
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 06 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.916340
This article is part of the Research TopicThe Genetics of Human Mendelian Skin DisordersView all 11 articles
 Yupei Wang1,2
Yupei Wang1,2 Chuan Zhang1,2
Chuan Zhang1,2 Bingbo Zhou1,2Ling Hui1,2Lei Zheng1,2Xue Chen1,2Shifan Wang1,2Lan Yang1,2Shengju Hao1,2
Bingbo Zhou1,2Ling Hui1,2Lei Zheng1,2Xue Chen1,2Shifan Wang1,2Lan Yang1,2Shengju Hao1,2 Qinghua Zhang1,2*
Qinghua Zhang1,2*Background: Ectodysplasin A (EDA) variations are major pathogenic factors for hypohidrotic ectodermal dysplasia (HED), the most common form of ectodermal dysplasia (ED), characterized by hypotrichosis, hypohidrosis, hypodontia, and other oral features.
Methods: Molecular genetic defects in three HED families were detected by whole-exome sequencing and confirmed by Sanger sequencing or multiplex ligation-dependent probe amplification. The effect of splicing variant was further verified by EDA minigene in vitro analysis. De novo deletion was confirmed by chromosomal microarray analysis.
Results: Three variants (c.396 + 1 G > C, c.171-173 del GTT, and exon 1 deletion) were identified, all affecting exon 1 of the EDA gene. Variants c.396 + 1 G > C and c.171-173 del GTT were first identified. Minigene analysis of the splicing variant (c.396 + 1 G > C) displayed a prolonged EDA-A1 transcript containing extra 699 bp at the start of intron 1, representing a functional cryptic splice site formation in vitro. Combining the results of chromosomal microarray analysis and whole-exome sequencing, the deletion variant was over 87 kb. Three variants were predicted to affect protein function to differing degrees, and were responsible for X-linked HED with varying phenotype.
Conclusion: Investigating the clinical and molecular characteristics of these variations broadens our understanding of EDA gene variants, supporting clinical diagnosis, genetic counseling, and prenatal diagnosis of HED.
Ectodermal dysplasia (ED) is a set of genetic diseases with two or more abnormally developed ectoderm-derived structures, including hair, teeth, nails, skin, and sweat glands (Pigno et al., 1996; Vaidya et al., 2013; Wright et al., 2019). Of the 200 different types of ED reported, ∼30 causative genes have been identified at the molecular level (Itin and Fistarol, 2004). Genetic variations in ectodysplasin A (EDA) pathway genes, such as EDA, ectodysplasin A receptor (EDAR), and EDAR-associated adapter protein (EDARADD) are known to be associated with hypohidrotic ED (HED); the prevalence of which is ∼1/100,000 (Khabour et al., 2010; Deshmukh and Prashanth, 2012; Okita et al., 2019). EDA is a unique gene involved in the pathogenesis of X-linked hypohidrotic ectodermal dysplasia (XLHED; OMIM 305100), which accounts for 95% of patients with HED, and the remaining 5% are mainly due to autosomal dominant or recessive inheritance (Clarke et al., 1987; Clauss et al., 2008; Deshmukh and Prashanth, 2012; Wang et al., 2020). XLHED is characterized by a clinical triad of hypodontia (congenital absence of teeth), hypoplasia of sweat glands, and hypotrichosis (sparse hair) (Moura et al., 2019).
The EDA gene is located on chromosome Xq12-q13.1, which has nine exons, and consists of four domains: a transmembrane domain, furin recognition sequences, a collagen domain, and β-sheets A−H of the tumor necrosis factor (TNF) homology domain, encoded by exon 1, 3, 5–6 and 7–9, respectively (Schneider et al., 2001). It was identified as a membrane-bound signaling molecule of the TNF superfamily (Trzeciak and Koczorowski, 2016). Variations in the EDA gene lead to loss or dysfunction of EDA1, associated with the signaling of the epithelial–mesenchymal transition during embryogenesis, as well as the initiation and development of ectodermal derivatives (Korber et al., 2020). At least 100 variants of the EDA gene have been identified as pathogenic mutations in the NCBI ClinVar database (http://www.ncbi.nlm.nih.gov) based on published papers and submissions (Liu et al., 2012; Huang et al., 2015).
In the present study, we identified three variants of the EDA gene in three Chinese families with XLHED, and demonstrated that variants led to varying clinical phenotypes through different molecular mechanisms affected by exon 1 of the EDA gene.
Patients and their parents attended the Medical Genetics Centre, Gansu Provincial Maternity, and Childcare Hospital (Lanzhou, China), seeking genetic diagnosis of congenital symptoms (no sweating, pyrexia, and dysplasia of hair and teeth). Proband 1 was a 30-year-oldman; proband 2 was a 10-month-old boy; and proband 3 was a 20-day-old boy. Their parents were healthy and unrelated. All participants gave their signed informed consent for genetic studies before collecting blood samples or performing clinical evaluations.
