from the most highly expressed transcripts and thus do not cause the phenotypic effect associated with deleterious variants (Begay et al., 2015; Akinrinade et al., 2019). Similarly, haploinsufficiency of TCF4 causes the highly penetrant Pitt-Hopkins syndrome (Kharbanda et al., 2016; Sirp et al., 2021), PTVs identified in these gene in unaffected individuals were all found to be located in minimally expressed exons (Aguet et al., 2017), suggesting that functional protein can be made in the presence of these variants. The expression of tissue-specific isoforms can also affect the penetrance of a genotype, potentially resulting in distinct disease subtypes. For example, CACNA1C has two clinically important isoforms with mutually exclusive exons that explain two different forms of Timothy syndrome; pathogenic variants across the widely expressed transcript produce a multi-system disorder (type 1), while pathogenic variants in the alternative exon of a transcript predominantly expressed in the heart are much rarer and result in more severe cardiac-specific defects and fewer syndromic phenotypes (type 2) (Dick et al., 2016). Further examples are likely to be uncovered through large-scale analysis of isoform expression in different tissues and at different times.
Variants in regulatory regions can affect the phenotypic presentation of disease by altering the gene expression and through modulation of deleterious genetic variants found in associated protein-coding regions (Scacheri and Scacheri, 2015), potentially affecting the penetrance and expressivity of the monogenic variant. Cis-acting elements are DNA sequences located on the same haplotype as the gene they affect, whereas trans-acting factors are proteins or elements that bind to the cis-acting sequences to affect the gene expression. Variants in these non-coding regions can have multiple downstream effects, through interactions with other genetic features or through effects on monogenic variants (van der Lee et al., 2020). Small changes within transcription factor binding or expression can lead to dysregulation that affects multiple genes within the same regulatory network (van der Lee et al., 2020) and therefore could potentially alter the final phenotypic presentation. Cis-regulatory variants have been identified that modify the penetrance of coding variants and therefore contribute to disease risk or presentation. Pathogenic coding variants are depleted from higher-expressed haplotypes with cis-regulatory variants in the general population (Castel et al., 2018), suggesting that individuals who present with a disease phenotype may have an enrichment of cis-regulatory variants that increase the expression of the pathogenic allele, compared to individuals who are asymptomatic who have an enrichment of ‘protective’ regulatory variants that decrease the expression and, therefore, penetrance of the pathogenic allele (Castel et al., 2018).
Upstream open reading frames (uORFs) are tissue-specific cis-regulators of protein translation found in the 5′UTR region of protein-coding genes, and variants that alter uORFs can affect whether a deleterious protein-coding variant causes a disease phenotype or not and may alter the phenotypic presentation of the disease (Silva et al., 2019). Active translation of a uORF can reduce downstream protein levels by up to 80% via several mechanisms, including the production of a peptide that stalls the translating ribosome (Young et al., 2016) and termination at a uORF stop codon that can trigger NMD (Lee et al., 2021a). Variation that either introduces or removes uORF start or stop codons can, therefore, affect the phenotypic presentation, and uORF variants may also have a role in disease pathology (Whiffin et al., 2020). Variants in the downstream 3′UTR region may also play a role in regulation of the gene expression through altering the mRNA stability or translational efficiency (Jansen, 2001; Mignone et al., 2002; Steri et al., 2018). For example, a common single nucleotide polymorphism (SNP) downstream of GATA6 has been shown to reduce its expression, potentially resulting in a more severe pancreatic agenesis phenotype when found in trans with a LoF variant in the same gene (Kishore et al., 2020). Similarly, polymorphisms in the 3′UTR region of KCNQ1 have been suggested to alter the expression of the cis allele, either increasing the severity of the disease or reducing it through an uneven expression of WT or variant alleles (Amin et al., 2012). However, an attempt to replicate this in a diverse group of population cohorts found no association between the identified polymorphisms and the severity of disease (Kolder et al., 2015), highlighting the difficulties with trying to identify non-coding modifiers of rare disease, both in clinical cohorts and population studies.
Approximately 400,000 candidate enhancer regions have been identified in the human genome, with an average of around 20 enhancers per gene (The ENCODE Project Consortium, 2012; Yokoshi et al., 2020). Non-coding variants within enhancer regions can be a cause of phenotypic diversity through alterations in gene expression, therefore affecting overall disease phenotype presentation (Sun et al., 2018). Although identifying non-coding variants that affect disease presentation can be very difficult, there are some notable examples. A large study identified an SNP in an intronic enhancer of RET that appeared to increase the penetrance of Hirschsprung disease in patients with rare RET/coding variants (Emison et al., 2010). Intronic variants have also been suggested to affect the penetrance of coding variants in patients with Stargardt disease, where a deep intronic variant has been shown to be a major cis-acting modifier of the most common pathogenic variant in ABCA4 (Zernant et al., 2018; Lee et al., 2021b). A small study also suggested that SNPs in promoter regions affect the severity of arrhythmias among individuals with LoF variants in SCN5A (Park et al., 2012). Variants that create novel binding sites for transcription factors have been implicated in affecting penetrance through altering the gene expression, including a common non-coding polymorphism that alters the hepatic expression of SORT1 (Musunuru et al., 2010), contributing to myocardial infarction. Further WGS research is needed to identify non-coding variants that affect gene expression levels.
Genes are often associated with multiple cis-regulatory elements through topologically associated domains (TADs) (Delaneau et al., 2019). These domains are thought to affect the gene expression and mediate the effects of cis- and trans-regulatory factors through the 3D conformation of chromatin, and therefore, variants in these domains can affect penetrance and expressivity of genotypes (Galupa and Heard, 2017; McArthur and Capra, 2021). Although the expression of some genes has been shown to be unaffected by changes in TADs (Williamson et al., 2019), the creation of new TADs has been implicated in the pathogenicity of rare duplications (Franke et al., 2016). Alterations to the 3D chromatin structure within and between TADs can lead to mis-alignment of genes, enhancers, and silencers, affecting transcriptional control of the gene expression (Boltsis et al., 2021). Variants in TAD loops may have no effect on healthy individuals but could affect disease presentation in those with an underlying monogenic variant (Lu et al., 2020). Common genetic variants in cis-regulatory domains can affect the gene expression, and rare variants have been identified that disrupt the structure of the domain (Epstein, 2009; van der Lee et al., 2020), and both could contribute to varying phenotypic expressivity of identical protein-coding sequences by causing changes in upstream mechanisms of gene regulation. Structural changes that affect transcription factor binding can lead to functional gene expression changes (McArthur and Capra, 2021), as seen in the EPHA4 locus, where deletions or duplications that overlap the TAD boundary can cause severe limb malformations (Lupiáñez et al., 2015), while deletion of the entire locus does not (Helmbacher et al., 2000), which is thought to be due to differential gene enhancer associations.
Postzygotic de novo mutations that occur during cell division can result in somatic genetic variation that differs between cells, leading to mosaicism (Biesecker and Spinner, 2013). Monogenic disease is usually less severe in mosaic individuals than those who have the same variant expressed constitutively and depending upon which cells or tissues contain the pathogenic variant, mosaicism can result in non-penetrance or reduced expressivity (Hervé et al., 2015). Somatic mosaicism is suspected to be more widespread than is usually detected, especially when testing only a single tissue sample that may or may not contain the clinically relevant variant(s), although NGS is making it easier to identify lower-level genetic changes (Domogala et al., 2021; Chen et al., 2022).
Mosaic somatic variants have been suggested to be more representative than germline variants of the true diversity and range of potential variation in human disease as genotypes that are lethal in the constitutive form can be identified when present as mosaic (Bickley et al., 2014; Alswied et al., 2021). These include variants that cause osteogenesis imperfecta, where a mosaic father presented with mild symptoms, but the constitutive form was incompatible with life (Wallis et al., 1990), Proteus syndrome (Cohen, 2014) and CLOVES syndrome (Ferreira et al., 2021), two overgrowth disorders that are lethal in the constitutive form, and various mosaic aneuploidies (Leon et al., 2011). Alternatively, mosaic individuals can display different or milder phenotypes than those with germline variants in the same gene. For example, mosaic individuals with a variant in HRAS present with benign keratinocytic epidermal nevi (“woolly hair”) (Honda et al., 2017), whereas those with the same constitutive variant have the more severe Costello syndrome (Gripp et al., 1993). Other diseases that have been demonstrated to show a milder phenotype when caused by somatic mosaicism include telangiectasis (Tørring et al., 2017) and polycystic kidney disease (Hopp et al., 2020). Mosaic genotypes can also display varying phenotypes that include segmental forms of the constitutive disease, such as segmental neurofibromatosis type 1, where clinical manifestations are only shown in certain parts of the body (Jindal et al., 2019). In addition to presenting with variable expressivity, mosaic variants can also be incompletely penetrant. In individuals with primary immunodeficiencies, 80% of mosaic individuals were clinically asymptomatic, with the remaining 20% exhibiting partial clinical symptoms (Mensa-Vilaró et al., 2019; Gruber and Bogunovic, 2020). Similarly, mosaic chromosomal aneuploidy has been shown to be incompletely penetrant in population cohorts, with women who had 45,X/46,XX mosaicism presenting with normal reproductive lifespan and birth-rate and no cardiovascular complications, compared to those with the non-mosaic genotype (Tuke et al., 2019). Unaffected parents with mosaic pathogenic variants can pass their genotype onto their offspring as a constitutive germline variant, so an incompletely penetrant or milder disease in one generation can cause a completely penetrant disease in the next (Campbell et al., 2014; Acuna-Hidalgo et al., 2015; Lauritsen et al., 2017; Wright et al., 2019b; Mastromoro et al., 2020).
Somatic mosaicism can also rescue an individual from disease, through cellular reversion that reduces the expressivity of a phenotype. For example, somatic reversions have been observed in several cell lineages from individuals with immunodeficiency caused by biallelic variants in DOCK8, including variants that correct or remove germline PTVs, and recombination events that attenuate or remove the deleterious variant from one allele. These somatic reversions improve overall survival time, but they are unable to completely eliminate the disease phenotype (Jing et al., 2014). Somatic reversion has been observed in other primary immunodeficiencies (Hou et al., 2021; Miyazawa and Wada, 2021) and may partially explain incomplete penetrance (Gruber and Bogunovic, 2020). Reversion of the clinical phenotype in individuals with recessive dystrophic epidermolysis (Pasmooij et al., 2010) and Fanconi anemia (Gross et al., 2002; Nicoletti et al., 2020) has also been identified. Remarkably, long-term remission from WHIM syndrome, caused by GoF variants in CXCR4, was seen in an adult who had undergone chromothripsis of chromosome 2 resulting in deletion of the disease allele in a single hematopoietic stem cell, leading to the repopulation of the bone marrow with the haploinsufficient CXCR4 cells (McDermott et al., 2015; Heusinkveld et al., 2017).
Epigenetic modifications are molecularly heritable changes that alter gene expression without altering the DNA sequence itself, including DNA methylation, histone modifications, and microRNA (miRNA) expression (Weinhold, 2006). Differential epigenetic modifications between individuals carrying the same pathogenic genotype can potentially account for incomplete penetrance and variable expressivity of the phenotype. DNA methylation is important in the control of tissue-specific gene expression, alternative splicing, prevention of cryptic initiation of transcription from alternative promoters, and X chromosome inactivation, all of which have been shown to affect the progression of disease (Velasco and Francastel, 2019). Studies of MZ twins that are discordant for disease phenotypes have highlighted how epigenetic mechanisms could affect the penetrance or expressivity of disease (Castillo-Fernandez et al., 2014). For example, MZ twins with neurofibromatosis, caused by variants in NF1, showed significant discordance in the presence of tumors and severity of scoliosis, suggesting that additional non-hereditary factors were modifying their phenotypes (Rieley et al., 2011). Similarly, one MZ twin with a pathogenic homozygous variant in GBA was diagnosed with Gaucher disease, while the other was clinically asymptomatic (Lachmann et al., 2004; Biegstraaten et al., 2011), and differences in their epigenome were posited as a mechanism to explain this discordance. However, epigenetic studies are generally more challenging than genetic studies as variation may be both tissue and time-specific, making it harder to elucidate how epigenetic mechanisms affect the penetrance of such genotypes. One suggested mechanism is that epigenetics may compensate for the presence of a deleterious variant, and segregate through several generations without any ill effects until the epigenetic modifications are no longer functional (Tolmacheva et al., 2020). This has been seen in Xq24 microdeletions that are inherited from mothers with extremely skewed X-chromosome inactivation, which modifies the penetrance (Tolmacheva et al., 2020). Skewed X inactivation is also suggested to be a cause behind the clinical heterogeneity in Klinefelter syndrome (Skakkebaek et al., 2020). Epigenetic mechanisms have also been suggested to partially compensate for deletions in healthy carriers of IMMP2L deletions, which cause ID and NDD, as reduced DNA methylation levels were seen in healthy carriers but not in affected offspring (Vasilyev et al., 2021).
Another mechanism by which epigenetic changes may affect the penetrance of monogenic diseases is via miRNAs, small non-coding RNAs that regulate the gene expression (Catalanotto et al., 2016). One miRNA can influence multiple genes, and a gene can be affected by several miRNAs, potentially highlighting how variants in one miRNA may lead to multiple downstream phenotypic effects (Wallace et al., 2020). Differential miRNA expressions can be caused by genetic variation, and variants within miRNA could, thus, affect the allelic expression and modify the penetrance or expressivity of monogenic diseases (Cammaerts et al., 2015). The expression of numerous miRNAs may affect the penetrance and expressivity in hereditary breast and ovarian cancer (HBOC) (Tommasi et al., 2021); incomplete and age-dependent penetrance is common in carriers of pathogenic variants in BRCA1 and BRCA2, and variation in several miRNAs that bind the 3′UTRs and downregulate the expression of both genes have been linked with an increased risk of earlier onset cancer (Chen and Parmigiani, 2007; Chang et al., 2011; Moskwa et al., 2011; Sun et al., 2013; Tommasi et al., 2021).
There may be a threshold that has to be met for the manifestation of a clinical disease phenotype, and genetic and other factors may vary in their relative contribution to meeting this threshold for different diseases and in different individuals (Figure 4) (Walsh et al., 2020). Some highly penetrant monogenic disease variants may always be sufficient to push the genetic burden above the threshold of the disease, although secondary variants may still contribute to severity (Pizzo et al., 2019). For example, Dravet syndrome (DS) is a highly penetrant and devastating form of childhood epilepsy caused by de novo LoF variants in SCN1A (Ding et al., 2021). Although DS displays considerable clinical heterogeneity within families and severity may relate to background genetic variation (Hammer et al., 2017), there are no known modifiers that protect against the effects of the primary causal variant; the LoF variant alone is sufficient to push the individual above the threshold for disease and other variants can only change the severity of the phenotype above this point. Individuals with monogenic variants that are causative of disease alone and, thus, are already above the threshold for disease can be further modulated by secondary monogenic variants in related genes that also cause the same phenotype, and the accumulation of these PTVs is associated with a more severe phenotype as the burden is pushed way beyond the threshold (Bertolini et al., 2020). For example, in monogenic polycystic kidney disease, individuals with PTVs in each of the causative genes, PKD1 and PKD2, present with a much more severe disease than those with just one PTV (Arora et al., 2020). Many monogenic disease-causing variants have been found to have secondary genes or loci that affect the severity of their related clinical phenotypes (Posey et al., 2017; Pizzo et al., 2019) (Table 3).
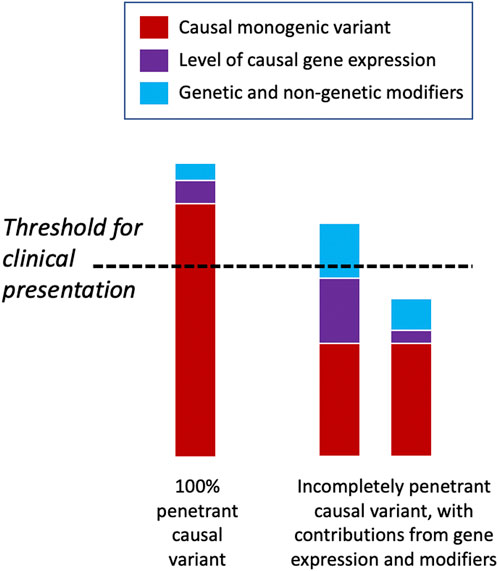
FIGURE 4. Threshold model of disease. Some deleterious monogenic variants are sufficient to cause the disease alone and do not need any genetic modifiers to cause the disease phenotype. Other monogenic variants may be incompletely penetrant and only display a disease phenotype when accompanied by other genetic or non-genetic factors that raise them above the clinical threshold for disease presentation. In the latter scenario, individuals may have the same underlying causal variant but have very different phenotypic presentations depending upon their modifying factors.
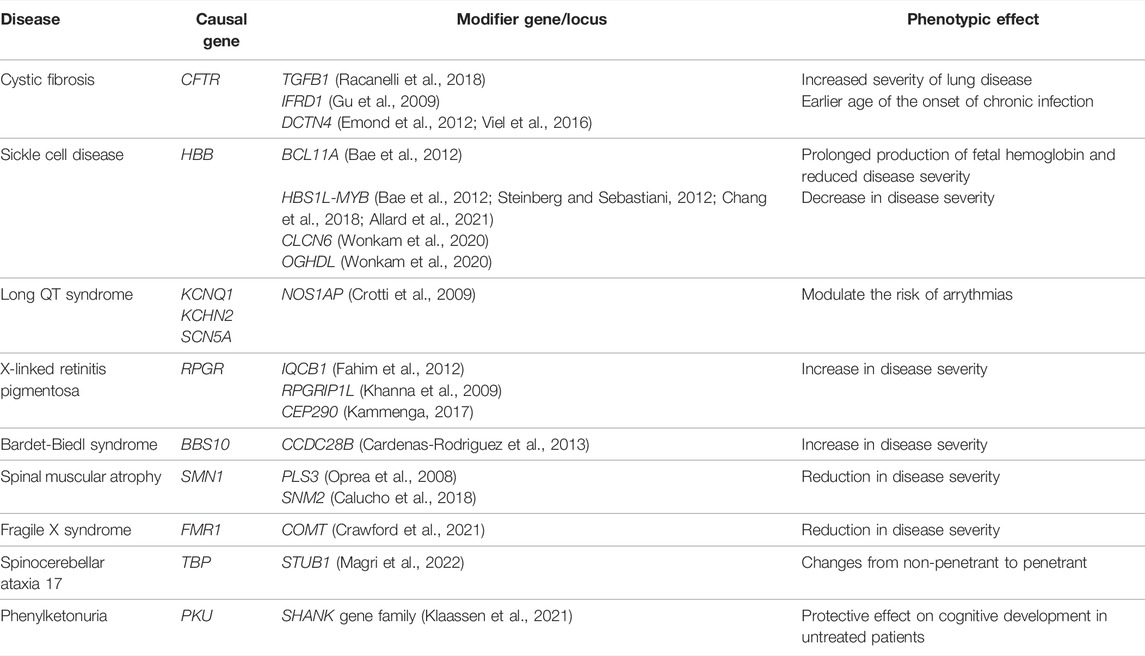
TABLE 3. Examples of monogenic conditions affected by a putative second genetic locus that modifies the phenotypic expression.