Peripheral blood (3–5 ml) was collected from proband family members for DNA extraction using a Genomic DNA Extraction kit (Tiangen Biotech, Beijing, China), and extracted genomic DNA was subsequently used for targeted whole-exome sequencing (WES) and Sanger sequencing.
Trio WES was carried out by MyGenostics Co., Ltd (Beijing, China). Briefly, qualified genomic DNA was fragmented randomly to an average size of 180 bp with a Bioruptor sonicator (Diagenode, Liege, NJ, United States). The fragmented DNA was then repaired and A-tails were ligated to the 3' end. Next, Illumina adapters (Illumina Inc., United States) were ligated to the fragments, and adapted DNA templates were amplified by PCR. DNA was then captured using a GenCap Custom Enrichment kit (MyGenosticsGenCap Enrichment Technologies, Beijing, China) and sequenced on an Illumina HiSeq 2500 platform (Illumina Inc.) as paired-end 90 bp reads. The mean sequencing depth was >100. N20 reads covered targeted bases by >95%.
Using the Trim Galore program, reads of low quality and adapters were filtered out after sequencing. SOAP aligner (SOAP v2.21) was used to align clean reads to the h19 human reference genome (UCSC Genome Browser hg19). Insertions, deletions, and single-nucleotide polymorphisms (SNPs) were identified by the Burrows–Wheeler alignment program (0.7.12-r1044) and tested using a GATK tool kit. The exome assistant program was used to annotate the locations of exonic, intronic, and intergenic regions, as well as protein-coding effects such as synonymous, missense, nonsense, and frameshift (Xu et al., 2015). Frequency and function were the main factors used to obtain candidate variants for further analysis. For the frequency filter, a 0.01 cut-off was applied according to allele frequency estimates from NCBI dbSNP (v152; http://www.ncbi.nlm.nih.gov/projects/SNP/), 1,000Gome (http://www.ncbi.nlm.nih.gov/Ftp/), and Exome Aggregation Consortium (http://gnomad.broadinstitute.org/) databases. Synonymous and missense variants, which were predicted to be benign or tolerated in Sorting Intolerant From Tolerant (SIFT), PolyPhen-2, Mutation Taster, and GERP++, were removed by the functional filter. Splicing variants were evaluated by MaxEntScan and dbscSNV11.
In order to identify the target variants associated with HED in family members, Sanger sequencing was performed. Direct PCR products were sequenced using BigDye Terminator v3.1 Cycle Sequencing kits (Applied Biosystems, Foster City, CA, United States) and analyzed on an ABI3500DX Genetic Analyzer (Applied Biosystems, Warrington, United Kingdom) using EDA primers (pedigree 1, Forward, 5′-actccactctgactcccaggac-3’; Reverse, 5′-ctggtcctgccctctaaattg-3’; pedigree 2, Forward, 5′-gcctcaagagagtgggtgtc-3’; Reverse, 5′-gtcctgggagtcagagtgga-3′).
In order to further investigate the pathogenic mechanism of the splicing variant at the 5’ (donor) splice site (5’ss; c.396 + 1 G > C), a minigene containing exon 1, partial intron 1, and exon 2 of the EDA gene were designed using exon-trapping pEGFP-C1 plasmids. Specifically, exon 1, the first and last 1,000 bp of intron 1 (the intermediate sequence of intron 1 was deleted), and exon 2 encoding wild-type (WT) or mutant type (MT) sequences were incorporated into the pEGFP-C1 vector within the 5′ Xhol and 3′ BamHI restriction enzyme sites using specific primers. WT and MT expression vector construction was performed by Hitrobio Tech, Beijing, China. All recombinant plasmids were validated by direct sequencing. Human embryonic kidney 293T (HEK 293T) cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, MA, United States), penicillin (100 U/L), and streptomycin (100 mg/L) at 37°C with 5% CO2. Transfection was performed using HEK 293T cells grown to 70–80% confluence in the serum-free medium by Lipofectamine 2000 Reagent (Thermo Fisher Scientific) following the manufacturer’s instructions, and cells were collected at 48 h after transfection. For RT-PCR analysis, a MiniBEST Universal RNA Extraction kit (Takara, Dalian, China) was used to extract total RNA and a PrimerScript RT Reagent kit (Takara) was used for reverse transcription. PCR amplification of minigene transcripts was conducted using vector-specific forward primer pEGFP-C-5 F (5′-CATGGTCCTGCTGGAGTTCGTG-3′) and reverse primer pEGFP-C-3 R (5′-ATCTCAGTGGTATTTGTGAGC-3′). PCR products were identified by 1% agarose gel electrophoresis, and Sanger sequencing was performed to analyze mutant patterns.