In contrast, some monogenic disease-causing variants may be partially tolerated and transmitted through unaffected generations unnoticed, until they surpass the threshold for causing disease in the presence of other contributory factors. For example, large copy number variants (CNVs) are well-known causes of NDDs, but some—such as recurrent 16p12.1 deletions (Hanson et al., 2015)—have been widely observed to be inherited from unaffected parents. In this case, the penetrance of a phenotype that is severe enough to present clinically requires an additional variant that modulates the primary genetic variant (Servetti et al., 2021) supporting a “two-hit” model of NDDs (Girirajan et al., 2010). Similarly, deleterious variants in CNTNAP2 and LRRC4C are insufficient to cause the disease alone but together may impair the development and function of synapses (Maussion et al., 2017; Um and Ko, 2017), suggesting a possible digenic mechanism for modulation of phenotypes (Poot, 2015). In many cases, however, there are likely to be numerous factors that affect whether an individual lies above or below the disease threshold, including the overall deleteriousness of the primary causal variant(s), the level of expression of the causal gene or isoform, and other genetic and non-genetic modifiers (Figure 4). Global modifiers that might affect penetrance and expressivity include polygenic risk, genetic compensation, variation in the NMD efficiency, family history, age, sex, and environmental factors.
The penetrance and expressivity of genotypes can be altered through the accumulated impact of many common genetic variants throughout the genome. The “omnigenic” model proposes that due to their interconnected nature, variants in gene-regulatory networks that are expressed in disease-relevant cells or tissues may affect the functioning of “core” disease-related genes due to effects on genes outside of the core pathways (Boyle et al., 2017), suggesting that many unrelated variants contribute to the presentation of a phenotype. Proposed as a factor in the inheritance of complex traits, this polygenic architecture could potentially also affect the presentation of monogenic conditions in a similar way, through non-coding variation that affects overall gene regulation, and many loci have been shown to additively affect expressivity and penetrance of monogenic variants in model organisms (Schell et al., 2022).
Genome-wide association studies (GWAS) have uncovered thousands of susceptibility loci for hundreds of diseases (Buniello et al., 2019), suggesting that the polygenic background can either predispose (Fahed et al., 2020) or protect individuals from diseases (Chami et al., 2020). Polygenic background can be quantified into a polygenic risk score (PRS) (Oetjens et al., 2019; Lewis and Vassos, 2020) and potentially used as a tool for the prediction of the overall disease risk in both monogenic and polygenic disorders (Khera et al., 2018). PRS associations highlight the additional risk of polygenic components in affecting the severity of monogenic disease, with the polygenic risk being shared across monogenic variant carriers and the general population (Kuchenbaecker et al., 2017). The effect of PRS has been widely explored to improve clinical interpretation of the penetrance of pathogenic variants across a range of monogenic conditions, including numerous familial cancer syndromes (Huyghe et al., 2019). The penetrance estimates for individuals with a pathogenic BRCA1 or BRCA2 variant range from 45 to 85% for breast cancer and from 10 to 65% for ovarian cancer (Petrucelli et al., 1993; van der Kolk et al., 2010), some of which can be explained by a polygenic background (Kuchenbaecker et al., 2017; Lee et al., 2019; Gallagher et al., 2020). Using a PRS generated from breast cancer GWAS, it has been shown that individual carriers of monogenic variants have risk differences of over 10% between the top and bottom PRS deciles (Kuchenbaecker et al., 2017). Interestingly, the majority of the SNPs identified as polygenic risk variants in breast cancer are common non-coding variants within regulatory regions, the target genes of which overlap with other known somatic cancer driver genes (Michailidou et al., 2017). Polygenic risk can also have a large effect on phenotypic diversity, even within individuals who have a known monogenic variant, illustrating that the genetic architecture for many diseases can be viewed as a spectrum rather than a binary classification of clinically symptomatic versus asymptomatic (Walsh et al., 2020). Although the overall polygenic contribution to the disease phenotype can be weaker in individuals with a monogenic variant (Harper et al., 2021), it can be useful in predicting overall penetrance and risk stratification.
The phenomenon of genetic compensation (or genetic buffering), where another gene or genes in a network can functionally compensate for LoF variants, has been shown in model organisms (Leopold et al., 2021) and hypothesized to play a role in incomplete penetrance in humans (Buglo et al., 2020). The upregulation of related genes or pathways or the differential expression of compensating alleles can help suppress a disease phenotype (Jordan et al., 2015), either through a small number of compensatory mechanisms or via a global shift in the gene expression. The functional redundancy of genes and rewiring of affected genetic networks may affect the penetrance and expressivity of corresponding phenotypes, and the consequence of a pathogenic variant may be influenced by variation across the genome (Payne and Wagner, 2015) and explain why certain LoF variants are tolerated by some individuals but not others (Subaran et al., 2015; Sulem et al., 2015). Haploinsufficiency can influence the expression of other genes in the same network, to maintain homeostasis or suppression of disease phenotypes (El-Brolosy and Stainier, 2017). The functional loss of one gene can be compensated for through functional redundancy (Chen et al., 2013). Genes that contain high numbers of PTVs in general population cohorts and thus are less likely to cause adverse phenotypes were found to belong to larger gene families than genes that contain known pathogenic PTVs (Ng et al., 2008), suggesting functional redundancy as a mechanism affecting penetrance (Hunter, 2022). Further research is needed to find robust evidence of this mechanism in humans.
The efficiency of NMD varies between individuals (Huang and Wilkinson, 2012), which could act as a potential modifier of penetrance and expressivity of PTVs targeted by NMD, irrespective of the specific causal variant(s) (Sarri et al., 2017). The variation in the NMD efficiency across codons, genes, cells, and tissues can affect disease pathology (Miller and Pearce, 2014; Sarkar et al., 2019; Sato and Singer, 2021). In studies of model organisms, the variant alleles that caused milder phenotypes were those that exhibited more NMD, with reduction in NMD being correlated with a more severe phenotype (El-Brolosy and Stainier, 2017). In this case, NMD could either help trigger a compensatory response, or haploinsufficiency could produce a milder phenotype than accumulation of truncated proteins. Variants in genes that encode the NMD machinery, or that either downregulate or remove NMD activity, have been linked to several NDD and ID syndromes, including variants in UPF2 (Hildebrand et al., 2020), UPF3A (Nguyen et al., 2012), EIF4A3 (Miller et al., 2017), SMG8 (Alzahrani et al., 2020), and RNPS1 (Nguyen et al., 2013), highlighting its importance in development and phenotypic expression. Common polymorphisms within the NMD pathway have been suggested to cause differences in NMD efficiency (Khajavi et al., 2006; Dyle et al., 2020), which could help explain differences in the expressivity of diseases caused by haploinsufficiency, with severity linked to whether they trigger NMD or not. Interindividual variability in NMD efficiency has the ability to alter the expressivity of genetic variants, by converting the cause of the disease phenotype from dominant-negative to haploinsufficiency, or vice versa (Supek et al., 2021). For example, two patients with the same PTV in the DMD gene displayed different clinical phenotypes, with one diagnosed with Duchenne muscular dystrophy, and the other with the milder Becker muscular dystrophy; here, the difference in the phenotype was suspected to be caused by weaker NMD efficiency in the less severely affected patient, which resulted in the production of the damaged but still partially functional DMD protein (Kerr et al., 2001; Torella et al., 2020).
Family history can be seen as a crude but effective proxy for the combined effect of many shared genetic and environmental modifiers of disease phenotypes. In many cases, the pathogenicity and penetrance of variants in monogenic diseases have only been determined through studies of large families with multiple affected individuals, which can make it difficult to disentangle the relative contribution of different modifiers. Family history is a well-known major risk factor for hereditary cancer syndromes, and the number of affected relatives increases the risk of a pathogenic variant carrier developing cancer (Brewer et al., 2017). Although the evidence base for estimating penetrance in individuals without a family history is currently very limited (Turner and Jackson, 2020), individuals identified with a pathogenic variant for a heritable monogenic disease but without a family history of that disease may have a lower penetrance than those with a family history (Moreno-De-Luca et al., 2015; Wright et al., 2019a, Jackson et al., 2022).
Evaluating genetic differences between affected and unaffected carriers in the same family—such as de novo variants or unique combinations of modifiers—can be informative for understanding penetrance. It has been shown that children with monogenic NDDs have an excess of other damaging genetic variants compared to their either mildly clinically affected or asymptomatic carrier parents, with the extra genetic burden being enriched in genes that are highly expressed within the brain and in neurodevelopmental pathways (Pizzo et al., 2019). Similarly, children with 22q11.2 deletion syndrome display a wide variability in IQ scores that is highly correlated with the scores of their immediate relatives (Olszewski et al., 2014). The IQ of individuals affected by 22q11.2 deletion syndrome follows a normal distribution curve, similar to that of the general population, only 30 points lower (De Smedt et al., 2007). The significant association seen between parental and proband IQ (Klaassen et al., 2016; Davies et al., 2020) suggests that inherited genetic variants associated with intelligence may alleviate some of the deleterious impact of the 22q11.2 deletion on phenotypic presentation. The heritability of intelligence may be driven either by the cumulative effect of many common small-effect variants, similar to the heritability within population cohorts (Davies et al., 2011), or by a small number of rare high-effect variants. Similarly, individuals carrying 16p11.2 deletions present with variable phenotypic diversities (Moreno-De-Luca et al., 2015; Fetit et al., 2020) and are frequently present in “healthy” general population cohorts (Rosenfeld et al., 2013), albeit with a range of cognitive and neuropsychiatric difficulties despite none of them reaching traditional clinical diagnosis threshold levels (Stefansson et al., 2014). Within these carrier individuals, the best overall predictor of the phenotype was that of the average of their parental phenotype for the traits of interest, with individuals displaying deleterious effects relative to their phenotypic family background (Polyak et al., 2015; Evans and Uljarević, 2018).
It can be argued that penetrance is an almost meaningless concept without specifying an age threshold as many diseases do not present until later in life. As we age, gene expression and chromatin structure across the genome change, which can increase the penetrance or expressivity of disease (Brookes and Shi, 2014; Bashkeel et al., 2019). Expression of certain genes can cause change in a predictable way throughout life, with some only being expressed in the foetus or during early childhood, and others only after this developmental period. For example, the relative proportion of two protein subunits in the NMDA receptor alters with age due to the varying expression levels of the two genes, GRIN2A and GRIN2B, which can alter phenotypic expression of deleterious variants in these genes; prenatally expressed GRIN2B is linked with severe cognitive defects from birth, while postnatally expressed GRIN2A is linked with epilepsies in childhood and schizophrenia in adults (Strehlow et al., 2019). Studies of individuals who are below the age-penetrant threshold for known age-dependent diseases could explain why some pathogenic variants are found in apparently asymptomatic population cohorts. Classical examples of conditions where penetrance increases with age include cancer predisposition syndromes such as Li-Fraumeni (Correa, 2016), Lynch Syndrome (Biller et al., 2019), and HBOC (Chen and Parmigiani, 2007), where penetrance is affected by the accumulation of DNA damage over time (White et al., 2014). Meta-analysis studies have shown that the cumulative breast cancer risks for BRCA1 and BRCA2 pathogenic variant carriers by age 70 are 57–65% and 45–49%, respectively (Antoniou et al., 2003; Chen and Parmigiani, 2007), highlighting the difficulties with predicting the course of disease even in known pathogenic variant carriers and the importance of considering family history as well as other genetic and environmental factors (Lee et al., 2019). Age-dependent penetrance of cognitive phenotypes is also seen in diseases caused by the slow accumulation of aberrant proteins, where variation can affect the rate at which the protein accumulates (Chiti and Dobson, 2017). For example, retinitis pigmentosa (RP) has been suggested to be caused by retention of misfolded proteins, which leads to upregulation of genes that encode for proapoptotic machinery, and leads to apoptosis of photoreceptor cells, accumulating damage over time and eventually reaching the disease threshold and causing penetrant disease (Rose and Bhattacharya, 2016). Age-dependent penetrance may also be caused through gradual loss of neurons, causing the associated disease phenotype when the number of surviving cells drops below a certain threshold or overcomes brain plasticity (Magrinelli et al., 2021). For example, progressive and late occurring neurological manifestations in patients with DNMT1 variants may originate from the gradual loss of DNA methylation over time, affecting adult neurogenesis (Velasco and Francastel, 2019).
The penetrance of age-dependent variants, present a diagnostic and prognostic challenge for individuals with such genotypes (Kalia et al., 2017). Previously, testing for many conditions early in life was not possible, and so little is known about long-term effects of mildly deleterious variants. Variants in HFE cause hereditary hemochromatosis, which can lead to iron overload in adulthood, and were previously thought to be an adult-onset condition. However, healthy cohort studies of children have shown that the effects of homozygous variants in HFE can be seen in childhood and that the cumulative effect of excess iron over a lifetime may affect the penetrance of numerous iron-related diseases (Kim and Connor, 2020). Recent population studies of adults have also shown substantially higher morbidity in homozygous HFE variant carriers with increasing age (Pilling et al., 2019). In this case early identification of individuals at risk can help with monitoring disease progression and introducing timely interventions (such as blood donation).
Sex can affect the penetrance and expressivity of some genetic disorders, most obviously when deleterious genetic variants occur on the X chromosome, with hemizygous males more phenotypically affected than heterozygous females. Although differences in the penetrance of inherited variants based on sex have been reported in a variety of disorders (Cooper et al., 2013), mechanisms behind sex-dependent penetrance outside those that occur on the X chromosome are mostly unknown. However, there are widespread sex-biased differences in gene expression (Oliva et al., 2020), so differences in penetrance of phenotypes are also likely to be common. Females are less likely to be diagnosed with neurodevelopmental disorders than males, with a fourfold increase in the number of males diagnosed with autism spectrum disorders (ASD) compared to females (Scott et al., 2002; Christensen et al., 2016), suggesting that there may be a female protective effect that affects the penetrance of such conditions (Jacquemont et al., 2014). Girls diagnosed with ASD have an increased number of CNVs compared to boys with the same diagnosis, and asymptomatic mothers with children diagnosed with NDDs or ASD had a higher genetic burden of deleterious variants than fathers (Polyak et al., 2015), suggesting there may be some other cause for the incomplete penetrance and variable expressivity in females compared to males. However, females are ascertained at a closer frequency to males when they are more severely affected, suggesting some bias in clinical ascertainment due to differing phenotypic presentations between the sexes (Ratto et al., 2018), supported by the fact that males were more likely to be referred for genetic testing than females carrying the same autosomal variant (Russell et al., 2011).
The environment can affect disease penetrance or expressivity in both a negative and positive manner and includes diet, drugs, alcohol intake, physical activity, ultraviolet light, in utero exposures, education, and socio-economic status, among many others factors. Epigenetic factors can provide a mechanistic link between the environment and gene expression (Dolinoy et al., 2007; Cavalli and Heard, 2019; Safi-Stibler and Gabory, 2020), and studies of the human microbiome can also explain some extreme variability in genotype–phenotype presentation (Sanna et al., 2019). However, although gene–environment interactions are likely to be widespread, they are often extremely hard to prove as the complete and systematic collection of an individual’s environment is almost impossible, and detailed relevant exposure data are rarely available alongside genetic data.
Inborn errors of metabolism perhaps provide the simplest examples of monogenic diseases where both a pathogenic genotype and an environmental exposure are required to cause disease (van Karnebeek and Stockler, 2012). A clear example of the dietary impact on phenotypic variation is phenylketonuria, a rare autosomal recessive disease that is usually detected through newborn screening, whereby individuals who have damaging biallelic variants in PAH can be put on a low phenylalanine diet to avoid serious disease progression (Flydal and Martinez, 2013; Al Hafid and Christodoulou, 2015). Later onset monogenic disease penetrance can also be affected by the environment, as seen in several cancer syndromes, including colorectal cancer, where inherited genetic variants interact with dietary variables and BMI to confer the overall risk (Lee et al., 2015). Cancer susceptibility can also be altered through gene–environment interactions such as smoking or sunburn, which can accelerate the accumulation of somatic variants that contribute toward tumorigenesis (Newcomb and Carbone, 1992; Wu et al., 2016). Similarly, environmental exposure to cigarette smoke, air pollution, and other airborne toxins can cause accumulation of unfolded or misfolded proteins and therefore affect the penetrance or expressivity of chronic lung disease (Wei et al., 2013). Individuals who carry a damaging monogenic variant may also be more susceptible to some environmental exposures, which can affect phenotypic severity (Tukker et al., 2021). For example, cystic fibrosis is characterized by progressive damage to the lungs, and non-genetic factors may account for up to 50% of the clinical variation seen (Collaco et al., 2010). Environmental factors such as smoking, air pollutants, temperature, and high-fat diets have all been shown to affect the severity and progression of disease (Collaco et al., 2010; Collaco et al., 2011; Schindler et al., 2015; Tukker et al., 2021), and the specific CFTR variant can also modulate how much environmental impact has on disease severity (Collaco and Cutting, 2008). Environmental factors can also affect the presentation of disease in primary atopic disorders, commonly seen as monogenic allergic disorders, where diet, microbiome at the epithelial-environment interface, presence/extent of infection, and psychological stress can all affect the penetrance or expressivity of the related phenotype (Sacco and Milner, 2019).
Determining the penetrance and expressivity of a variant can be difficult because it is sensitive to ascertainment context, and many studies are designed to enable the discovery of causative pathogenic variants in clinically affected individuals rather than to analyze effect sizes in populations (Manrai et al., 2016). This has been demonstrated in recent studies that stress the importance of cohort background for the determination of penetrance (Goodrich et al., 2021; Mirshahi et al., 2021). Investigating clinically classified pathogenic variants in large population cohorts can provide additional information about penetrance and expressivity (Kingdom et al., 2022), or determine whether variants or genes have been misclassified (Wright et al., 2019a). However, finding low penetrance pathogenic variants in large numbers of asymptomatic individuals challenges the concept of pathogenicity, particularly in the absence of known modifiers. What does it mean to describe a genotype as pathogenic if it is frequently found in individuals without disease and no explanation as to why? Reclassification of previously reported pathogenic variants occurs frequently, with variants first classified prior to the release of large population datasets showing a higher rate of reclassification (Harrison and Rehm, 2019). A study reappraising pathogenic variants in Brugada syndrome showed that only one gene (SCN5A) out of 21 could be definitively identified as causal (Hosseini et al., 2018), and another study has raised doubt over the involvement of 11/58 genes thought to cause inherited monogenic retinal disease (Hanany and Sharon, 2019). Variants that show low penetrance or a wide range of expressivity can also be potentially classified as risk alleles rather than causative variants. Some CFTR variants have been classified this way, with variations in cystic fibrosis phenotypes from very mild to very severe, and over 1900 different genotypes have been reported (Collaco and Cutting, 2008; Guillot et al., 2014; Pereira et al., 2019). Many genotype–phenotype associations are only reported once, or they are reported several times but with inconsistent results due to differences in data collection, differences in methods, or differences in cohort ascertainment. Associations can also differ due to poor annotation of coding genes, lack of relevant functional information for non-coding regions, sequencing and annotation errors, and varying penetrance and expressivity, making a simple binary classification of many genetic variants very difficult.
An overlapping genetic basis between complex traits and monogenic conditions is becoming increasingly apparent across the genome. Deleterious variants in genes causative of monogenic disease can be further dysregulated by non-coding variants that are associated with common traits, and monogenic forms of numerous common complex diseases have been identified (Peltonen et al., 2006; Chami et al., 2020; Hassanin et al., 2021). This overlap can cause considerable complexity when it comes to determining genotype–phenotype relationships (Freund et al., 2018). The prevalence of incomplete penetrance and variable expressivity raises questions as to what constitutes a disease state as opposed to extremes of normal phenotypic variation, especially within conditions that show significant clinical heterogeneity (Moreno-De-Luca et al., 2015), with many traits that constitute a clinical phenotype being the extreme end of either side of the bell curve of continuous distribution in the general population. Therefore, defining the penetrance of a genotype can be difficult, especially when there is ambiguity as to what defines the “disease state,” particularly for disorders where clinical features are only identified when they reach above a certain threshold (Senol-Cosar et al., 2019).
Relatively few studies have investigated low penetrant rare variants in detail or identified why such variants cause disease in one individual and not another. Despite increasing numbers of sequenced individuals, identification of genetic modifiers for monogenic conditions remains challenging. By definition, carriers of rare variants that cause monogenic conditions will be rare, with even fewer individuals having identical genetic modifiers that explain incomplete penetrance or variable expression. NGS approaches involving bioinformatic algorithms, including pathogenicity score-based prioritizations, can produce conflicting results and often need manual curation to identify candidate variants. A computational approach that could comprehensively analyze and prioritize candidate variants and potential modifiers would be a great advantage. Even in large population cohorts genome-wide analysis of genetic interactions lacks statistical power and can be easily affected by confounders (Wei et al., 2014). Many genetic modifiers are likely to be located in non-coding regions, making it challenging to determine their direct functional effect on the gene expression, especially as much of the genome is found to be bound by at least one transcription factor, many of which have no known function yet (The ENCODE Project Consortium, 2012). Improved computational approaches to identify candidate modifier gene interactions across the genome are needed (Lee et al., 2010), as well as identification of functional non-coding regions and the genes that they affect (Petrovski et al., 2015), and machine learning approaches such as DeepSEA and Enformer (Avsec et al., 2021) could improve annotation of these regions (Zhou and Troyanskaya, 2015).
Participants in population studies are usually investigated in a research-based environment rather than a clinical context, and despite rigorous phenotypic collection in some population studies, individuals involved may have subclinical manifestations of disease phenotypes that were unnoticed at the time of recruitment, or were not recorded in their medical histories (van Rooij et al., 2020). Lack of comprehensive phenotypic data can make using population cohorts to calculate the penetrance of genotypes very difficult but can at least provide a lower boundary of penetrance, with small clinical studies providing the upper boundary (Elfatih et al., 2021). Variant interpretation guidelines suggest that the penetrance of pathogenic variants in general population cohorts should be taken into account when calculating the overall penetrance of such variants (Kalia et al., 2017); however, even within healthy population cohorts there have been individuals identified with the associated phenotype but who have previously been described as unaffected (Chen et al., 2016), as well as individuals who display symptoms but are below the clinical threshold for classification. This is further complicated by conditions that are late-onset. In addition, genetic studies of human disease currently fail to capture the diversity that exists across the world, with most studies involving individuals of European descent (Lawson et al., 2020). This issue directly affects penetrance estimates, particularly as GWAS results and PRS may not be transferrable across diverse populations due to differing allele frequencies (Sirugo et al., 2019). Many deleterious variants may not be sufficient alone to cause disease, and therefore, estimates of penetrance need to consider the presence of other genetic variants and potential environmental effects (Figure 4). Calculating the etiological fraction of rare variants in specific conditions may provide a useful way to evaluate the probability that a variant detected in an individual with disease is causative (Walsh et al., 2017; Walsh et al., 2019), and disease-specific variant classifiers may also be of use (Zhang et al., 2021).
As WGS becomes more common, individuals at risk of genetic disease will be identified earlier in life, potentially even from birth (Holm et al., 2018) and often prior to the appearance of relevant phenotypes. This can have a positive impact on overall health, with individuals who have no family history but a previously unknown high risk of disease being identified, enabling preventative screening or early treatment interventions. However, it can also cause harm through overdiagnosis; As seen across a number of population cohort studies, healthy individuals can harbor many potentially deleterious variants without ever developing any clinical symptoms. The effective use of genomic data requires a comprehensive understanding of functional genotype–phenotype correlations, which goes beyond that of Mendelian inheritance patterns. The increased sequencing of unselected populations, linked with electronic health records or other longitudinal phenotypic data, gives us an unprecedented ability to identify and reclassify rare variants and calculate penetrance estimates for a wide range of diseases and genotypes. These large-scale studies are crucial to inform the development of genomic screening programs (Holm et al., 2018; Wojcik et al., 2021) and the management of incidental or secondary findings. Discovery estimates of secondary findings vary from 1–3% of the population, with the majority of identified variants being those that confer susceptibility to cancer (Hart et al., 2019; Gordon et al., 2020). Incidental findings are predicted to be detectable at an appreciable level in individuals in the general population, many of whom may never develop the corresponding disease, suggesting that more robust determinations of pathogenicity are needed, including penetrance estimates for those without a family history of the disease (Johnston et al., 2012).
Incomplete penetrance and variable expressivity are a significant concern for the correct interpretation of genetic variation and of diagnosing genetic disease. Correctly estimating penetrance and expressivity is challenging, with clinical cohorts and population studies both offering a different insight into its quantification. Although many monogenic disease-causing variants are fully penetrant, many are not, and improving our knowledge will involve WGS of population cohorts of increasing size and diversity, as well as functional studies of individual patients with specific clinical phenotypes. Achieving a mechanistic understanding of how incomplete penetrance and variable expressivity occur will help inform diagnostic and prognostic testing, clinical management, and accurate genetic counseling. To improve diagnostics and clinical interpretation of incompletely penetrant genotypes, a more sophisticated approach to disease genetics may be needed that integrates disease mechanism and specific variants with variation in levels of gene and tissue-specific isoform expressions and other genetic and non-genetic modifiers. Improving our knowledge of how variants exert their effects on genes, cellular pathways, and overall phenotypes will improve our understanding of disease and facilitate the development of new therapeutic interventions.
RK: literature review and drafting of the article. CW: critical revision and additions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acuna-Hidalgo, R., Bo, T., Kwint, M. P., van de Vorst, M., Pinelli, M., Veltman, J. A., et al. (2015). Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am. J. Hum. Genet. 97, 67–74. doi:10.1016/j.ajhg.2015.05.008
Aguet, F., Brown, A. A., Castel, S. E., Davis, J. R., He, Y., Jo, B., et al. (2017). Genetic Effects on Gene Expression across Human Tissues. Nature 550, 204–213.
Ahluwalia, J. K., Hariharan, M., Bargaje, R., Pillai, B., and Brahmachari, V. (2009). Incomplete Penetrance and Variable Expressivity: Is There a microRNA Connection? BioEssays 31, 981–992. doi:10.1002/bies.200900066
Akinrinade, O., Heliö, T., Lekanne Deprez, R. H., Jongbloed, J. D. H., Boven, L. G., van den Berg, M. P., et al. (2019). Relevance of Titin Missense and Non-frameshifting Insertions/Deletions Variants in Dilated Cardiomyopathy. Sci. Rep. 9, 4093. doi:10.1038/s41598-019-39911-x
Akiyama, M. (2010). FLG Mutations in Ichthyosis Vulgaris and Atopic Eczema: Spectrum of Mutations and Population Genetics. Br. J. Dermatol. 162, 472–477. doi:10.1111/j.1365-2133.2009.09582.x
Al Hafid, N., and Christodoulou, J. (2015). Phenylketonuria: a Review of Current and Future Treatments. Transl. Pediatr. 4, 304–317. doi:10.3978/j.issn.2224-4336.2015.10.07
Allard, P., Alhaj, N., Lobitz, S., Cario, H., Jarisch, A., Grosse, R., et al. (2021). Genetic Modifiers of Fetal Hemoglobin Affect the Course of Sickle Cell Disease in Patients Treated with Hydroxyurea. Haematologica.
Alswied, A., Rehman, A., Lai, L.-W., Duran, J., Sardar, M., and Proytcheva, M. A. (2021). Rare Monosomy 7 and Deletion 7p at Diagnosis of Chronic Myeloid Leukemia in Accelerated Phase. Cancer Genet. 252-253, 111–114. doi:10.1016/j.cancergen.2021.01.006
Alzahrani, F., Kuwahara, H., Long, Y., Al-Owain, M., Tohary, M., AlSayed, M., et al. (2020). Recessive, Deleterious Variants in SMG8 Expand the Role of Nonsense-Mediated Decay in Developmental Disorders in Humans. Am. J. Hum. Genet. 107, 1178–1185. doi:10.1016/j.ajhg.2020.11.007
Amin, A. S., Giudicessi, J. R., Tijsen, A. J., Spanjaart, A. M., Reckman, Y. J., Klemens, C. A., et al. (2012). Variants in the 3′ Untranslated Region of the KCNQ1-Encoded Kv7.1 Potassium Channel Modify Disease Severity in Patients with Type 1 Long QT Syndrome in an Allele-specific Manner. Eur. Heart J. 33, 714–723. doi:10.1093/eurheartj/ehr473
Antoniou, A., Pharoah, P. D. P., Narod, S., Risch, H. A., Eyfjord, J. E., Hopper, J. L., et al. (2003). Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am. J. Hum. Genet. 72, 1117–1130. doi:10.1086/375033
Arning, L. (2016). The Search for Modifier Genes in Huntington Disease - Multifactorial Aspects of a Monogenic Disorder. Mol. Cell. Probes 30, 404–409. doi:10.1016/j.mcp.2016.06.006
Arora, V., Bijarnia-Mahay, S., Tiwari, V., Bansal, S., Gupta, P., Setia, N., et al. (2020). Co-inheritance of Pathogenic Variants in PKD1 and PKD2 Genes Presenting as Severe Antenatal Phenotype of Autosomal Dominant Polycystic Kidney Disease. Eur. J. Med. Genet. 63, 103734. doi:10.1016/j.ejmg.2019.103734
Aubart, M., Gazal, S., Arnaud, P., Benarroch, L., Gross, M.-S., Buratti, J., et al. (2018). Association of Modifiers and Other Genetic Factors Explain Marfan Syndrome Clinical Variability. Eur. J. Hum. Genet. 26, 1759–1772. doi:10.1038/s41431-018-0164-9
Aubart, M., Gross, M.-S., Hanna, N., Zabot, M.-T., Sznajder, M., Detaint, D., et al. (2015). The Clinical Presentation of Marfan Syndrome Is Modulated by Expression of Wild-type FBN1 Allele. Hum. Mol. Genet. 24, 2764–2770. doi:10.1093/hmg/ddv037
Austin, E. D., Phillips, J. A., Cogan, J. D., Hamid, R., Yu, C., Stanton, K. C., et al. (2009). Truncating and Missense BMPR2 Mutations Differentially Affect the Severity of Heritable Pulmonary Arterial Hypertension. Respir. Res. 10, 87. doi:10.1186/1465-9921-10-87
Auton, A., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A Global Reference for Human Genetic Variation. Nature 526, 68–74. doi:10.1038/nature15393
Avsec, Ž., Agarwal, V., Visentin, D., Ledsam, J. R., Grabska-Barwinska, A., Taylor, K. R., et al. (2021). Effective Gene Expression Prediction from Sequence by Integrating Long-Range Interactions. Nat. Methods 18, 1196–1203. doi:10.1038/s41592-021-01252-x