MLPA was performed on the pedigree 3 using a SALSA MLPA Probemix P183 kit (MRC-Holland, Amsterdam, the Netherlands; http://www.mlpa.com). This kit included probes from all exons of EDARR, EDAR, EDA, and WINT10A, all associated with HED. The assay was performed according to the manufacturer’s protocol. Briefly, 100 ng genomic DNA was denatured at 98°C for 5 min then allowed to hybridize to the MLPA probe overnight. Ligation reactions were then performed using Ligase-65 enzyme and PCR was carried out with SALSA PCR primers. PCR products were separated by capillary electrophoresis on an ABI 3500 Genetic Analyzer (Applied Biosystems). Original data were analyzed by Gene mapper 4.0 and Coffalyser.Net software, and copy number was calculated according to the MLPA kit instructions.
CMA was performed with a CytoScan 750K array (Affymetrix, Santa Clara, CA, United States) according to the manufacturer’s recommendations. Genomic DNA of 40 ng/μL was digested, ligated with adaptors, amplified, purified, labeled, and then hybridized into the array. After the completion of hybridization, the array was washed with buffer, stained, and scanned with a laser scanner. Data were analyzed with Chromosome Analysis Suite (ChAS) (version 4.2.0.80) software. The hg19/GRCh37 genome was used for genomic assembly. All identified variants were further analyzed with reference to public databases including Database of Genomic Variants (DGV, http://projects.tcag.ca/variation), the 1,000 Genome Project (http://browser.1000genomes.org), DECIPHER (http://decipher.sanger.ac.uk/), gnomAD (http://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and OMIM (https://www.omim.org).
Proband 1 presented a distinctive facial appearance with sparse hair and eyelashes, and no eyebrows, along with a flat nose, thick lips, and albino around the lips (Figure 1A). His uncle (III3) and uncle’s grandpa (II7) were both HED patients presenting the same symptoms. Probands 2 and 3 showed a similar phenotype with sparse hair, dry skin, and frequent fever. As early as the fetus stage, alveolar bone dysplasia was identified in proband 3 by sonographic examination, indicating thinner upper alveolar bones and fewer tooth germs compared to normal fetus, as we reported previously (Li et al., 2021). After birth, his skin appeared abnormally dry and wrinkled (Figure 3B) and his upper alveolar bones were thinner compared with normal. Proband 2 also appeared to have thinner alveolar bone at 10 months. A preliminary diagnosis of HED was made by a dermatologist based on the clinical manifestations presented by all probands (Julia et al., 2018). The varying symptoms of the three patients are shown in Table 1.
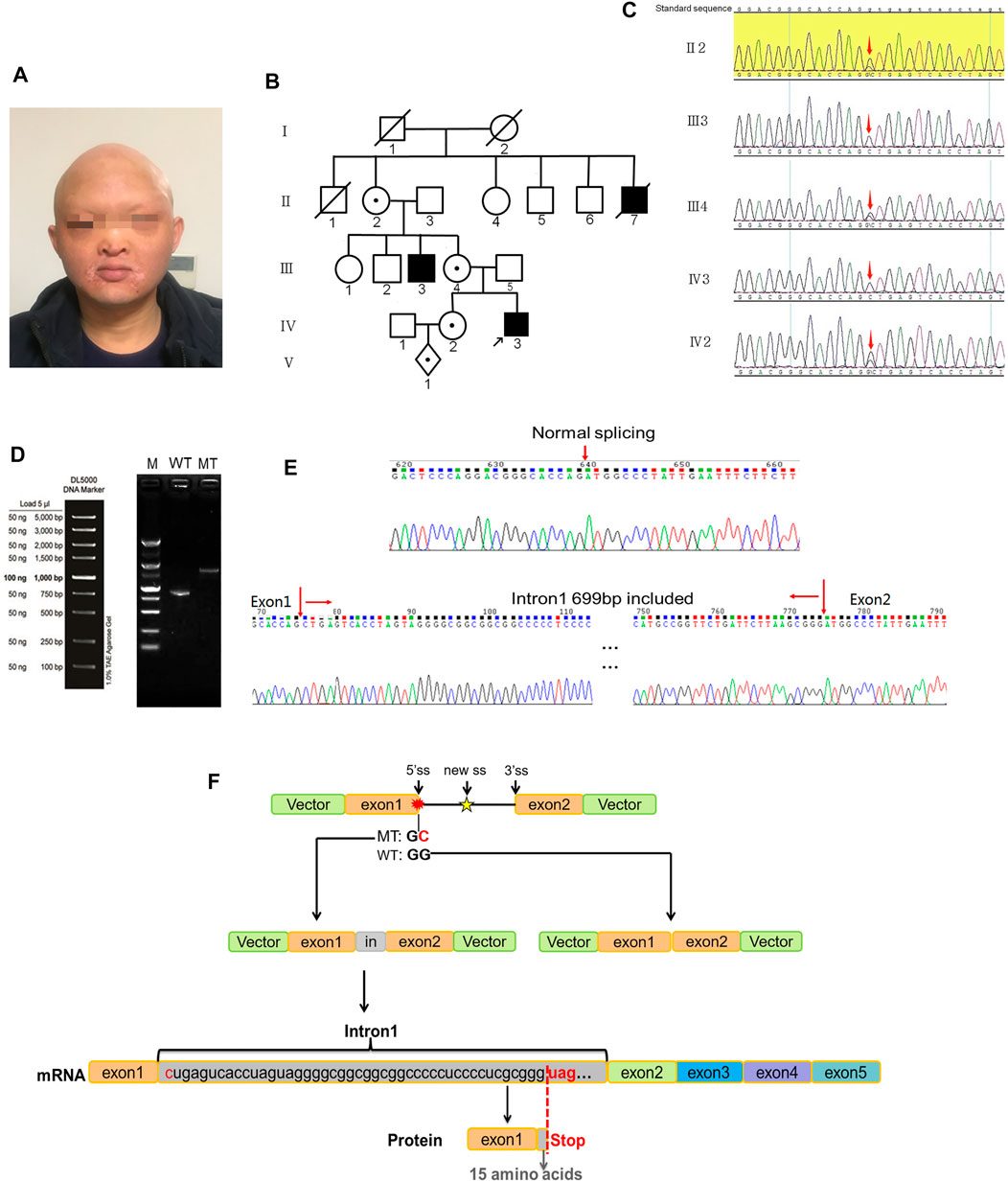
FIGURE 1. Variants in case 1. (A) Frontal photograph of proband 1 showing various developmental defects of ectodermal appendages including protuberant lips, short chin, and flat nose. (B) Family tree showing that the proband’s uncle (III3) and uncle’s grandpa (II7) were HED positive. (C) Validation of the mutation site by Sanger sequencing. The red arrow indicates the mutated base. (D) Electrophoretogram results of transcripts generated from the transfected WT and MT EDA minigenes. Lane M, 5,000 bp markers; Lane WT, WT EDA minigene transcripts showing a single band of 821 bp; Lane MT, MT minigenes containing mutant alleles showing a longer band (the empty lane has been cropped). (E) Inclusion of an extra 699 nucleotides in intron 1 validated by Sanger sequencing. (F) The splicing and transcription pattern of MT EDA pre-mRNA.
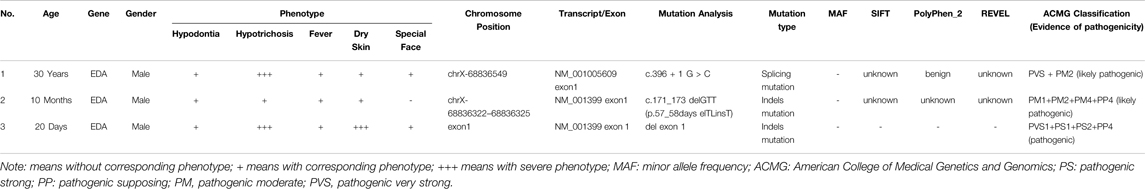
TABLE 1. The genetic characteristics of EDA gene in patients.
A hemizygous splicing variant of the EDA gene (c.396 + 1 G > C) was identified in pedigree 1 (Figure 1C). The variant resulted in the destruction of the splicing donor. There is no information in the 1,000 Genomes, MyGenosticsInhouse, ESP6500, EXAC, or ExAC_EAS population databases about this variant. It was predicted to be deleterious by MaxEntScan following analysis of variants near the 5′ and 3′ splice sites. Additionally, dbscSNV11 predicted that the variant was deleterious. The hemizygous variant c.396 + 1 G > C of the EDA gene was verified by Sanger sequencing, and it was judged to be pathogenic based on American College of Medical Genetics and Genomics (ACMG) Guidelines. Furthermore, II2, III4, and IV2 carried the same variant, while III3 was a patient and died (Figure 1B). Employing amniotic fluid puncture for prenatal diagnosis, fetus V2 was detected as a carrier with EDA gene heterozygous variant c.396 + 1 G > C. Similarly, proband 2 and proband 3 were identified as c.171-173 del GTT and exon 1 deletion hemizygous variants of the EDA gene respectively. Sanger sequencing was then performed on available lineage members (Figure 2B). The results showed that the variant in proband 2 (c.171-173 del GTT) was inherited from his mother (Figure 2A). However, MLPA showed the mother of proband 3 did not carry the same variant, indicating a de novo deletion variant (Figure 3D). Based on raw data of WES and probe coverage of MLPA, boundaries of this deletion established at the upstream and downstream regions of exon 1. The length of the deletion can be roughly estimated by CytoScan750k gene chip for its composition of 550,000 non-polymorphic CNV probes and more than 200,000 SNP probes. According to the position of the missing SNP, there is a deletion of 82,724 bp in the X chromosome (chrX: 68,748,640–68,831,364), which belong to the upstream region of EDA gene (Figure 3E). At downstream of missing SNP region, there are 6314 bp length without SNP coverage which contain upstream of exon 1, exon 1 and partial intron 1 of EDA gene. Combining the results of CMA, MLPA, and WES, the absolute deletion length extended to downstream of exon 1, which is 87,938 bp (chrX: 68,748,640–68,836,578) including upstream of exon 1 (87,513 bp) and exon 1 (396 bp), and downstream of exon 1 (29 bp). In fact, the deletion length might be longer. Detailed information for EDA variations is shown in Table 1. Variants c.396 + 1 G > C and c.171-173 del GTT are reported herein for the first time.