Bae, H. T., Baldwin, C. T., Sebastiani, P., Telen, M. J., Ashley-Koch, A., Garrett, M., et al. (2012). Meta-analysis of 2040 Sickle Cell Anemia Patients: BCL11A and HBS1L-MYB Are the Major Modifiers of HbF in African Americans. Blood 120, 1961–1962. doi:10.1182/blood-2012-06-432849
Bahlo, M., Bennett, M. F., Degorski, P., Tankard, R. M., Delatycki, M. B., and Lockhart, P. J. (2018). Recent Advances in the Detection of Repeat Expansions with Short-Read Next-Generation Sequencing. F1000Res 7, F1000. doi:10.12688/f1000research.13980.1
Baranzini, S. E., Mudge, J., van Velkinburgh, J. C., Khankhanian, P., Khrebtukova, I., Miller, N. A., et al. (2010). Genome, Epigenome and RNA Sequences of Monozygotic Twins Discordant for Multiple Sclerosis. Nature 464, 1351–1356. doi:10.1038/nature08990
Bashkeel, N., Perkins, T. J., Kærn, M., and Lee, J. M. (2019). Human Gene Expression Variability and its Dependence on Methylation and Aging. BMC Genomics 20, 941. doi:10.1186/s12864-019-6308-7
Begay, R. L., Graw, S., Sinagra, G., Merlo, M., Slavov, D., Gowan, K., et al. (2015). Role of Titin Missense Variants in Dilated Cardiomyopathy. J. Am. Heart Assoc. 4, e002645. doi:10.1161/JAHA.115.002645
Bertolini, S., Calandra, S., Arca, M., Averna, M., Catapano, A. L., Tarugi, P., et al. (2020). Homozygous Familial Hypercholesterolemia in Italy: Clinical and Molecular Features. Atherosclerosis 312, 72–78. doi:10.1016/j.atherosclerosis.2020.08.027
Bickley, V. M., Parker, E. M., Taylor, J. C., Watt, J. E., Robertson, M. D., Doudney, K. W. E., et al. (2014). Rare Complete and Partial Monosomy 7 Mosaicism Detected in a Case with FTT and Borderline Motor Delay, Subsequently Diagnosed with Juvenile MDS: An Exposition of This Case and Other Interesting Mosaic Cancer Case Studies. Cancer Genet. 207, 286. doi:10.1016/j.cancergen.2014.06.009
Biegstraaten, M., van Schaik, I. N., Aerts, J. M. F. G., Langeveld, M., Mannens, M. M. A. M., Bour, L. J., et al. (2011). A Monozygotic Twin Pair with Highly Discordant Gaucher Phenotypes. Blood Cells, Mol. Dis. 46, 39–41. doi:10.1016/j.bcmd.2010.10.007
Biesecker, L. G., and Spinner, N. B. (2013). A Genomic View of Mosaicism and Human Disease. Nat. Rev. Genet. 14, 307–320. doi:10.1038/nrg3424
Biller, L. H., Syngal, S., and Yurgelun, M. B. (2019). Recent Advances in Lynch Syndrome. Fam. Cancer 18, 211–219. doi:10.1007/s10689-018-00117-1
Binder, B. J., Landman, K. A., Newgreen, D. F., and Ross, J. V. (2015). Incomplete Penetrance: The Role of Stochasticity in Developmental Cell Colonization. J. Theor. Biol. 380, 309–314. doi:10.1016/j.jtbi.2015.05.028
Boltsis, I., Grosveld, F., Giraud, G., and Kolovos, P. (2021). Chromatin Conformation in Development and Disease. Front. Cell. Dev. Biol. 9, 723859. doi:10.3389/fcell.2021.723859
Boycott, K. M., Rath, A., Chong, J. X., Hartley, T., Alkuraya, F. S., Baynam, G., et al. (2017). International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 100, 695–705. doi:10.1016/j.ajhg.2017.04.003
Boyle, E. A., Li, Y. I., and Pritchard, J. K. (2017). An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 169, 1177–1186. doi:10.1016/j.cell.2017.05.038
Bozhilov, Y. K., Downes, D. J., Telenius, J., Marieke Oudelaar, A., Olivier, E. N., Mountford, J. C., et al. (2021). A Gain-Of-Function Single Nucleotide Variant Creates a New Promoter Which Acts as an Orientation-dependent Enhancer-Blocker. Nat. Commun. 12, 3806. doi:10.1038/s41467-021-23980-6
Brewer, H. R., Jones, M. E., Schoemaker, M. J., Ashworth, A., and Swerdlow, A. J. (2017). Family History and Risk of Breast Cancer: an Analysis Accounting for Family Structure. Breast Cancer Res. Treat. 165, 193–200. doi:10.1007/s10549-017-4325-2
Brookes, E., and Shi, Y. (2014). Diverse Epigenetic Mechanisms of Human Disease. Annu. Rev. Genet. 48, 237–268. doi:10.1146/annurev-genet-120213-092518
Buglo, E., Sarmiento, E., Martuscelli, N. B., Sant, D. W., Danzi, M. C., Abrams, A. J., et al. (2020). Genetic Compensation in a Stable Slc25a46 Mutant Zebrafish: A Case for Using F0 CRISPR Mutagenesis to Study Phenotypes Caused by Inherited Disease. PLoS ONE 15, e0230566. doi:10.1371/journal.pone.0230566
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of Published Genome-wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 47, D1005–D1012. doi:10.1093/nar/gky1120
Cabal-Herrera, A. M., Tassanakijpanich, N., Salcedo-Arellano, M. J., and Hagerman, R. J. (2020). Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 21, E4391. doi:10.3390/ijms21124391
Calucho, M., Bernal, S., Alías, L., March, F., Venceslá, A., Rodríguez-Álvarez, F. J., et al. (2018). Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 28, 208–215. doi:10.1016/j.nmd.2018.01.003
Cammaerts, S., Strazisar, M., De Rijk, P., and Del Favero, J. (2015). Genetic Variants in microRNA Genes: Impact on microRNA Expression, Function, and Disease. Front. Genet. 6, 186. doi:10.3389/fgene.2015.00186
Campbell, I. M., Yuan, B., Robberecht, C., Pfundt, R., Szafranski, P., McEntagart, M. E., et al. (2014). Parental Somatic Mosaicism Is Underrecognized and Influences Recurrence Risk of Genomic Disorders. Am. J. Hum. Genet. 95, 173–182. doi:10.1016/j.ajhg.2014.07.003
Cardenas-Rodriguez, M., Osborn, D. P. S., Irigoín, F., Graña, M., Romero, H., Beales, P. L., et al. (2013). Characterization of CCDC28B Reveals its Role in Ciliogenesis and Provides Insight to Understand its Modifier Effect on Bardet-Biedl Syndrome. Hum. Genet. 132, 91–105. doi:10.1007/s00439-012-1228-5
Carelle-Calmels, N., Saugier-Veber, P., Girard-Lemaire, F., Rudolf, G., Doray, B., Guérin, E., et al. (2009). Genetic Compensation in a Human Genomic Disorder. N. Engl. J. Med. 360, 1211–1216. doi:10.1056/nejmoa0806544
Castel, S. E., Cervera, A., Mohammadi, P., Aguet, F., Reverter, F., Wolman, A., et al. (2018). Modified Penetrance of Coding Variants by Cis-Regulatory Variation Contributes to Disease Risk. Nat. Genet. 50, 1327–1334. doi:10.1038/s41588-018-0192-y
Castillo-Fernandez, J. E., Spector, T. D., and Bell, J. T. (2014). Epigenetics of Discordant Monozygotic Twins: Implications for Disease. Genome Med. 6, 60. doi:10.1186/s13073-014-0060-z
Catalanotto, C., Cogoni, C., and Zardo, G. (2016). MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Ijms 17, 1712. doi:10.3390/ijms17101712
Cavalli, G., and Heard, E. (2019). Advances in Epigenetics Link Genetics to the Environment and Disease. Nature 571, 489–499. doi:10.1038/s41586-019-1411-0
Chami, N., Preuss, M., Walker, R. W., Moscati, A., and Loos, R. J. F. (2020). The Role of Polygenic Susceptibility to Obesity Among Carriers of Pathogenic Mutations in MC4R in the UK Biobank Population. PLOS Med. 17, e1003196. doi:10.1371/journal.pmed.1003196
Chang, A. K., Ginter Summarell, C. C., Birdie, P. T., and Sheehan, V. A. (2018). Genetic Modifiers of Severity in Sickle Cell Disease. Chem. 68, 147–164. doi:10.3233/ch-189004
Chang, S., Wang, R.-H., Wang, R.-H., Akagi, K., Kim, K.-A., Martin, B. K., et al. (2011). Tumor Suppressor BRCA1 Epigenetically Controls Oncogenic microRNA-155. Nat. Med. 17, 1275–1282. doi:10.1038/nm.2459
Chen, D., Xu, Y., Ding, C., Wang, Y., Fu, Y., Cai, B., et al. (2022). The Inconsistency between Two Major Aneuploidy-Screening Platforms-Single-Nucleotide Polymorphism Array and Next-Generation Sequencing-In the Detection of Embryo Mosaicism. BMC Genomics 23, 62. doi:10.1186/s12864-022-08294-1
Chen, R., Morgan, A. A., Dudley, J., Deshpande, T., Li, L., Kodama, K., et al. (2008). FitSNPs: Highly Differentially Expressed Genes Are More Likely to Have Variants Associated with Disease. Genome Biol. 9, R170. doi:10.1186/gb-2008-9-12-r170
Chen, R., Shi, L., Hakenberg, J., Naughton, B., Sklar, P., Zhang, J., et al. (2016). Analysis of 589,306 Genomes Identifies Individuals Resilient to Severe Mendelian Childhood Diseases. Nat. Biotechnol. 34, 531–538. doi:10.1038/nbt.3514
Chen, S., and Parmigiani, G. (2007). Meta-Analysis of BRCA1 and BRCA2 Penetrance. Jco 25, 1329–1333. doi:10.1200/jco.2006.09.1066
Chen, W.-H., Zhao, X.-M., van Noort, V., and Bork, P. (2013). Human Monogenic Disease Genes Have Frequently Functionally Redundant Paralogs. PLoS Comput. Biol. 9, e1003073. doi:10.1371/journal.pcbi.1003073
Chiti, F., and Dobson, C. M. (2017). Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 86, 27–68. doi:10.1146/annurev-biochem-061516-045115
Chong, J. X., Buckingham, K. J., Jhangiani, S. N., Boehm, C., Sobreira, N., Smith, J. D., et al. (2015). The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 97, 199–215. doi:10.1016/j.ajhg.2015.06.009
Christensen, D. L., Baio, J., Braun, K. V. N., Bilder, D., Charles, J., Constantino, J. N., et al. (2016). Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill. Summ. 65, 1–23. doi:10.15585/mmwr.ss6503a1
Claussnitzer, M., Cho, J. H., Collins, R., Cox, N. J., Dermitzakis, E. T., Hurles, M. E., et al. (2020). A Brief History of Human Disease Genetics. Nature 577, 179–189. doi:10.1038/s41586-019-1879-7
Coban-Akdemir, Z., White, J. J., Song, X., Jhangiani, S. N., Fatih, J. M., Gambin, T., et al. (2018). Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-Of-Function Alleles. Am. J. Hum. Genet. 103, 171–187. doi:10.1016/j.ajhg.2018.06.009
Cohen, M. M. (2014). Proteus Syndrome Review: Molecular, Clinical, and Pathologic Features. Clin. Genet. 85, 111–119. doi:10.1111/cge.12266
Collaco, J. M., Blackman, S. M., McGready, J., Naughton, K. M., and Cutting, G. R. (2010). Quantification of the Relative Contribution of Environmental and Genetic Factors to Variation in Cystic Fibrosis Lung Function. J. Pediatr. 157, 802–807. doi:10.1016/j.jpeds.2010.05.018
Collaco, J. M., and Cutting, G. R. (2008). Update on Gene Modifiers in Cystic Fibrosis. Curr. Opin. Pulm. Med. 14, 559–566. doi:10.1097/mcp.0b013e3283121cdc
Collaco, J. M., McGready, J., Green, D. M., Naughton, K. M., Watson, C. P., Shields, T., et al. (2011). Effect of Temperature on Cystic Fibrosis Lung Disease and Infections: a Replicated Cohort Study. PloS One 6, e27784. doi:10.1371/journal.pone.0027784
Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., and Kehrer-Sawatzki, H. (2013). Where Genotype Is Not Predictive of Phenotype: towards an Understanding of the Molecular Basis of Reduced Penetrance in Human Inherited Disease. Hum. Genet. 132, 1077–1130. doi:10.1007/s00439-013-1331-2
Crawford, H., Scerif, G., Wilde, L., Beggs, A., Stockton, J., Sandhu, P., et al. (2021). Genetic Modifiers in Rare Disorders: the Case of Fragile X Syndrome. Eur. J. Hum. Genet. 29, 173–183. doi:10.1038/s41431-020-00711-x
Crotti, L., Monti, M. C., Insolia, R., Peljto, A., Goosen, A., Brink, P. A., et al. (2009). NOS1AP Is a Genetic Modifier of the Long-QT Syndrome. Circulation 120, 1657–1663. doi:10.1161/circulationaha.109.879643
Cummings, B. B., Karczewski, K. J., Kosmicki, J. A., Seaby, E. G., Watts, N. A., Singer-Berk, M., et al. (2020). Transcript Expression-Aware Annotation Improves Rare Variant Interpretation. Nature 581, 452–458. doi:10.1038/s41586-020-2329-2
Cummings, B. B., Marshall, J. L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A. R., et al. (2017). Improving Genetic Diagnosis in Mendelian Disease with Transcriptome Sequencing. Sci. Transl. Med. 9, eaal5209. doi:10.1126/scitranslmed.aal5209
Davies, G., Tenesa, A., Payton, A., Yang, J., Harris, S. E., Liewald, D., et al. (2011). Genome-wide Association Studies Establish that Human Intelligence Is Highly Heritable and Polygenic. Mol. Psychiatry 16, 996–1005. doi:10.1038/mp.2011.85
Davies, R. W., Fiksinski, A. M., Fiksinski, A. M., Breetvelt, E. J., Williams, N. M., Hooper, S. R., et al. (2020). Using Common Genetic Variation to Examine Phenotypic Expression and Risk Prediction in 22q11.2 Deletion Syndrome. Nat. Med. 26, 1912–1918. doi:10.1038/s41591-020-1103-1
de Marvao, A., McGurk, K. A., Zheng, S. L., Thanaj, M., Bai, W., Duan, J., et al. (2021). Phenotypic Expression and Outcomes in Individuals with Rare Genetic Variants of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 78, 1097–1110. doi:10.1016/j.jacc.2021.07.017
De Smedt, B., Devriendt, K., Fryns, J.-P., Vogels, A., Gewillig, M., and Swillen, A. (2007). Intellectual Abilities in a Large Sample of Children with Velo?Cardio?Facial Syndrome: an Update. J. Intellect. Disabil. Res. 51, 666–670. doi:10.1111/j.1365-2788.2007.00955.x
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. doi:10.1016/j.neuron.2011.09.011
Delaneau, O., Zazhytska, M., Borel, C., Giannuzzi, G., Rey, G., Howald, C., et al. (2019). Chromatin Three-Dimensional Interactions Mediate Genetic Effects on Gene Expression. Science 364, eaat8266. doi:10.1126/science.aat8266
Díaz de Bustamante, A., Ruiz-Casares, E., Darnaude, M. T., Perucho, T., and Martínez-Quesada, G. (2012). Phenotypic Variability in Marfan Syndrome in a Family with a Novel Nonsense FBN1 Gene Mutation. Rev. Esp. Cardiol. Engl. Ed. 65, 380–381. doi:10.1016/j.recesp.2011.05.027
Dick, I. E., Joshi-Mukherjee, R., Yang, W., and Yue, D. T. (2016). Arrhythmogenesis in Timothy Syndrome Is Associated with Defects in Ca2+-dependent Inactivation. Nat. Commun. 7, 10370. doi:10.1038/ncomms10370
Dickinson, M. E., Flenniken, A. M., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., et al. (2016). High-throughput Discovery of Novel Developmental Phenotypes. Nature 537, 508–514. doi:10.1038/nature19356
Dietz, H. (1993). “FBN1-Related Marfan Syndrome,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Ding, J., Li, X., Tian, H., Wang, L., Guo, B., Wang, Y., et al. (2021). SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 12, 743726. doi:10.3389/fneur.2021.743726
Dolinoy, D., Weidman, J., and Jirtle, R. (2007). Epigenetic Gene Regulation: Linking Early Developmental Environment to Adult Disease. Reprod. Toxicol. 23, 297–307. doi:10.1016/j.reprotox.2006.08.012
Domogala, D. D., Gambin, T., Zemet, R., Wu, C. W., Schulze, K. V., Yang, Y., et al. (2021). Detection of Low-Level Parental Somatic Mosaicism for Clinically Relevant SNVs and Indels Identified in a Large Exome Sequencing Dataset. Hum. Genomics 15, 72. doi:10.1186/s40246-021-00369-6
Downs, B., Sherman, S., Cui, J., Kim, Y. C., Snyder, C., Christensen, M., et al. (2019). Common Genetic Variants Contribute to Incomplete Penetrance: Evidence from Cancer-free BRCA1 Mutation Carriers. Eur. J. Cancer 107, 68–78. doi:10.1016/j.ejca.2018.10.022
Dyle, M. C., Kolakada, D., Cortazar, M. A., and Jagannathan, S. (2020). How to Get Away with Nonsense: Mechanisms and Consequences of Escape from Nonsense-Mediated RNA Decay. Wiley Interdiscip. Rev. RNA 11, e1560. doi:10.1002/wrna.1560
Eckersley-Maslin, M. A., and Spector, D. L. (2014). Random Monoallelic Expression: Regulating Gene Expression One Allele at a Time. Trends Genet. 30, 237–244. doi:10.1016/j.tig.2014.03.003
El-Brolosy, M. A., and Stainier, D. Y. R. (2017). Genetic Compensation: A Phenomenon in Search of Mechanisms. PLOS Genet. 13, e1006780. doi:10.1371/journal.pgen.1006780
Elfatih, A., Mohammed, I., Abdelrahman, D., and Mifsud, B. (2021). Frequency and Management of Medically Actionable Incidental Findings from Genome and Exome Sequencing Data: a Systematic Review. Physiol. Genomics 53, 373–384. doi:10.1152/physiolgenomics.00025.2021
Ellingford, J. M., Ahn, J. W., Bagnall, R. D., Baralle, D., Barton, S., Campbell, C., et al. (2021). Recommendations for Clinical Interpretation of Variants Found in Non-coding Regions of the Genome. Search life-sciences lit. 12 (28), 21267792.