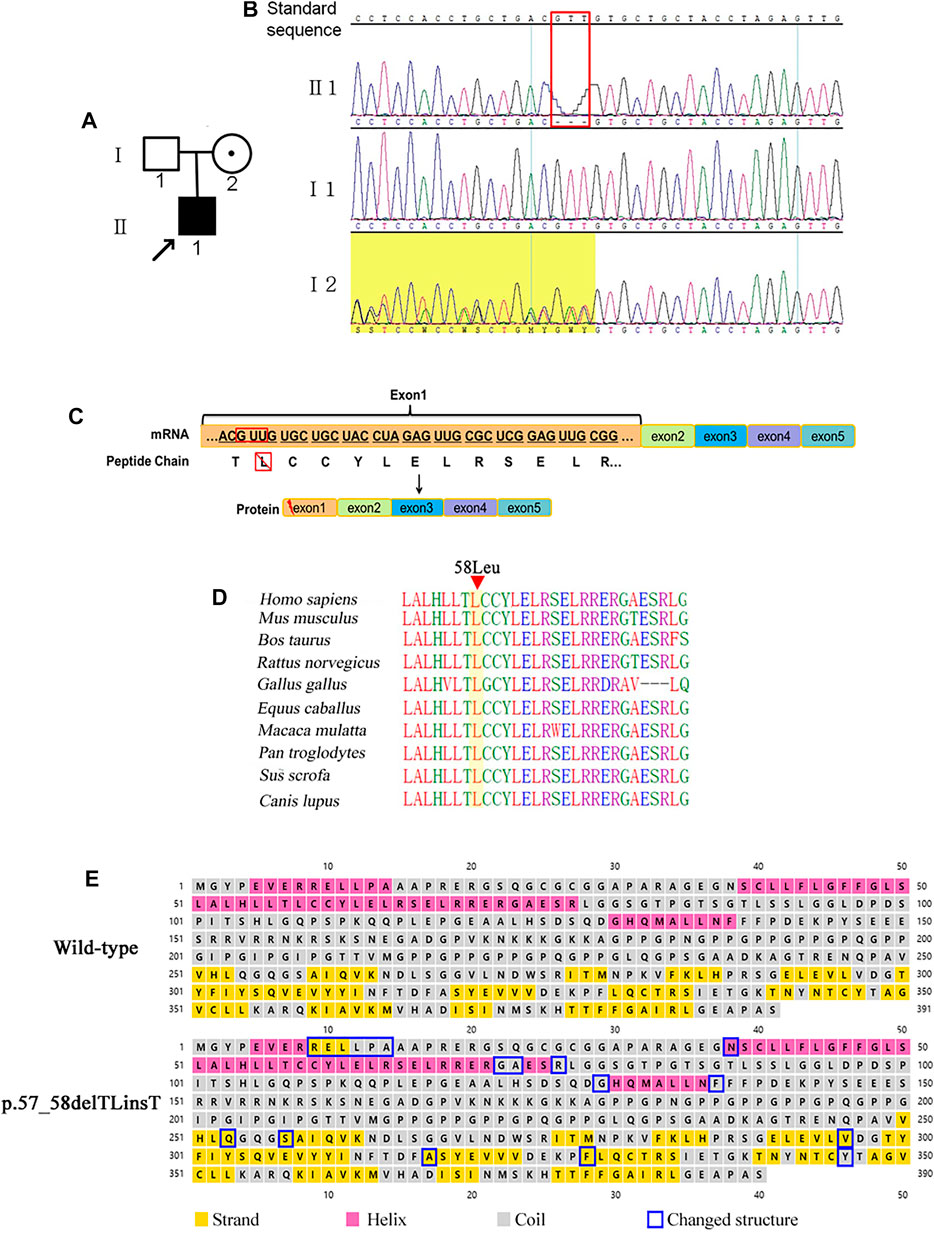
FIGURE 2. Variants in case 2. (A) Family tree showing the mutations inherited from I2. (B) Validation of the mutation site (c.171_173delGTT) by Sanger sequencing. (C) Transcription pattern. (D) Conservation analysis of affected amino acids in the EDA protein among 10 different mammalian species. (E) Secondary structure prediction of the WT EDA protein and the p.57_58delTLinsT mutant.
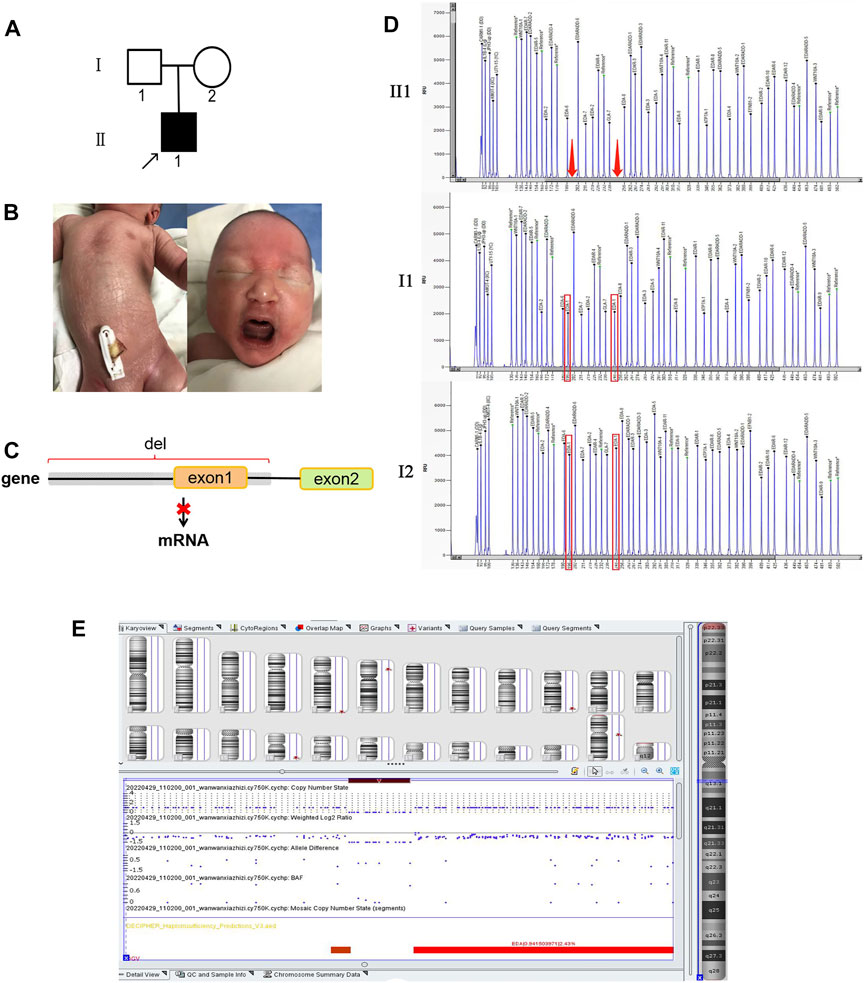
FIGURE 3. Variants in case 3. (A) Family tree showing a de novo mutation. (B) Dry skin of proband 3. (C) Transcription pattern. (D) MLPA results show deletion of exon 1. (E) CMA profile showing a loss of 82 kb of Xq (chrX: 68,831,364–68,748,640; hg19) (dark red box) which is upstream of the EDA gene. Images were generated using the Chromosome Analysis Suite (ChAS) software.
RT-PCR assays were performed to investigate transcripts generated from transfected WT and MT EDA minigenes. The WT EDA minigenes produced a single band of 821 bp comprising the expected pEGFP-C-5′, exon 1, exon 2, and pEGFP-C-3′ regions. However, the MT RT-PCR products showed a longer band (Figure 1D). Sequence analysis of the abnormal products revealed the inclusion of the first 699 nucleotides from intron 1, which may be attributed to the presence of a cryptic 5′ splice site in intron 1 because the AG sequence was observed at positions 700 and 701 of intron 1 (Figure 1E).
For proband 1, inclusion of the first 699 nucleotides of intron 1 could potentially result in a redundant reading frame in exon 1 of the EDA gene, and ultimately generated a longer peptide. However, there was a stop codon at positions c.396 + 46–48 that could lead to abnormal truncated EDA-A1 proteins or nonsense-mediated mRNA decay (NMD; Figure 2F). Therefore, c.396 + 1 G > C was judged to be a loss-of-function variant.
For proband 2, variants of c.171-173 del GTT resulted in a single amino acid deletion of the leucine codon (Figure 2C). The mutation taster tool predicted this to be a disease-causing variation. Comparative sequence alignment was performed across most mammals using the T-Coffee Multiple Sequence Alignment Program (http://www.ebi.ac.uk/Tools/msa/tcoffee). The results showed that this amino acid has been conserved in mammals during evolution, and is therefore important for protein structure and/or function (Figure 2D). Furthermore, this variation might cause changes in the α-helical and β-sheet secondary structure components of the EDA protein predicted by Psipred 4.0 software (Figure 2E).
For proband 3, MLPA was applied to confirm the absence of exon 1 of the EDA gene (Figure 3D). The results showed that two peaks representing exon 1 were absent in proband 3 but not in his parents. The detection of CMA indicated that there was a large fragment deletion, including the upstream region (87 kb) of the EDA gene, which completely destroyed the initiation of transcription of EDA and caused the failure of EDA protein synthesis (Figure 3C).