Emison, E. S., Garcia-Barcelo, M., Grice, E. A., Lantieri, F., Amiel, J., Burzynski, G., et al. (2010). Differential Contributions of Rare and Common, Coding and Noncoding Ret Mutations to Multifactorial Hirschsprung Disease Liability. Am. J. Hum. Genet. 87, 60–74. doi:10.1016/j.ajhg.2010.06.007
Emond, M. J., Louie, T., Louie, T., Emerson, J., Zhao, W., Mathias, R. A., et al. (2012). Exome Sequencing of Extreme Phenotypes Identifies DCTN4 as a Modifier of Chronic Pseudomonas aeruginosa Infection in Cystic Fibrosis. Nat. Genet. 44, 886–889. doi:10.1038/ng.2344
Epstein, D. J. (2009). Cis-regulatory Mutations in Human Disease. Briefings Funct. Genomics Proteomics 8, 310–316. doi:10.1093/bfgp/elp021
Erikson, G. A., Bodian, D. L., Rueda, M., Molparia, B., Scott, E. R., Scott-Van Zeeland, A. A., et al. (2016). Whole-Genome Sequencing of a Healthy Aging Cohort. Cell. 165, 1002–1011. doi:10.1016/j.cell.2016.03.022
Evans, D. W., and Uljarević, M. (2018). Parental Education Accounts for Variability in the IQs of Probands with Down Syndrome: A Longitudinal Study. Am. J. Med. Genet. 176, 29–33. doi:10.1002/ajmg.a.38519
Fahed, A. C., Wang, M., Homburger, J. R., Patel, A. P., Bick, A. G., Neben, C. L., et al. (2020). Polygenic Background Modifies Penetrance of Monogenic Variants for Tier 1 Genomic Conditions. Nat. Commun. 11, 3635. doi:10.1038/s41467-020-17374-3
Fahim, A. T., Bowne, S. J., Sullivan, L. S., Webb, K. D., Williams, J. T., Wheaton, D. K., et al. (2012). Polymorphic Variation of RPGRIP1L and IQCB1 as Modifiers of X-Linked Retinitis Pigmentosa Caused by Mutations in RPGR. Adv. Exp. Med. Biol. 723, 313–320. doi:10.1007/978-1-4614-0631-0_41
Falkenberg, K. D., Braverman, N. E., Moser, A. B., Steinberg, S. J., Klouwer, F. C. C., Schlüter, A., et al. (2017). Allelic Expression Imbalance Promoting a Mutant PEX6 Allele Causes Zellweger Spectrum Disorder. Am. J. Hum. Genet. 101, 965–976. doi:10.1016/j.ajhg.2017.11.007
Faure, A. J., Domingo, J., Schmiedel, J. M., Hidalgo-Carcedo, C., Diss, G., and Lehner, B. (2022). Mapping the Energetic and Allosteric Landscapes of Protein Binding Domains. Nature 604, 175–183. doi:10.1038/s41586-022-04586-4
Ferreira, J., Gutiérrez, J. C. L., Carneiro, A., Araújo, A., Sousa, P. P., Braga, S., et al. (2021). CLOVES Syndrome Diagnosis and Treatment in an Adult Patient. Ann. Vasc. Surg. 75, 533.e5–e9. doi:10.1016/j.avsg.2021.03.044
Fetit, R., Price, D. J., Lawrie, S. M., and Johnstone, M. (2020). Understanding the Clinical Manifestations of 16p11.2 Deletion Syndrome: a Series of Developmental Case Reports in Children. Psychiatr. Genet. 30, 136–140. doi:10.1097/ypg.0000000000000259
Fink, D. A., Nelson, L. M., Pyeritz, R., Johnson, J., Sherman, S. L., Cohen, Y., et al. (2018). Fragile X Associated Primary Ovarian Insufficiency (FXPOI): Case Report and Literature Review. Front. Genet. 9, 529. doi:10.3389/fgene.2018.00529
Flannick, J., Beer, N. L., Bick, A. G., Agarwala, V., Molnes, J., Gupta, N., et al. (2013). Assessing the Phenotypic Effects in the General Population of Rare Variants in Genes for a Dominant Mendelian Form of Diabetes. Nat. Genet. 45, 1380–1385. doi:10.1038/ng.2794
Flower, M., Lomeikaite, V., Ciosi, M., Cumming, S., Morales, F., Lo, K., et al. (2019). MSH3 Modifies Somatic Instability and Disease Severity in Huntington's and Myotonic Dystrophy Type 1. Brain 142, 1876–1886. doi:10.1093/brain/awz115
Flydal, M. I., and Martinez, A. (2013). Phenylalanine Hydroxylase: Function, Structure, and Regulation. IUBMB Life 65, 341–349. doi:10.1002/iub.1150
Franke, M., Ibrahim, D. M., Andrey, G., Schwarzer, W., Heinrich, V., Schöpflin, R., et al. (2016). Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 538, 265–269. doi:10.1038/nature19800
Freund, M. K., Burch, K. S., Shi, H., Mancuso, N., Kichaev, G., Garske, K. M., et al. (2018). Phenotype-Specific Enrichment of Mendelian Disorder Genes Near GWAS Regions across 62 Complex Traits. Am. J. Hum. Genet. 103, 535–552. doi:10.1016/j.ajhg.2018.08.017
Fry, A., Littlejohns, T. J., Sudlow, C., Doherty, N., Adamska, L., Sprosen, T., et al. (2017). Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants with Those of the General Population. Am. J. Epidemiol. 186, 1026–1034. doi:10.1093/aje/kwx246
Gallagher, S., Hughes, E., Wagner, S., Tshiaba, P., Rosenthal, E., Roa, B. B., et al. (2020). Association of a Polygenic Risk Score with Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw. Open 3, e208501. doi:10.1001/jamanetworkopen.2020.8501
Galupa, R., and Heard, E. (2017). Topologically Associating Domains in Chromosome Architecture and Gene Regulatory Landscapes during Development, Disease, and Evolution. Cold Spring Harb. Symp. Quant. Biol. 82, 267–278. doi:10.1101/sqb.2017.82.035030
Gerrits, M. C. F., Foncke, E. M. J., Koelman, J. H. T. M., and Tijssen, M. a. J. (2009). Pediatric Writer's Cramp in Myoclonus-Dystonia: Maternal Imprinting Hides Positive Family History. Eur. J. Paediatr. Neurology 13, 178–180. doi:10.1016/j.ejpn.2008.03.007
Girirajan, S., Rosenfeld, J. A., Cooper, G. M., Antonacci, F., Siswara, P., Itsara, A., et al. (2010). A Recurrent 16p12.1 Microdeletion Supports a Two-Hit Model for Severe Developmental Delay. Nat. Genet. 42, 203–209. doi:10.1038/ng.534
Glazier, A. A., Thompson, A., and Day, S. M. (2019). Allelic Imbalance and Haploinsufficiency in MYBPC3-Linked Hypertrophic Cardiomyopathy. Pflugers Arch. - Eur. J. Physiol. 471, 781–793. doi:10.1007/s00424-018-2226-9
Goodrich, J. K., Singer-Berk, M., Son, R., Sveden, A., Wood, J., England, E., et al. (2021). Determinants of Penetrance and Variable Expressivity in Monogenic Metabolic Conditions across 77,184 Exomes. Nat. Commun. 12, 3505. doi:10.1038/s41467-021-23556-4
Gordon, A. S., Zouk, H., Venner, E., Eng, C. M., Funke, B. H., Amendola, L. M., et al. (2020). Frequency of Genomic Secondary Findings Among 21,915 eMERGE Network Participants. Genet. Med. 22, 1470–1477. doi:10.1038/s41436-020-0810-9
Goula, A.-V., and Merienne, K. (2013). Abnormal Base Excision Repair at Trinucleotide Repeats Associated with Diseases: A Tissue-Selective Mechanism. Genes. 4, 375–387. doi:10.3390/genes4030375
Green, D. J., Sallah, S. R., Ellingford, J. M., Lovell, S. C., and Sergouniotis, P. I. (2020). Variability in Gene Expression Is Associated with Incomplete Penetrance in Inherited Eye Disorders. Genes. 11, 179. doi:10.3390/genes11020179
Gripp, K. W., and Rauen, K. A. (1993). “Costello Syndrome,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Gripp, G. M. Mirzaa, and A. Amemiya (Seattle (WA): University of Washington, Seattle).
Gross, M., Hanenberg, H., Lobitz, S., Friedl, R., Herterich, S., Dietrich, R., et al. (2002). Reverse Mosaicism in Fanconi Anemia: Natural Gene Therapy via Molecular Self-Correction. Cytogenet. Genome Res. 98, 126–135. doi:10.1159/000069805
Gruber, C., and Bogunovic, D. (2020). Incomplete Penetrance in Primary Immunodeficiency: a Skeleton in the Closet. Hum. Genet. 139, 745–757. doi:10.1007/s00439-020-02131-9
Gu, Y., Harley, I. T. W., Henderson, L. B., Aronow, B. J., Vietor, I., Huber, L. A., et al. (2009). Identification of IFRD1 as a Modifier Gene for Cystic Fibrosis Lung Disease. Nature 458, 1039–1042. doi:10.1038/nature07811
Gudmundsson, S., Singer-Berk, M., Watts, N. A., Phu, W., Goodrich, J. K., Solomonson, M., et al. (2021). Genome Aggregation Database Consortium. doi:10.1002/humu.24309Variant Interpretation Using Population Databases: Lessons from gnomADHum. Mutat.
Gui, B., Slone, J., and Huang, T. (2017). Perspective: Is Random Monoallelic Expression a Contributor to Phenotypic Variability of Autosomal Dominant Disorders? Front. Genet. 8, 191. doi:10.3389/fgene.2017.00191
Guillot, L., Beucher, J., Tabary, O., Le Rouzic, P., Clement, A., and Corvol, H. (2014). Lung Disease Modifier Genes in Cystic Fibrosis. Int. J. Biochem. Cell. Biol. 52, 83–93. doi:10.1016/j.biocel.2014.02.011
Haendel, M., Vasilevsky, N., Unni, D., Bologa, C., Harris, N., Rehm, H., et al. (2020). How Many Rare Diseases Are There? Nat. Rev. Drug Discov. 19, 77–78. doi:10.1038/d41573-019-00180-y
Haeri, M., and Knox, B. E. (2012). Endoplasmic Reticulum Stress and Unfolded Protein Response Pathways: Potential for Treating Age-Related Retinal Degeneration. J. Ophthalmic Vis. Res. 7, 45–59.
Hagerman, R. J., Berry-Kravis, E., Hazlett, H. C., Bailey, D. B., Moine, H., Kooy, R. F., et al. (2017). Fragile X Syndrome. Nat. Rev. Dis. Prim. 3, 17065. doi:10.1038/nrdp.2017.65
Hammer, M. F., Ishii, A., Johnstone, L., Tchourbanov, A., Lau, B., Sprissler, R., et al. (2017). Rare Variants of Small Effect Size in Neuronal Excitability Genes Influence Clinical Outcome in Japanese Cases of SCN1A Truncation-Positive Dravet Syndrome. PLOS ONE 12, e0180485. doi:10.1371/journal.pone.0180485
Hanany, M., and Sharon, D. (2019). Allele Frequency Analysis of Variants Reported to Cause Autosomal Dominant Inherited Retinal Diseases Question the Involvement of 19% of Genes and 10% of Reported Pathogenic Variants. J. Med. Genet. 56, 536–542. doi:10.1136/jmedgenet-2018-105971
Hanson, E., Bernier, R., Porche, K., Jackson, F. I., Goin-Kochel, R. P., Snyder, L. G., et al. (2015). The Cognitive and Behavioral Phenotype of the 16p11.2 Deletion in a Clinically Ascertained Population. Biol. Psychiatry 77, 785–793. doi:10.1016/j.biopsych.2014.04.021
Harper, A. R., Goel, A., Goel, A., Grace, C., Thomson, K. L., Petersen, S. E., et al. (2021). Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 53, 135–142. doi:10.1038/s41588-020-00764-0
Harrison, S. M., and Rehm, H. L. (2019). Is 'likely Pathogenic' Really 90% Likely? Reclassification Data in ClinVar. Genome Med. 11, 72. doi:10.1186/s13073-019-0688-9
Hart, M. R., Biesecker, B. B., Blout, C. L., Christensen, K. D., Amendola, L. M., Bergstrom, K. L., et al. (2019). Secondary Findings from Clinical Genomic Sequencing: Prevalence, Patient Perspectives, Family History Assessment, and Health-Care Costs from a Multisite Study. Genet. Med. 21, 1100–1110. doi:10.1038/s41436-018-0308-x
Hassanin, E., May, P., Aldisi, R., Krawitz, P., Maj, C., and Bobbili, D. R. (2021). Assessing the Role of Polygenic Background on the Penetrance of Monogenic Forms in Parkinson’s Disease. Medrxiv. doi:10.1101/2021.06.06.21253270
Helmbacher, F., Schneider-Maunoury, S., Topilko, P., Tiret, L., and Charnay, P. (2000). Targeting of the EphA4 Tyrosine Kinase Receptor Affects Dorsal/ventral Pathfinding of Limb Motor Axons. Dev. Camb. Engl. 127, 3313–3324. doi:10.1242/dev.127.15.3313
Hervé, B., Roume, J., Cognard, S., Fauvert, D., Molina-Gomes, D., and Vialard, F. (2015). Low-level Mosaicism of a De Novo Derivative Chromosome 9 from a t(5;9)(q35.1;q34.3) Has a Major Phenotypic Impact. Eur. J. Med. Genet. 58, 346–350. doi:10.1016/j.ejmg.2015.04.005
Hesson, L. B., Packham, D., Kwok, C. T., Nunez, A. C., Ng, B., Schmidt, C., et al. (2015). Lynch Syndrome Associated with Two MLH1 Promoter Variants and Allelic Imbalance of MLH1 Expression. Hum. Mutat. 36, 622–630. doi:10.1002/humu.22785
Heusinkveld, L. E., Yim, E., Yang, A., Azani, A. B., Liu, Q., Gao, J.-L., et al. (2017). Pathogenesis, Diagnosis and Therapeutic Strategies in WHIM Syndrome Immunodeficiency. Expert Opin. Orphan Drugs 5, 813–825. doi:10.1080/21678707.2017.1375403
Hildebrand, M. S., Jackson, V. E., Scerri, T. S., Van Reyk, O., Coleman, M., Braden, R. O., et al. (2020). Severe Childhood Speech Disorder. Neurology 94, e2148–e2167. doi:10.1212/wnl.0000000000009441
Høie, M. H., Cagiada, M., Beck Frederiksen, A. H., Stein, A., and Lindorff-Larsen, K. (2022). Predicting and Interpreting Large-Scale Mutagenesis Data Using Analyses of Protein Stability and Conservation. Cell. Rep. 38, 110207. doi:10.1016/j.celrep.2021.110207
Holm, I. A., Agrawal, P. B., Agrawal, P. B., Ceyhan-Birsoy, O., Christensen, K. D., Fayer, S., et al. (2018). The BabySeq Project: Implementing Genomic Sequencing in Newborns. BMC Pediatr. 18, 225. doi:10.1186/s12887-018-1200-1
Holmans, P. A., Massey, T. H., and Jones, L. (2017). Genetic Modifiers of Mendelian Disease: Huntington's Disease and the Trinucleotide Repeat Disorders. Hum. Mol. Genet. 26, R83–R90. doi:10.1093/hmg/ddx261
Honda, A., Umegaki-Arao, N., Sasaki, T., Nakabayashi, K., Hata, K., Matsubara, Y., et al. (2017). Somatic HRAS p.G12S Mosaic Mutation Causes Unilaterally Distributed Epidermal Nevi, Woolly Hair and Palmoplantar Keratosis. J. Dermatol. 44, e109–e110. doi:10.1111/1346-8138.13801
Hopp, K., Cornec-Le Gall, E., Senum, S. R., Te Paske, I. B. A. W., Raj, S., Lavu, S., et al. (2020). Detection and Characterization of Mosaicism in Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 97, 370–382. doi:10.1016/j.kint.2019.08.038
Hosseini, S. M., Kim, R., Udupa, S., Costain, G., Jobling, R., Liston, E., et al. (2018). Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation 138, 1195–1205. doi:10.1161/circulationaha.118.035070
Hou, Y., Gratz, H. P., Ureña-Bailén, G., Gratz, P. G., Schilbach-Stückle, K., Renno, T., et al. (2021). Somatic Reversion of a Novel IL2RG Mutation Resulting in Atypical X-Linked Combined Immunodeficiency. Genes. 13, 35. doi:10.3390/genes13010035
Huang, L., and Wilkinson, M. F. (2012). Regulation of Nonsense-Mediated mRNA Decay. WIREs RNA 3, 807–828. doi:10.1002/wrna.1137
Hunter, P. (2022). Understanding Redundancy and Resilience. EMBO Rep. 23, e54742. doi:10.15252/embr.202254742
Huyghe, J. R., Bien, S. A., Harrison, T. A., Kang, H. M., Chen, S., Schmit, S. L., et al. (2019). Discovery of Common and Rare Genetic Risk Variants for Colorectal Cancer. Nat. Genet. 51, 76–87. doi:10.1038/s41588-018-0286-6
Ibrahim, D. M., Tayebi, N., Knaus, A., Stiege, A. C., Sahebzamani, A., Hecht, J., et al. (2016). A Homozygous HOXD13 Missense Mutation Causes a Severe Form of Synpolydactyly with Metacarpal to Carpal Transformation. Am. J. Med. Genet. 170, 615–621. doi:10.1002/ajmg.a.37464
Inoue, K., Khajavi, M., Ohyama, T., Hirabayashi, S.-i., Wilson, J., Reggin, J. D., et al. (2004). Molecular Mechanism for Distinct Neurological Phenotypes Conveyed by Allelic Truncating Mutations. Nat. Genet. 36, 361–369. doi:10.1038/ng1322
Ittisoponpisan, S., Alhuzimi, E., Sternberg, M. J. E., and David, A. (2017). Landscape of Pleiotropic Proteins Causing Human Disease: Structural and System Biology Insights. Hum. Mutat. 38, 289–296. doi:10.1002/humu.23155
Ittisoponpisan, S., Islam, S. A., Khanna, T., Alhuzimi, E., David, A., and Sternberg, M. J. E. (2019). Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated?. J. Mol. Biol. 431 (11), 2197–2212. doi:10.1016/j.jmb.2019.04.009
Jackson, L., Weedon, M. N., Harrison, J., Wood, A. R., Ruth, K. S., Tyrrell, J., and Wright, C. (2022). Influence of family history on penetrance of hereditary cancers in a population setting. Medrxiv. 94, 415–425. doi:10.1101/2022.07.08.22277415
Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., et al. (2014). A Higher Mutational Burden in Females Supports a “Female Protective Model” in Neurodevelopmental Disorders. Am. J. Hum. Genet. 94, 415–425. doi:10.1016/j.ajhg.2014.02.001
Jansen, R.-P. (2001). mRNA Localization: Message on the Move. Nat. Rev. Mol. Cell. Biol. 2, 247–256. doi:10.1038/35067016
Jindal, R., Shirazi, N., and Rawat, K. (2019). Hereditary Segmental Neurofibromatosis: a Report of Three Cases in a Family. BMJ Case Rep. 12, e228826. doi:10.1136/bcr-2018-228826
Jing, H., Zhang, Q., Zhang, Y., Hill, B. J., Dove, C. G., Gelfand, E. W., et al. (2014). Somatic Reversion in Dedicator of Cytokinesis 8 Immunodeficiency Modulates Disease Phenotype. J. Allergy Clin. Immunol. 133, 1667–1675. doi:10.1016/j.jaci.2014.03.025
Johnston, J. J., Lewis, K. L., Ng, D., Singh, L. N., Wynter, J., Brewer, C., et al. (2015). Individualized Iterative Phenotyping for Genome-wide Analysis of Loss-Of-Function Mutations. Am. J. Hum. Genet. 96, 913–925. doi:10.1016/j.ajhg.2015.04.013
Johnston, J. J., Rubinstein, W. S., Facio, F. M., Ng, D., Singh, L. N., Teer, J. K., et al. (2012). Secondary Variants in Individuals Undergoing Exome Sequencing: Screening of 572 Individuals Identifies High-Penetrance Mutations in Cancer-Susceptibility Genes. Am. J. Hum. Genet. 91, 97–108. doi:10.1016/j.ajhg.2012.05.021
Jordan, D. M., Frangakis, S. G., Frangakis, S. G., Golzio, C., Cassa, C. A., Kurtzberg, J., et al. (2015). Identification of Cis-Suppression of Human Disease Mutations by Comparative Genomics. Nature 524, 225–229. doi:10.1038/nature14497
Jordan, W., Rieder, L. E., and Larschan, E. (2019). Diverse Genome Topologies Characterize Dosage Compensation across Species. Trends Genet. 35, 308–315. doi:10.1016/j.tig.2019.02.001
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing, 2016 Update (ACMG SF v2.0): a Policy Statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255. doi:10.1038/gim.2016.190
Kammenga, J. E. (2017). The Background Puzzle: How Identical Mutations in the Same Gene Lead to Different Disease Symptoms. FEBS J. 284, 3362–3373. doi:10.1111/febs.14080
Kay, C., Collins, J. A., Miedzybrodzka, Z., Madore, S. J., Gordon, E. S., Gerry, N., et al. (2016). Huntington Disease Reduced Penetrance Alleles Occur at High Frequency in the General Population. Neurology 87, 282–288. doi:10.1212/wnl.0000000000002858
Kennedy, J., Goudie, D., Blair, E., Chandler, K., Joss, S., McKay, V., et al. (2019). KAT6A Syndrome: Genotype-Phenotype Correlation in 76 Patients with Pathogenic KAT6A Variants. Genet. Med. 21, 850–860. doi:10.1038/s41436-018-0259-2
Kerr, T. P., Sewry, C. A., Robb, S. A., and Roberts, R. G. (2001). Long Mutant Dystrophins and Variable Phenotypes: Evasion of Nonsense-Mediated Decay? Hum. Genet. 109, 402–407. doi:10.1007/s004390100598
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). Nonsense-mediated mRNA Decay Modulates Clinical Outcome of Genetic Disease. Eur. J. Hum. Genet. 14, 1074–1081. doi:10.1038/sj.ejhg.5201649
Khanna, H., Davis, E. E., Murga-Zamalloa, C. A., Estrada-Cuzcano, A., Lopez, I., den Hollander, A. I., et al. (2009). A Common Allele in RPGRIP1L Is a Modifier of Retinal Degeneration in Ciliopathies. Nat. Genet. 41, 739–745. doi:10.1038/ng.366
Kharbanda, M., Kannike, K., Lampe, A., Berg, J., Timmusk, T., and Sepp, M. (2016). Partial Deletion of TCF4 in Three Generation Family with Non-syndromic Intellectual Disability, without Features of Pitt-Hopkins Syndrome. Eur. J. Med. Genet. 59, 310–314. doi:10.1016/j.ejmg.2016.04.003
Khera, A. V., Chaffin, M., Aragam, K. G., Haas, M. E., Roselli, C., Choi, S. H., Natarajan, P., Lander, E. S., Lubitz, S. A., Ellinor, P. T., and Kathiresan, S. (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224. doi:10.1038/s41588-018-0183-z
Kim, E., Napierala, M., and Dent, S. Y. R. (2011). Hyperexpansion of GAA Repeats Affects Post-initiation Steps of FXN Transcription in Friedreich's Ataxia. Nucleic Acids Res. 39, 8366–8377. doi:10.1093/nar/gkr542
Kim, Y., and Connor, J. R. (2020). The Roles of Iron and HFE Genotype in Neurological Diseases. Mol. Aspects Med. 75, 100867. doi:10.1016/j.mam.2020.100867
Kingdom, R., Tuke, M., Wood, A., Beaumont, R. N., Frayling, T., Weedon, M. N., et al. (2022). Rare Genetic Variants in Dominant Developmental Disorder Loci Cause Milder Related Phenotypes in the General Population. AJHG in Press.