According to the HGMD Professional database (2017.2), there are 355 registered variations of the EDA1 gene, of which 31 occur in the intron region. In the three HED pedigrees, an unreported 5’ss variant (c.396 + 1 G > C) of the EDA gene was found, and the splicing alteration mechanism was confirmed by minigene in vitro. The variant led to aberrant pre-mRNA splicing in exon 1 of the EDA gene, which generates a longer transcript with an extra 699 nucleotides in intron 1. In most cases (98.7%), GT and AG sequences are canonical splice sequences at the 5′ and 3′ ends of the intron, respectively, that define exon–intron boundaries for spliceosome recognition. The 5′ donor splice site variants at + 1 and +2 positions, as well as the 3′ acceptor splice site variants at–1 and −2 positions, are considered pathogenic (Caridi et al., 2016; Anna and Monika, 2018; Ma et al., 2019). Furthermore, variant c.526+1G > A in the EDA splice donor site caused the complete omission of exon 3, which resulted in abnormal truncated EDA-A1 proteins (Liu et al., 2018). In addition, c.396 + 5 G > A and c.396 + 2 T > C variants were also reported in HED cases without further research.
Splicing variants can be summarized into four types according to the impact on the final composition of mRNA: 1) Exon skipping, in which an authentic splice site variant and a variant within an exon usually result in whole-exon skipping; 2) cryptic exon inclusion, in which inclusion of a subsequent intron (pseudo exon) is caused by a deeply intronic nucleotide variant; 3) exon sequence removal, in which activation of cryptic splice sites caused by a single-nucleotide variant in an exon results in exclusion of exonic sequences; and 4) intron inclusion, in which intronic sequence inclusion generated by a variant in the intron or authentic site leads to the generation of a cryptic intronic splice site (Wimmer et al., 2007; Anna and Monika, 2018). Variants at the canonical splice sequence either result in skipping of one or more adjacent exons, or activation of a cryptic splice site of the same type in a neighboring exon site. In case 1, the positions of c.369 + 700–701 present alternative GT nucleotides as a stronger cryptic intronic splice site described in type 4, resulting in inclusion of an intron fragment instead of exon skipping. For 5’ss, a highly significant correlation was observed between the mutational consequences at the RNA level and the number of potentially utilizable cryptic donor splice sites in the region around the affected splice site (Buratti et al., 2007). More potential splice sites in the region around the affected splice site mean an increased probability of inclusion of introns (Kovacova et al., 2020). In case 1, 32 potentially utilizable GT nucleotides upstream of the chosen splice site were ignored, and only one aberrant transcript was generated in the minigene assay. Studies show that recognition of the 5’ss in a pre-mRNA is initiated by the formation of a base pairing interaction between the splice site sequence and particular sequences in the U1 snRNA of the spliceosome (Liu et al., 2017). Hence, the chosen cryptic donor splice site may be associated with a strong splicing motif for greater sequence complementarity, leading to the 5′ end of U1 snRNP binding with higher affinity.
Variant c.171-173 del GTT leads to p.57-58 del TL ins T in the EDA gene, predicted as unknown by PolyPhen_2, SIFT, and REVEL. In addition, the HGMD database has no relevant reports for this locus, hence it was evaluated as uncertain by ACMG. However, the symptoms of hypohidrosis, hypotrichosis, and thickness of the alveolar arch strongly support the clinical diagnosis of XLHED. This represents new pathogenic evidence for this variant. Variant with full deletion of exon 1 was first reported in Finland (Pääkkönen et al., 2001). In China, we previously reported the ultrasonic phenotype of this XLHED with thinner alveolar bone than a normal fetus. After birth, the infant’s upper alveolar bones were also thinner compared with normal. Hence, it is important to take the thickness of the alveolar arch into consideration for investigating intrauterine or infancy dental development.
Although all three variants affected exon 1, the results were different between variants, as we predicted. For proband 1, there was an extra stop codon in intron 1 that could lead to NMD and decreased expression of the abnormal truncated EDA-A1 protein. For proband 2, the variant led to loss of leucine in exon 1, which could affect the transmembrane transportation domain of the EDA-A1 protein. For proband 3, the variant led to loss of exon 1 and upstream region, which completely destroyed the initiation of transcription and translation. The wild-type EDA protein could not be produced normally. Correspondingly, the symptoms in proband 3 were the most severe in terms of skin and hypotrichosis, whereas proband 2 did not exhibit severe dry skin.
In conclusion, we investigated the genetic and clinical features of patients with XLHED. Three variants located in or affecting exon 1 of the EDA gene were identified, including two novel variants of the splicing donor site (c.396 + 1 G > C) and c.171-173 del GTT. We further demonstrated the role of c.396 + 1 G > C in altering gene transcription (creating a cryptic 5′ splice site in exon 1) in vitro, which facilitates accurate prenatal diagnosis and genetic counseling for other family members of pedigree 1. As there is still no effective treatment for XLHED, our findings also broaden our knowledge of the EDA gene in HED patients, and can be used as a reference for clinical disease screening, diagnosis, and genetic counseling.
The datasets for this article are not publicly available due to participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethical Review Committee of Gansu Provincial Maternity and Child-Care Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YW: conceptualization, methodology, writing (original draft preparation); CZ, BZ, LH, LZ: software, data mining, revising and approval of the version; XC, SW, LY, SH: experiment conduction, data mining, investigation; QZ: supervision, writing (reviewing), editing.