Kishore, S., De Franco, E., Cardenas-Diaz, F. L., Letourneau-Freiberg, L. R., Sanyoura, M., Osorio-Quintero, C., et al. (2020). A Non-coding Disease Modifier of Pancreatic Agenesis Identified by Genetic Correction in a Patient-Derived iPSC Line. Cell. Stem Cell. 27, 137–146. e6. doi:10.1016/j.stem.2020.05.001
Klaassen, K., Djordjevic, M., Skakic, A., Kecman, B., Drmanac, R., Pavlovic, S., et al. (2021). Untreated PKU Patients without Intellectual Disability: SHANK Gene Family as a Candidate Modifier. Mol. Genet. Metabolism Rep. 29, 100822. doi:10.1016/j.ymgmr.2021.100822
Klaassen, P., Duijff, S., Swanenburg de Veye, H., Beemer, F., Sinnema, G., Breetvelt, E., et al. (2016). Explaining the Variable Penetrance of CNVs: Parental Intelligence Modulates Expression of Intellectual Impairment Caused by the 22q11.2 Deletion. Am. J. Med. Genet. 171, 790–796. doi:10.1002/ajmg.b.32441
Kolder, I. C. R. M., Tanck, M. W. T., Postema, P. G., Barc, J., Sinner, M. F., Zumhagen, S., et al. (2015). Analysis for Genetic Modifiers of Disease Severity in Patients with Long-QT Syndrome Type 2. Circ. Cardiovasc. Genet. 8, 447–456. doi:10.1161/circgenetics.114.000785
Kraemer, K. H., and DiGiovanna, J. J. (1993). “Xeroderma Pigmentosum,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Kuchenbaecker, K. B., McGuffog, L., Barrowdale, D., Lee, A., Soucy, P., Dennis, J., et al. (2017). Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 109. doi:10.1093/jnci/djw302
Kumar, K. R., Davis, R. L., Tchan, M. C., Wali, G. M., Mahant, N., Ng, K., et al. (2019). Whole Genome Sequencing for the Genetic Diagnosis of Heterogenous Dystonia Phenotypes. Park. Relat. Disord. 69, 111–118. doi:10.1016/j.parkreldis.2019.11.004
Lacaze, P., Sebra, R., Riaz, M., Tiller, J., Revote, J., Phung, J., et al. (2020). Medically Actionable Pathogenic Variants in a Population of 13,131 Healthy Elderly Individuals. Genet. Med. 22, 1883–1886. doi:10.1038/s41436-020-0881-7
Lachmann, R. H., Grant, I. R., Halsall, D., and Cox, T. M. (2004). Twin Pairs Showing Discordance of Phenotype in Adult Gaucher's Disease. Qjm 97, 199–204. doi:10.1093/qjmed/hch036
Lareau, L. F., and Brenner, S. E. (2015). Regulation of Splicing Factors by Alternative Splicing and NMD Is Conserved between Kingdoms yet Evolutionarily Flexible. Mol. Biol. Evol. 32, 1072–1079. doi:10.1093/molbev/msv002
Laskaratos, A., Breza, M., Karadima, G., and Koutsis, G. (2021). Wide Range of Reduced Penetrance Alleles in Spinal and Bulbar Muscular Atrophy: a Model-Based Approach. J. Med. Genet. 58, 385–391. doi:10.1136/jmedgenet-2020-106963
Lauritsen, K. F., Lildballe, D. L., Coucke, P. J., Monrad, R., Larsen, D. A., and Gregersen, P. A. (2017). A Mild Form of Stickler Syndrome Type II Caused by Mosaicism of COL11A1. Eur. J. Med. Genet. 60, 275–278. doi:10.1016/j.ejmg.2017.03.005
Lawson, D. J., Davies, N. M., Haworth, S., Ashraf, B., Howe, L., Crawford, A., et al. (2020). Is Population Structure in the Genetic Biobank Era Irrelevant, a Challenge, or an Opportunity? Hum. Genet. 139, 23–41. doi:10.1007/s00439-019-02014-8
Lee, A., Mavaddat, N., Wilcox, A. N., Cunningham, A. P., Carver, T., Hartley, S., et al. (2019). BOADICEA: a Comprehensive Breast Cancer Risk Prediction Model Incorporating Genetic and Nongenetic Risk Factors. Genet. Med. 21, 1708–1718. doi:10.1038/s41436-018-0406-9
Lee, D. S. M., Park, J., Kromer, A., Baras, A., Rader, D. J., Ritchie, M. D., et al. (2021). Disrupting Upstream Translation in mRNAs Is Associated with Human Disease. Nat. Commun. 12, 1515. doi:10.1038/s41467-021-21812-1
Lee, I., Lehner, B., Vavouri, T., Shin, J., Fraser, A. G., and Marcotte, E. M. (2010). Predicting Genetic Modifier Loci Using Functional Gene Networks. Genome Res. 20, 1143–1153. doi:10.1101/gr.102749.109
Lee, J., Meyerhardt, J. A., Giovannucci, E., and Jeon, J. Y. (2015). Association between Body Mass Index and Prognosis of Colorectal Cancer: A Meta-Analysis of Prospective Cohort Studies. PLoS ONE 10, e0120706. doi:10.1371/journal.pone.0120706
Lee, W., Zernant, J., Nagasaki, T., Molday, L. L., Su, P.-Y., Fishman, G. A., et al. (2021). Cis-acting Modifiers in the ABCA4 Locus Contribute to the Penetrance of the Major Disease-Causing Variant in Stargardt Disease. Hum. Mol. Genet. 30, 1293–1304. doi:10.1093/hmg/ddab122
Lek, M., Karczewski, K. J., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., et al. (2016). Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 536, 285–291. doi:10.1038/nature19057
Leon, E., Jamal, S. M., Zou, Y. S., and Milunsky, J. M. (2011). Partial Trisomy 8 Mosaicism Due to a Pseudoisodicentric Chromosome 8. Am. J. Med. Genet. 155, 1740–1744. doi:10.1002/ajmg.a.34073
Leopold, P., Amandine, B., Maykel, L., Michael, W. Y., Meredith, L., Bryan-Joseph, S. L., et al. (2021). Natural Variants Suppress Mutations in Hundreds of Essential Genes. Mol. Syst. Biol. 17, e10138. doi:10.15252/msb.202010138
Lewis, C. M., and Vassos, E. (2020). Polygenic Risk Scores: from Research Tools to Clinical Instruments. Genome Med. 12, 44. doi:10.1186/s13073-020-00742-5
Li, Q., Liang, P., Wang, S., Li, W., Wang, J., Yang, Y., et al. (2021). A Novel KCNQ4 Gene Variant (c.857A>G; p.Tyr286Cys) in an Extended Family with Non-syndromic D-eafness 2A. Mol. Med. Rep. 23, 420. doi:10.3892/mmr.2021.12059
Li, X.-H., and Babu, M. M. (2018). Human Diseases from Gain-Of-Function Mutations in Disordered Protein Regions. Cell. 175, 40–42. doi:10.1016/j.cell.2018.08.059
Li, Y., Wang, Y., Ming, Y., Chaolan, P., Jia, Z., Cheng, N., et al. (2021). A KRT6A Mutation p.Ile462Asn in a Chinese Family with Pachyonychia Congenita, and Identification of Maternal Mosaicism: a Case Report. BMC Med. Genomics 14, 259. doi:10.1186/s12920-021-01109-4
Lindeboom, R. G. H., Supek, F., and Lehner, B. (2016). The Rules and Impact of Nonsense-Mediated mRNA Decay in Human Cancers. Nat. Genet. 48, 1112–1118. doi:10.1038/ng.3664
Liu, X.-R., Xu, X.-X., Lin, S.-M., Fan, C.-Y., Ye, T.-T., Tang, B., et al. (2021). GRIN2A Variants Associated with Idiopathic Generalized Epilepsies. Front. Mol. Neurosci. 14, 720984. doi:10.3389/fnmol.2021.720984
Lu, L., Liu, X., Huang, W.-K., Giusti-Rodríguez, P., Cui, J., Zhang, S., et al. (2020). Robust Hi-C Maps of Enhancer-Promoter Interactions Reveal the Function of Non-coding Genome in Neural Development and Diseases. Mol. Cell. 79, 521–534. e15. doi:10.1016/j.molcel.2020.06.007
Lu, Y.-Y., and Krebber, H. (2021). Nuclear mRNA Quality Control and Cytoplasmic NMD Are Linked by the Guard Proteins Gbp2 and Hrb1. Int. J. Mol. Sci. 22, 11275. doi:10.3390/ijms222011275
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell. 161, 1012–1025. doi:10.1016/j.cell.2015.04.004
Magri, S., Nanetti, L., Gellera, C., Sarto, E., Rizzo, E., Mongelli, A., et al. (2022). Digenic Inheritance of STUB1 Variants and TBP Polyglutamine Expansions Explains the Incomplete Penetrance of SCA17 and SCA48. Genet. Med. 24, 29–40. doi:10.1016/j.gim.2021.08.003
Magrinelli, F., Balint, B., and Bhatia, K. P. (2021). Challenges in Clinicogenetic Correlations: One Gene - Many Phenotypes. Mov. Disord. Clin. Pract. 8, 299–310. doi:10.1002/mdc3.13165
Mahat, U., Ambani, N. M., Rotz, S. J., and Radhakrishnan, K. (2021). Heterozygous CTLA4 Splice Site Mutation c.458-1G > C Presenting with Immunodeficiency and Variable Degree of Immune Dysregulation in Three Generation Kindred of Caribbean Descent. Pediatr. Hematol. Oncol. 38, 658–662. doi:10.1080/08880018.2021.1906802
Manrai, A. K., Funke, B. H., Rehm, H. L., Olesen, M. S., Maron, B. A., Szolovits, P., et al. (2016). Genetic Misdiagnoses and the Potential for Health Disparities. N. Engl. J. Med. 375, 655–665. doi:10.1056/nejmsa1507092
Marian, A. J., and Braunwald, E. (2017). Hypertrophic Cardiomyopathy. Circ. Res. 121, 749–770. doi:10.1161/circresaha.117.311059
Maroilley, T., and Tarailo-Graovac, M. (2019). Uncovering Missing Heritability in Rare Diseases. Genes. 10, 275. doi:10.3390/genes10040275
Masnada, S., Hedrich, U. B. S., Gardella, E., Schubert, J., Kaiwar, C., Klee, E. W., et al. (2017). Clinical Spectrum and Genotype-Phenotype Associations of KCNA2-Related Encephalopathies. Brain J. Neurol. 140, 2337–2354. doi:10.1093/brain/awx184
Massey, T. H., and Jones, L. (2018). The Central Role of DNA Damage and Repair in CAG Repeat Diseases. Dis. Model. Mech. 11, dmm031930. doi:10.1242/dmm.031930
Mastromoro, G., Capalbo, A., Guido, C. A., Torres, B., Fabbretti, M., Traversa, A., et al. (2020). Small 7p22.3 Microdeletion: Case Report of Snx8 Haploinsufficiency and Neurological Findings. Eur. J. Med. Genet. 63, 103772. doi:10.1016/j.ejmg.2019.103772
Maussion, G., Cruceanu, C., Rosenfeld, J. A., Bell, S. C., Jollant, F., Szatkiewicz, J., et al. (2017). Implication of LRRC4C and DPP6 in Neurodevelopmental Disorders. Am. J. Med. Genet. 173, 395–406. doi:10.1002/ajmg.a.38021
McArthur, E., and Capra, J. A. (2021). Topologically Associating Domain Boundaries that Are Stable across Diverse Cell Types Are Evolutionarily Constrained and Enriched for Heritability. Am. J. Hum. Genet. 108, 269–283. doi:10.1016/j.ajhg.2021.01.001
McDermott, D. H., Gao, J.-L., Liu, Q., Siwicki, M., Martens, C., Jacobs, P., et al. (2015). Chromothriptic Cure of WHIM Syndrome. Cell. 160, 686–699. doi:10.1016/j.cell.2015.01.014
McDermott, J. H., Study, D. D. D., and Clayton-Smith, J. (2017). Sibling Recurrence of Total Anomalous Pulmonary Venous Drainage. Eur. J. Med. Genet. 60, 265–267. doi:10.1016/j.ejmg.2017.03.003
Mensa-Vilaró, A., Bravo García-Morato, M., de la Calle-Martin, O., Franco-Jarava, C., Martínez-Saavedra, M. T., González-Granado, L. I., et al. (2019). Unexpected Relevant Role of Gene Mosaicism in Patients with Primary Immunodeficiency Diseases. J. Allergy Clin. Immunol. 143, 359–368.
Michailidou, K., Lindström, S., Dennis, J., Beesley, J., Hui, S., Kar, S., et al. (2017). Association Analysis Identifies 65 New Breast Cancer Risk Loci. Nature 551, 92–94. doi:10.1038/nature24284
Mignone, F., Gissi, C., Liuni, S., and Pesole, G. (2002). Untranslated Regions of mRNAs. Genome Biol. 3, REVIEWS0004. doi:10.1186/gb-2002-3-3-reviews0004
Miller, E. E., Kobayashi, G. S., Musso, C. M., Allen, M., Ishiy, F. A. A., de Caires, L. C., et al. (2017). EIF4A3 Deficient Human iPSCs and Mouse Models Demonstrate Neural Crest Defects that Underlie Richieri-Costa-Pereira Syndrome. Hum. Mol. Genet. 26, 2177–2191. doi:10.1093/hmg/ddx078
Miller, J. N., and Pearce, D. A. (2014). Nonsense-mediated Decay in Genetic Disease: Friend or Foe? Mutat. Research/Reviews Mutat. Res. 762, 52–64. doi:10.1016/j.mrrev.2014.05.001
Minikel, E. V., Vallabh, S. M., Lek, M., Estrada, K., Samocha, K. E., Sathirapongsasuti, J. F., et al. (2016). Quantifying Penetrance in a Dominant Disease Gene Using Large Population Control Cohorts. Sci. Transl. Med. 8, 322ra9. doi:10.1126/scitranslmed.aad5169
Mirshahi, U. L., Colclough, K., Wright, C. F., Wood, A. R., Beaumont, R. N., Tyrrell, J., et al. (2021). The Penetrance of Age-Related Monogenic Disease Depends on Ascertainment Context. MedRxiv 2021, 21259641. doi:10.1101/2021.06.28.21259641
Miyazawa, H., and Wada, T. (2021). Reversion Mosaicism in Primary Immunodeficiency Diseases. Front. Immunol. 12, 783022. doi:10.3389/fimmu.2021.783022
Morales, F., Vásquez, M., Santamaría, C., Cuenca, P., Corrales, E., and Monckton, D. G. (2016). A Polymorphism in the MSH3 Mismatch Repair Gene Is Associated with the Levels of Somatic Instability of the Expanded CTG Repeat in the Blood DNA of Myotonic Dystrophy Type 1 Patients. DNA Repair 40, 57–66. doi:10.1016/j.dnarep.2016.01.001
Moreno-De-Luca, A., Evans, D. W., Boomer, K. B., Hanson, E., Bernier, R., Goin-Kochel, R. P., et al. (2015). The Role of Parental Cognitive, Behavioral, and Motor Profiles in Clinical Variability in Individuals with Chromosome 16p11.2 Deletions. JAMA Psychiatry 72, 119–126. doi:10.1001/jamapsychiatry.2014.2147
Moskwa, P., Buffa, F. M., Pan, Y., Panchakshari, R., Gottipati, P., Muschel, R. J., et al. (2011). miR-182-mediated Downregulation of BRCA1 Impacts DNA Repair and Sensitivity to PARP Inhibitors. Mol. Cell. 41, 210–220. doi:10.1016/j.molcel.2010.12.005
Muse, E. D., Chen, S.-F., and Torkamani, A. (2021). Monogenic and Polygenic Models of Coronary Artery Disease. Curr. Cardiol. Rep. 23, 107. doi:10.1007/s11886-021-01540-0
Musunuru, K., Strong, A., Frank-Kamenetsky, M., Lee, N. E., Ahfeldt, T., Sachs, K. V., et al. (2010). From Noncoding Variant to Phenotype via SORT1 at the 1p13 Cholesterol Locus. Nature 466, 714–719. doi:10.1038/nature09266
Nagy, E., and Maquat, L. E. (1998). A Rule for Termination-Codon Position within Intron-Containing Genes: when Nonsense Affects RNA Abundance. Trends Biochem. Sci. 23, 198–199. doi:10.1016/s0968-0004(98)01208-0
Nathan, N., Berdah, L., Delestrain, C., Sileo, C., and Clement, A. (20201983). Interstitial Lung Diseases in Children. La Presse Médicale 49, 103909. doi:10.1016/j.lpm.2019.06.007
Neeve, V. C. M., Samuels, D. C., Bindoff, L. A., van den Bosch, B., Van Goethem, G., Smeets, H., et al. (2012). What Is Influencing the Phenotype of the Common Homozygous Polymerase-γ Mutation p.Ala467Thr? Brain J. Neurol. 135, 3614–3626. doi:10.1093/brain/aws298
Newcomb, P. A., and Carbone, P. P. (1992). The Health Consequences of Smoking: Cancer. Med. Clin. N. Am. 76, 305–331. doi:10.1016/s0025-7125(16)30355-8
Ng, P. C., Levy, S., Huang, J., Stockwell, T. B., Walenz, B. P., Li, K., et al. (2008). Genetic Variation in an Individual Human Exome. PLOS Genet. 4, e1000160. doi:10.1371/journal.pgen.1000160
Nguengang Wakap, S., Lambert, D. M., Olry, A., Rodwell, C., Gueydan, C., Lanneau, V., et al. (2020). Estimating Cumulative Point Prevalence of Rare Diseases: Analysis of the Orphanet Database. Eur. J. Hum. Genet. 28, 165–173. doi:10.1038/s41431-019-0508-0
Nguyen, L. S., Jolly, L., Shoubridge, C., Chan, W. K., Huang, L., Laumonnier, F., et al. (2012). Transcriptome Profiling of UPF3B/NMD-Deficient Lymphoblastoid Cells from Patients with Various Forms of Intellectual Disability. Mol. Psychiatry 17, 1103–1115. doi:10.1038/mp.2011.163
Nguyen, L. S., Kim, H.-G., Rosenfeld, J. A., Shen, Y., Gusella, J. F., Lacassie, Y., et al. (2013). Contribution of Copy Number Variants Involving Nonsense-Mediated mRNA Decay Pathway Genes to Neuro-Developmental Disorders. Hum. Mol. Genet. 22, 1816–1825. doi:10.1093/hmg/ddt035
Nguyen, L. S., Wilkinson, M. F., and Gecz, J. (2014). Nonsense-mediated mRNA Decay: Inter-individual Variability and Human Disease. Neurosci. Biobehav. Rev. 46, 175–186. doi:10.1016/j.neubiorev.2013.10.016
Nicoletti, E., Rao, G., Bueren, J. A., Río, P., Navarro, S., Surrallés, J., et al. (2020). Mosaicism in Fanconi Anemia: Concise Review and Evaluation of Published Cases with Focus on Clinical Course of Blood Count Normalization. Ann. Hematol. 99, 913–924. doi:10.1007/s00277-020-03954-2
Niday, Z., and Tzingounis, A. V. (2018). Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox. Neuroscientist 24, 368–380. doi:10.1177/1073858418763752
Niemi, M. E. K., Martin, H. C., Rice, D. L., Gallone, G., Gordon, S., Kelemen, M., et al. (2018). Common Genetic Variants Contribute to Risk of Rare Severe Neurodevelopmental Disorders. Nature 562, 268–271. doi:10.1038/s41586-018-0566-4
Oetjens, M. T., Kelly, M. A., Sturm, A. C., Martin, C. L., and Ledbetter, D. H. (2019). Quantifying the Polygenic Contribution to Variable Expressivity in Eleven Rare Genetic Disorders. Nat. Commun. 10, 4897. doi:10.1038/s41467-019-12869-0
Oliva, M., Muñoz-Aguirre, M., Kim-Hellmuth, S., Wucher, V., Gewirtz, A. D. H., Cotter, D. J., et al. (2020). The Impact of Sex on Gene Expression across Human Tissues. Science 369, eaba3066. doi:10.1126/science.aba3066
Olszewski, A. K., Radoeva, P. D., Fremont, W., Kates, W. R., and Antshel, K. M. (2014). Is Child Intelligence Associated with Parent and Sibling Intelligence in Individuals with Developmental Disorders? an Investigation in Youth with 22q11.2 Deletion (Velo-Cardio-Facial) Syndrome. Res. Dev. Disabil. 35, 3582–3590. doi:10.1016/j.ridd.2014.08.034
Omim, (2022). OMIM Gene Map Statistics. Available at: https://www.omim.org/statistics/geneMap (Accessed 04 11, 2022).
Oprea, G. E., Kröber, S., McWhorter, M. L., Rossoll, W., Müller, S., Krawczak, M., et al. (2008). Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy. Science 320, 524–527. doi:10.1126/science.1155085
Park, J. K., Martin, L. J., Zhang, X., Jegga, A. G., and Benson, D. W. (2012). Genetic Variants in SCN5A Promoter Are Associated with Arrhythmia Phenotype Severity in Patients with Heterozygous Loss-Of-Function Mutation. Heart rhythm. 9, 1090–1096. doi:10.1016/j.hrthm.2012.02.023
Pasmooij, A. M. G., Garcia, M., Escamez, M. J., Miranda Nijenhuis, A., Azon, A., Cuadrado-Corrales, N., et al. (2010). Revertant Mosaicism Due to a Second-Site Mutation in COL7A1 in a Patient with Recessive Dystrophic Epidermolysis Bullosa. J. Investigative Dermatology 130, 2407–2411. doi:10.1038/jid.2010.163
Paulson, H. (2018). Repeat Expansion Diseases. Handb. Clin. Neurol. 147, 105–123. doi:10.1016/b978-0-444-63233-3.00009-9
Payán-Gómez, C., Ramirez-Cheyne, J., and Saldarriaga, W. (2021). Variable Expressivity in Fragile X Syndrome: Towards the Identification of Molecular Characteristics that Modify the Phenotype. Appl. Clin. Genet. 14, 305–312.
Payne, J. L., and Wagner, A. (2015). Mechanisms of Mutational Robustness in Transcriptional Regulation. Front. Genet. 6, 322. doi:10.3389/fgene.2015.00322
Peltonen, L., Perola, M., Naukkarinen, J., and Palotie, A. (2006). Lessons from Studying Monogenic Disease for Common Disease. Hum. Mol. Genet. 15, R67–R74. doi:10.1093/hmg/ddl060
Pereira, S. V.-N., Ribeiro, J. D., Ribeiro, A. F., Bertuzzo, C. S., and Marson, F. A. L. (2019). Novel, Rare and Common Pathogenic Variants in the CFTR Gene Screened by High-Throughput Sequencing Technology and Predicted by In Silico Tools. Sci. Rep. 9, 6234. doi:10.1038/s41598-019-42404-6
Perez, B. A., Shorrock, H. K., Banez-Coronel, M., Zu, T., El Romano, L., Laboissonniere, L. A., et al. (2021). CCG•CGG Interruptions in High-Penetrance SCA8 Families Increase RAN Translation and Protein Toxicity. EMBO Mol. Med. 13, e14095. doi:10.15252/emmm.202114095
Petrovski, S., Gussow, A. B., Wang, Q., Halvorsen, M., Han, Y., Weir, W. H., et al. (2015). The Intolerance of Regulatory Sequence to Genetic Variation Predicts Gene Dosage Sensitivity. PLOS Genet. 11, e1005492. doi:10.1371/journal.pgen.1005492
Petrucelli, N., Daly, M. B., and Pal, T. (1993). BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Pilling, L. C., Tamosauskaite, J., Jones, G., Wood, A. R., Jones, L., Kuo, C.-L., et al. (2019). Common Conditions Associated with Hereditary Haemochromatosis Genetic Variants: Cohort Study in UK Biobank. BMJ 364, k5222. doi:10.1136/bmj.k5222
Pinter, S. F., Colognori, D., Beliveau, B. J., Sadreyev, R. I., Payer, B., Yildirim, E., et al. (2015). Allelic Imbalance Is a Prevalent and Tissue-specific Feature of the Mouse Transcriptome. Genetics 200, 537–549. doi:10.1534/genetics.115.176263
Pipis, M., Cortese, A., Polke, J. M., Poh, R., Vandrovcova, J., Laura, M., et al. (2022). Charcot-Marie-Tooth Disease Type 2CC Due to NEFH Variants Causes a Progressive, Non-length-dependent, Motor-Predominant Phenotype. J. Neurol. Neurosurg. Psychiatry 93, 48–56. doi:10.1136/jnnp-2021-327186
Pizzo, L., Jensen, M., Polyak, A., Rosenfeld, J. A., Mannik, K., Krishnan, A., et al. (2019). Rare Variants in the Genetic Background Modulate Cognitive and Developmental Phenotypes in Individuals Carrying Disease-Associated Variants. Genet. Med. 21, 816–825. doi:10.1038/s41436-018-0266-3
Platzer, K., Yuan, H., Schütz, H., Winschel, A., Chen, W., Hu, C., et al. (2017). GRIN2Bencephalopathy: Novel Findings on Phenotype, Variant Clustering, Functional Consequences and Treatment Aspects. J. Med. Genet. 54, 460–470. doi:10.1136/jmedgenet-2016-104509
Polyak, A., Rosenfeld, J. A., and Girirajan, S. (2015). An Assessment of Sex Bias in Neurodevelopmental Disorders. Genome Med. 7, 94. doi:10.1186/s13073-015-0216-5
Poot, M. (2015). Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders. Mol. Syndromol. 6, 7–22. doi:10.1159/000371594
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 376, 21–31. doi:10.1056/nejmoa1516767
Qiagen, (2022). Human Gene Mutation Database (HGMD) Professional | QIAGEN. Available at: http://www.hgmd.cf.ac.uk/ac/index.php (Accessed 04 11, 2022).
Racanelli, A. C., Kikkers, S. A., Choi, A. M. K., and Cloonan, S. M. (2018). Autophagy and Inflammation in Chronic Respiratory Disease. Autophagy 14, 221–232. doi:10.1080/15548627.2017.1389823
Rahit, K. M. T. H., and Tarailo-Graovac, M. (2020). Genetic Modifiers and Rare Mendelian Disease. Genes. 11, 239. doi:10.3390/genes11030239
Ratto, A. B., Kenworthy, L., Yerys, B. E., Bascom, J., Wieckowski, A. T., White, S. W., et al. (2018). What about the Girls? Sex-Based Differences in Autistic Traits and Adaptive Skills. J. Autism Dev. Disord. 48, 1698–1711. doi:10.1007/s10803-017-3413-9
Raymond, D., Saunders-Pullman, R., and Ozelius, L. (1993). “SGCE Myoclonus-Dystonia,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle).
Rieley, M. B., Stevenson, D. A., Viskochil, D. H., Tinkle, B. T., Martin, L. J., and Schorry, E. K. (2011). Variable Expression of Neurofibromatosis 1 in Monozygotic Twins. Am. J. Med. Genet. 155, 478–485. doi:10.1002/ajmg.a.33851
Rose, A. m., and Bhattacharya, S. s. (2016). Variant Haploinsufficiency and Phenotypic Non-penetrance inPRPF31-Associated Retinitis Pigmentosa. Clin. Genet. 90, 118–126. doi:10.1111/cge.12758
Rosenfeld, J. A., Coe, B. P., Eichler, E. E., Cuckle, H., and Shaffer, L. G. (2013). Estimates of Penetrance for Recurrent Pathogenic Copy-Number Variations. Genet. Med. 15, 478–481. doi:10.1038/gim.2012.164
Russell, G., Steer, C., and Golding, J. (2011). Social and Demographic Factors that Influence the Diagnosis of Autistic Spectrum Disorders. Soc. Psychiatry Psychiatr. Epidemiol. 46, 1283–1293. doi:10.1007/s00127-010-0294-z
Sacco, K. A., and Milner, J. D. (2019). Gene-environment Interactions in Primary Atopic Disorders. Curr. Opin. Immunol. 60, 148–155. doi:10.1016/j.coi.2019.06.002
Safi-Stibler, S., and Gabory, A. (2020). Epigenetics and the Developmental Origins of Health and Disease: Parental Environment Signalling to the Epigenome, Critical Time Windows and Sculpting the Adult Phenotype. Seminars Cell. & Dev. Biol. 97, 172–180. doi:10.1016/j.semcdb.2019.09.008
Sanna, S., van Zuydam, N. R., Mahajan, A., Kurilshikov, A., Vich Vila, A., Võsa, U., et al. (2019). Causal Relationships Among the Gut Microbiome, Short-Chain Fatty Acids and Metabolic Diseases. Nat. Genet. 51, 600–605. doi:10.1038/s41588-019-0350-x
Sarkar, H., Mitsios, A., Smart, M., Skinner, J., Welch, A. A., Kalatzis, V., et al. (2019). Nonsense-mediated mRNA Decay Efficiency Varies in Choroideremia Providing a Target to Boost Small Molecule Therapeutics. Hum. Mol. Genet. 28, 1865–1871. doi:10.1093/hmg/ddz028
Sarri, C. A., Roussaki-Schulze, A., Vasilopoulos, Y., Zafiriou, E., Patsatsi, A., Stamatis, C., et al. (2017). Netherton Syndrome: A Genotype-Phenotype Review. Mol. Diagn. Ther. 21, 137–152. doi:10.1007/s40291-016-0243-y
Sato, H., and Singer, R. H. (2021). Cellular Variability of Nonsense-Mediated mRNA Decay. Nat. Commun. 12, 7203. doi:10.1038/s41467-021-27423-0
Scacheri, C. A., and Scacheri, P. C. (2015). Mutations in the Noncoding Genome. Curr. Opin. Pediatr. 27, 659–664. doi:10.1097/mop.0000000000000283
Schacherer, J. (2016). Beyond the Simplicity of Mendelian Inheritance. Comptes Rendus Biol. 339, 284–288. doi:10.1016/j.crvi.2016.04.006
Schell, R., Hale, J. J., Mullis, M. N., Matsui, T., Foree, R., and Ehrenreich, I. M. (2022). Genetic Basis of a Spontaneous Mutation's Expressivity. Genetics 220, iyac013. doi:10.1093/genetics/iyac013
Schindler, T., Michel, S., and Wilson, A. W. M. (2015). Nutrition Management of Cystic Fibrosis in the 21st Century. Nutr. Clin. Pract. 30, 488–500. doi:10.1177/0884533615591604
Schmidt, L. S., and Linehan, W. M. (2018). FLCN : The Causative Gene for Birt-Hogg-Dubé Syndrome. Gene 640, 28–42. doi:10.1016/j.gene.2017.09.044
Scott, F. J., Baron-Cohen, S., Bolton, P., and Brayne, C. (2002). Brief Report Prevalence of Autism Spectrum Conditions in Children Aged 5-11 Years in Cambridgeshire, UK. Autism 6, 231–237. doi:10.1177/1362361302006003002
Senol-Cosar, O., Schmidt, R. J., Qian, E., Hoskinson, D., Mason-Suares, H., Funke, B., et al. (2019). Considerations for Clinical Curation, Classification, and Reporting of Low-Penetrance and Low Effect Size Variants Associated with Disease Risk. Genet. Med. 21, 2765–2773. doi:10.1038/s41436-019-0560-8
Servetti, M., Pisciotta, L., Tassano, E., Cerminara, M., Nobili, L., Boeri, S., et al. (2021). Neurodevelopmental Disorders in Patients with Complex Phenotypes and Potential Complex Genetic Basis Involving Non-coding Genes, and Double CNVs. Front. Genet. 12, 732002. doi:10.3389/fgene.2021.732002
Shawky, R. M. (2014). Reduced Penetrance in Human Inherited Disease. Egypt. J. Med. Hum. Genet. 15, 103–111. doi:10.1016/j.ejmhg.2014.01.003
Shieh, J. T. C. (2019). Genomic Sequencing Expansion and Incomplete Penetrance. Pediatrics 143, S22–S26. doi:10.1542/peds.2018-1099e
Silva, J., Fernandes, R., and Romão, L. (2019). Translational Regulation by Upstream Open Reading Frames and Human Diseases. Adv. Exp. Med. Biol. 1157, 99–116. doi:10.1007/978-3-030-19966-1_5
Sirp, A., Roots, K., Nurm, K., Tuvikene, J., Sepp, M., and Timmusk, T. (2021). Functional Consequences of TCF4 Missense Substitutions Associated with Pitt-Hopkins Syndrome, Mild Intellectual Disability, and Schizophrenia. J. Biol. Chem. 297, 101381. doi:10.1016/j.jbc.2021.101381
Sirugo, G., Williams, S. M., and Tishkoff, S. A. (2019). The Missing Diversity in Human Genetic Studies. Cell. 177, 26–31. doi:10.1016/j.cell.2019.02.048
Skakkebaek, A., Viuff, M., Nielsen, M. M., and Gravholt, C. H. (2020). Epigenetics and Genomics in Klinefelter Syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 184, 216–225. doi:10.1002/ajmg.c.31802
Smith, F. J., Hansen, C. D., Hull, P. R., Kaspar, R. L., McLean, W. I., O’Toole, E., et al. (1993). “Pachyonychia Congenita,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle).
Somaschini, M., Cavazza, A., Riva, S., Sassi, I., and Carrera, P. (2005). Genetic Basis in Chronic Interstitial Familial Pneumopathy. Familial Study of SFTPC. Pediatr. Med. Chir. 27, 103–107.
Speletas, M., Szilagyi, A., Psarros, F., Moldovan, D., Magerl, M., Kompoti, M., et al. (2015). Hereditary Angioedema: Molecular and Clinical Differences Among European Populations. J. Allergy Clin. Immunol. 135, 570–573. e10. doi:10.1016/j.jaci.2014.08.007
Stefansson, H., Meyer-Lindenberg, A., Steinberg, S., Magnusdottir, B., Morgen, K., Arnarsdottir, S., et al. (2014). CNVs Conferring Risk of Autism or Schizophrenia Affect Cognition in Controls. Nature 505, 361–366. doi:10.1038/nature12818
Stefl, S., Nishi, H., Petukh, M., Panchenko, A. R., and Alexov, E. (2013). Molecular Mechanisms of Disease-Causing Missense Mutations. J. Mol. Biol. 425, 3919–3936. doi:10.1016/j.jmb.2013.07.014
Steinberg, M. H., and Sebastiani, P. (2012). Genetic Modifiers of Sickle Cell Disease. Am. J. Hematol. 87, 795–803. doi:10.1002/ajh.23232
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: Towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and Next-Generation Sequencing Studies. Hum. Genet. 136 (6), 665–677. doi:10.1007/s00439-017-1779-6
Stephanou, C., Tamana, S., Minaidou, A., Papasavva, P., Kleanthous, M., and Kountouris, P. (2019). Genetic Modifiers at the Crossroads of Personalised Medicine for Haemoglobinopathies. J. Clin. Med. 8, 1927. doi:10.3390/jcm8111927
Steri, M., Idda, M. L., Whalen, M. B., and Orrù, V. (2018). Genetic Variants in mRNA Untranslated Regions. Wiley Interdiscip. Rev. RNA 9, e1474. doi:10.1002/wrna.1474
Strehlow, V., Heyne, H. O., Vlaskamp, D. R. M., Marwick, K. F. M., Rudolf, G., de Bellescize, J., et al. (2019). GRIN2A-Related Disorders: Genotype and Functional Consequence Predict Phenotype. Brain. 142 (1), 80–92. doi:10.1093/brain/awy304
Stoeklé, H.-C., Mamzer-Bruneel, M.-F., Vogt, G., and Hervé, C. (2016). 23andMe: a New Two-Sided Data-Banking Market Model. BMC Med. Ethics 17, 19.
Subaran, R. L., Conte, J. M., Stewart, W. C. L., and Greenberg, D. A. (2015). Pathogenic EFHC 1 Mutations Are Tolerated in Healthy Individuals Dependent on Reported Ancestry. Epilepsia 56, 188–194. doi:10.1111/epi.12864
Sulem, P., Helgason, H., Oddson, A., Stefansson, H., Gudjonsson, S. A., Zink, F., et al. (2015). Identification of a Large Set of Rare Complete Human Knockouts. Nat. Genet. 47, 448–452. doi:10.1038/ng.3243
Sun, C., Li, N., Yang, Z., Zhou, B., He, Y., Weng, D., et al. (2013). miR-9 Regulation of BRCA1 and Ovarian Cancer Sensitivity to Cisplatin and PARP Inhibition. J. Natl. Cancer Inst. 105, 1750–1758. doi:10.1093/jnci/djt302
Sun, J. H., Zhou, L., Emerson, D. J., Phyo, S. A., Titus, K. R., Gong, W., et al. (2018). Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell. 175, 224–238. e15. doi:10.1016/j.cell.2018.08.005
Supek, F., Lehner, B., and Lindeboom, R. G. H. (2021). To NMD or Not to NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 37, 657–668. doi:10.1016/j.tig.2020.11.002
Syrbe, S., Hedrich, U. B. S., Hedrich, U. B. S., Riesch, E., Djémié, T., Müller, S., et al. (2015). De Novo loss- or Gain-Of-Function Mutations in KCNA2 Cause Epileptic Encephalopathy. Nat. Genet. 47, 393–399. doi:10.1038/ng.3239
Taniguchi, Y., Takeda, N., Inuzuka, R., Matsubayashi, Y., Kato, S., Doi, T., et al. (2021). Impact of Pathogenic FBN1 Variant Types on the Development of Severe Scoliosis in Patients with Marfan Syndrome. J. Med. Genet.
Tarailo-Graovac, M., Zhu, J. Y. A., Matthews, A., van Karnebeek, C. D. M., and Wasserman, W. W. (2017). Assessment of the ExAC Data Set for the Presence of Individuals with Pathogenic Genotypes Implicated in Severe Mendelian Pediatric Disorders. Genet. Med. 19, 1300–1308. doi:10.1038/gim.2017.50
The Encode Project Consortium, (2012). An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 489, 57–74.
Tolmacheva, E. N., Kashevarova, A. A., Nazarenko, L. P., Minaycheva, L. I., Skryabin, N. A., Lopatkina, M. E., et al. (2020). Delineation of Clinical Manifestations of the Inherited Xq24 Microdeletion Segregating with sXCI in Mothers: Two Novel Cases with Distinct Phenotypes Ranging from UBE2A Deficiency Syndrome to Recurrent Pregnancy Loss. Cytogenet. Genome Res. 160, 245–254. doi:10.1159/000508050
Tommasi, C., Pellegrino, B., Boggiani, D., Sikokis, A., Michiara, M., Uliana, V., et al. (2021). Biological Role and Clinical Implications of microRNAs in BRCA Mutation Carriers. Front. Oncol. 11, 700853. doi:10.3389/fonc.2021.700853
Torella, A., Zanobio, M., Zeuli, R., del Vecchio Blanco, F., Savarese, M., Giugliano, T., et al. (2020). The Position of Nonsense Mutations Can Predict the Phenotype Severity: A Survey on the DMD Gene. PLOS ONE 15, e0237803. doi:10.1371/journal.pone.0237803
Tørring, P. M., Kjeldsen, A. D., Ousager, L. B., and Brusgaard, K. (2017). ENG Mutational Mosaicism in a Family with Hereditary Hemorrhagic Telangiectasia. Mol. Genet. Genomic Med. 6, 121–125. doi:10.1002/mgg3.361
Tuke, M. A., Ruth, K. S., Wood, A. R., Beaumont, R. N., Tyrrell, J., Jones, S. E., et al. (2019). Mosaic Turner Syndrome Shows Reduced Penetrance in an Adult Population Study. Genet. Med. 21, 877–886. doi:10.1038/s41436-018-0271-6
Tukker, A. M., Royal, C. D., Bowman, A. B., and McAllister, K. A. (2021). The Impact of Environmental Factors on Monogenic Mendelian Diseases. Toxicol. Sci. 181, 3–12. doi:10.1093/toxsci/kfab022
Turner, H., and Jackson, L. (2020). Evidence for Penetrance in Patients without a Family History of Disease: a Systematic Review. Eur. J. Hum. Genet. 28, 539–550. doi:10.1038/s41431-019-0556-5
Um, J. W., and Ko, J. (2017). Neural Glycosylphosphatidylinositol-Anchored Proteins in Synaptic Specification. Trends Cell. Biol. 27, 931–945. doi:10.1016/j.tcb.2017.06.007
van der Kolk, D. M., de Bock, G. H., Leegte, B. K., Schaapveld, M., Mourits, M. J. E., de Vries, J., et al. (2010). Penetrance of Breast Cancer, Ovarian Cancer and Contralateral Breast Cancer in BRCA1 and BRCA2 Families: High Cancer Incidence at Older Age. Breast Cancer Res. Treat. 124, 643–651. doi:10.1007/s10549-010-0805-3
van der Lee, R., Correard, S., and Wasserman, W. W. (2020). Deregulated Regulators: Disease-Causing Cis Variants in Transcription Factor Genes. Trends Genet. 36, 523–539. doi:10.1016/j.tig.2020.04.006
van Karnebeek, C. D. M., and Stockler, S. (2012). Treatable Inborn Errors of Metabolism Causing Intellectual Disability: A Systematic Literature Review. Mol. Genet. Metabolism 105, 368–381. doi:10.1016/j.ymgme.2011.11.191
van Leeuwen, J., Pons, C., Boone, C., and Andrews, B. J. (2017). Mechanisms of Suppression: The Wiring of Genetic Resilience. BioEssays 39, 1700042. doi:10.1002/bies.201700042
van Moorsel, C. H. M., van der Vis, J. J., and Grutters, J. C. (2021). Genetic Disorders of the Surfactant System: Focus on Adult Disease. Eur. Respir. Rev. 30. doi:10.1183/16000617.0085-2020
van Rooij, J., Arp, P., Broer, L., Verlouw, J., van Rooij, F., Kraaij, R., et al. (2020). Reduced Penetrance of Pathogenic ACMG Variants in a Deeply Phenotyped Cohort Study and Evaluation of ClinVar Classification over Time. Genet. Med. doi:10.1038/s41436-020-0900-8
Vasilyev, S. A., Skryabin, N. A., Kashevarova, A. A., Tolmacheva, E. N., Savchenko, R. R., Vasilyeva, O. Y., et al. (2021). Differential DNA Methylation of the IMMP2L Gene in Families with Maternally Inherited 7q31.1 Microdeletions Is Associated with Intellectual Disability and Developmental Delay. Cytogenet. Genome Res. 161, 105–119. doi:10.1159/000514491
Velasco, G., and Francastel, C. (2019). Genetics Meets DNA Methylation in Rare Diseases. Clin. Genet. 95, 210–220. doi:10.1111/cge.13480
Viel, M., Hubert, D., Burgel, P.-R., Génin, E., Honoré, I., Martinez, B., et al. (2016). DCTN4 as a Modifier of chronicPseudomonas Aeruginosainfection in Cystic Fibrosis. Clin. Respir. J. 10, 777–783. doi:10.1111/crj.12288
Vu, V., Verster, A. J., Schertzberg, M., Chuluunbaatar, T., Spensley, M., Pajkic, D., et al. (2015). Natural Variation in Gene Expression Modulates the Severity of Mutant Phenotypes. Cell. 162, 391–402. doi:10.1016/j.cell.2015.06.037
Vuckovic, D., Bao, E. L., Akbari, P., Lareau, C. A., Mousas, A., Jiang, T., et al. (2020). The Polygenic and Monogenic Basis of Blood Traits and Diseases (Genetic and Genomic Medicine).
Wallace, D. R., Taalab, Y. M., Heinze, S., Tariba Lovaković, B., Pizent, A., Renieri, E., et al. (2020). Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 9, E901. doi:10.3390/cells9040901
Wallis, G. A., Starman, B. J., Zinn, A. B., and Byers, P. H. (1990). Variable Expression of Osteogenesis Imperfecta in a Nuclear Family Is Explained by Somatic Mosaicism for a Lethal Point Mutation in the Alpha 1(I) Gene (COL1A1) of Type I Collagen in a Parent. Am. J. Hum. Genet. 46, 1034–1040.
Walsh, R., Mazzarotto, F., Whiffin, N., Buchan, R., Midwinter, W., Wilk, A., et al. (2019). Quantitative Approaches to Variant Classification Increase the Yield and Precision of Genetic Testing in Mendelian Diseases: the Case of Hypertrophic Cardiomyopathy. Genome Med. 11, 5. doi:10.1186/s13073-019-0616-z
Walsh, R., Tadros, R., and Bezzina, C. R. (2020). When Genetic Burden Reaches Threshold. Eur. Heart J. 41, 3849–3855. doi:10.1093/eurheartj/ehaa269
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., McGuire, K. J., et al. (2017). Reassessment of Mendelian Gene Pathogenicity Using 7,855 Cardiomyopathy Cases and 60,706 Reference Samples. Genet. Med. 19, 192–203. doi:10.1038/gim.2016.90
Weedon, M. N., Cebola, I., Cebola, I., Patch, A.-M., Flanagan, S. E., De Franco, E., et al. (2014). Recessive Mutations in a Distal PTF1A Enhancer Cause Isolated Pancreatic Agenesis. Nat. Genet. 46, 61–64. doi:10.1038/ng.2826
Wei, J., Rahman, S., Ayaub, E. A., Dickhout, J. G., and Ask, K. (2013). Protein Misfolding and Endoplasmic Reticulum Stress in Chronic Lung Disease. Chest 143, 1098–1105. doi:10.1378/chest.12-2133
Wei, W.-H., Hemani, G., and Haley, C. S. (2014). Detecting Epistasis in Human Complex Traits. Nat. Rev. Genet. 15, 722–733. doi:10.1038/nrg3747
Weinhold, B. (2006). Epigenetics: The Science of Change. Environ. Health Perspect. 114, A160–A167. doi:10.1289/ehp.114-a160
Whiffin, N., Karczewski, K. J., Karczewski, K. J., Zhang, X., Chothani, S., Smith, M. J., et al. (2020). Characterising the Loss-Of-Function Impact of 5' Untranslated Region Variants in 15,708 Individuals. Nat. Commun. 11, 2523. doi:10.1038/s41467-019-10717-9
White, M. C., Holman, D. M., Boehm, J. E., Peipins, L. A., Grossman, M., and Jane Henley, S. (2014). Age and Cancer Risk. Am. J. Prev. Med. 46, S7–S15. doi:10.1016/j.amepre.2013.10.029
Williamson, I., Kane, L., Devenney, P. S., Flyamer, I. M., Anderson, E., Kilanowski, F., et al. (2019). Developmentally Regulated Shh Expression Is Robust to TAD Perturbations. Development 146, dev179523. doi:10.1242/dev.179523
Wojcik, M. H., Zhang, T., Ceyhan-Birsoy, O., Genetti, C. A., Lebo, M. S., Yu, T. W., et al. (2021). Discordant Results between Conventional Newborn Screening and Genomic Sequencing in the BabySeq Project. Genet. Med. 23, 1372–1375. doi:10.1038/s41436-021-01146-5
Wonkam, A., Chimusa, E. R., Mnika, K., Pule, G. D., Ngo Bitoungui, V. J., Mulder, N., et al. (2020). Genetic Modifiers of Long-Term Survival in Sickle Cell Anemia. Clin. Transl. Med. 10, e152. doi:10.1002/ctm2.152
Wright, C. F., FitzPatrick, D. R., and Firth, H. V. (2018). Paediatric Genomics: Diagnosing Rare Disease in Children. Nat. Rev. Genet. 19, 253–268. doi:10.1038/nrg.2017.116
Wright, C. F., Prigmore, E., Rajan, D., Handsaker, J., McRae, J., Kaplanis, J., et al. (2019b). Clinically-relevant Postzygotic Mosaicism in Parents and Children with Developmental Disorders in Trio Exome Sequencing Data. Nat. Commun. 10, 2985. doi:10.1038/s41467-019-11059-2
Wright, C. F., Quaife, N. M., Ramos-Hernández, L., Danecek, P., Ferla, M. P., Samocha, K. E., et al. (2021). Non-coding Region Variants Upstream of MEF2C Cause Severe Developmental Disorder through Three Distinct Loss-Of-Function Mechanisms. Am. J. Hum. Genet. 108, 1083–1094. doi:10.1016/j.ajhg.2021.04.025
Wright, C. F., West, B., Tuke, M., Jones, S. E., Patel, K., Laver, T. W., et al. (2019a). Assessing the Pathogenicity, Penetrance, and Expressivity of Putative Disease-Causing Variants in a Population Setting. Am. J. Hum. Genet. 104, 275–286. doi:10.1016/j.ajhg.2018.12.015
Wu, S., Cho, E., Li, W.-Q., Weinstock, M. A., Han, J., and Qureshi, A. A. (2016). History of Severe Sunburn and Risk of Skin Cancer Among Women and Men in 2 Prospective Cohort Studies. Am. J. Epidemiol. 183, 824–833. doi:10.1093/aje/kwv282
Xue, Y., Chen, Y., Ayub, Q., Huang, N., Ball, E. V., Mort, M., et al. (2012). Deleterious- and Disease-Allele Prevalence in Healthy Individuals: Insights from Current Predictions, Mutation Databases, and Population-Scale Resequencing. Am. J. Hum. Genet. 91, 1022–1032. doi:10.1016/j.ajhg.2012.10.015
Yokoshi, M., Segawa, K., and Fukaya, T. (2020). Visualizing the Role of Boundary Elements in Enhancer-Promoter Communication. Mol. Cell. 78, 224–235. e5. doi:10.1016/j.molcel.2020.02.007
Young, S. K., Palam, L. R., Wu, C., Sachs, M. S., and Wek, R. C. (2016). Ribosome Elongation Stall Directs Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 291, 6546–6558. doi:10.1074/jbc.m115.705640
Zernant, J., Lee, W., Nagasaki, T., Collison, F. T., Fishman, G. A., Bertelsen, M., et al. (2018). Extremely Hypomorphic and Severe Deep Intronic Variants in the ABCA4 Locus Result in Varying Stargardt Disease Phenotypes. Cold Spring Harb. Mol. Case Stud. 4, a002733. doi:10.1101/mcs.a002733
Zhang, L. X., Lemire, G., Gonzaga-Jauregui, C., Molidperee, S., Galaz-Montoya, C., Liu, D. S., et al. (2020). Further Delineation of the Clinical Spectrum of KAT6B Disorders and Allelic Series of Pathogenic Variants. Genet. Med. 22, 1338–1347. doi:10.1038/s41436-020-0811-8
Zhang, M., Lu, L., Wei, B., Zhang, Y., Li, X., Shi, Y., et al. (2020). Brachydactyly Type A3 Is Caused by a Novel 13 Bp HOXD13 Frameshift Deletion in a Chinese Family. Am. J. Med. Genet. 182, 2432–2436. doi:10.1002/ajmg.a.61788
Zhang, X., Walsh, R., Whiffin, N., Buchan, R., Midwinter, W., Wilk, A., et al. (2021). Disease-specific Variant Pathogenicity Prediction Significantly Improves Variant Interpretation in Inherited Cardiac Conditions. Genet. Med. 23, 69–79. doi:10.1038/s41436-020-00972-3
Keywords: penetrance, expressivity, variant intepretation, genomic sequencing, rare disease
Citation: Kingdom R and Wright CF (2022) Incomplete Penetrance and Variable Expressivity: From Clinical Studies to Population Cohorts. Front. Genet. 13:920390. doi: 10.3389/fgene.2022.920390
Received: 14 April 2022; Accepted: 09 June 2022;
Published: 25 July 2022.
Edited by:
Laura V Milko, University of North Carolina at Chapel Hill, United StatesReviewed by:
Rachita Yadav, Harvard Medical School, United StatesCopyright © 2022 Kingdom and Wright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline F. Wright, Y2Fyb2xpbmUud3JpZ2h0QGV4ZXRlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.