This work was supported by the National Natural Science Foundation of China (12005042), the Gansu Provincial Natural Science Foundation (21JR1RA045, 21JR7RA680), Gansu Provincial Health Industry Scientific Research Program (GSWSKY2021-022), and the Lanzhou Talent Innovation Project (2018-RC-95).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all patients and their family members for their participation in this study.
Anna, A., and Monika, G. (2018). Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 59, 253–268. doi:10.1007/s13353-018-0444-7
Buratti, E., Chivers, M., Královičová, J., Romano, M., Baralle, M., Krainer, A. R., et al. (2007). Aberrant 5′ Splice Sites in Human Disease Genes: Mutation Pattern, Nucleotide Structure and Comparison of Computational Tools that Predict Their Utilization. Nucleic Acids Res. 35, 4250–4263. doi:10.1093/nar/gkm402
Caridi, G., Thomas, W., Campagnoli, M., Lugani, F., Galliano, M., and Minchiotti, L. (2016). A Novel Splicing Mutation in the Albumin Gene (c.270+1G>T) Causes Analbuminaemia in a German Infant. Ann. Clin. Biochem. 53, 615–619. doi:10.1177/0004563215618223
Clarke, A., Phillips, D. I., Brown, R., and Harper, P. S. (1987). Clinical Aspects of X-Linked Hypohidrotic Ectodermal Dysplasia. Archives Dis. Child. 62, 989–996. doi:10.1136/adc.62.10.989
Clauss, F., Manière, M.-C., Obry, F., Waltmann, E., Hadj-Rabia, S., Bodemer, C., et al. (2008). Dento-craniofacial Phenotypes and Underlying Molecular Mechanisms in Hypohidrotic Ectodermal Dysplasia (HED): a Review. J. Dent. Res. 87, 1089–1099. doi:10.1177/154405910808701205
Deshmukh, S., and Prashanth, S. (2012). Ectodermal Dysplasia: a Genetic Review. Int. J. Clin. Pediatr. Dent. 5, 197–202. doi:10.5005/jp-journals-10005-1165
Huang, S. X., Liang, J. L., Sui, W. G., Lin, H., Xue, W., Chen, J. J., et al. (2015). EDA Mutation as a Cause of Hypohidrotic Ectodermal Dysplasia: a Case Report and Review of the Literature. Genet. Mol. Res. 14, 10344–10351. doi:10.4238/2015.august.28.21
Itin, P. H., and Fistarol, S. K. (2004). Ectodermal Dysplasias. Am. J. Med. Genet. 131C, 45–51. doi:10.1002/ajmg.c.30033
Julia, R. R., Isabel, M.-R. M., Efraín, G.-G., and Glustein, P. M. (2018). Hypohidrotic Ectodermal Dysplasia: Clinical and Molecular Review. Int. J. Dermatology 57, 965–972. doi:10.1111/ijd.14048
Khabour, O. F., Mesmar, F. S., Al-Tamimi, F., Al-Batayneh, O. B., and Owais, A. I. (2010). Missense Mutation of the EDA Gene in a Jordanian Family with X-Linked Hypohidrotic Ectodermal Dysplasia: Phenotypic Appearance and Speech Problems. Genet. Mol. Res. 9, 941–948. doi:10.4238/vol9-2gmr810
Korber, I., Klein, O. D., Morhart, P., Faschingbauer, F., Grange, D. K., Clarke, A., et al. (2020). Safety and Immunogenicity of Fc-EDA, a Recombinant Ectodysplasin A1 Replacement Protein, in Human Subjects. British: J Clin Pharmacol.
Kovacova, T., Soucek, P., Hujova, P., Freiberger, T., and Grodecka, L. (2020). Splicing Enhancers at Intron-Exon Borders Participate in Acceptor Splice Sites Recognition. Int. J. Mol. Sci. 21.
Li, T. g., Ma, B., Tie, H. x., Zhang, Q. h., Hao, S. j., and Guan, C. l. (2021). Prenatal Sonographic Diagnosis of X‐linked Hypohidrotic Ectodermal Dysplasia: An Unusual Case. J. Clin. Ultrasound 49, 838–840. doi:10.1002/jcu.23020
Liu, G., Wang, X., Qin, M., Sun, L., and Zhu, J. (2018). A Novel Splicing Mutation of Ectodysplasin A Gene Responsible for Hypohidrotic Ectodermal Dysplasia. Oral Dis. 24, 1101–1106. doi:10.1111/odi.12874
Liu, W., Li, X., Liao, S., Dou, K., and Zhang, Y. (2017). Activation of the Intronic Cryptic 5′ Splice Site Depends on its Distance to the Upstream Cassette Exon. Gene 619, 30–36. doi:10.1016/j.gene.2017.03.023
Liu, Y., Yu, X., Wang, L., Li, C., Archacki, S., Huang, C., et al. (2012). Mutation p.Leu354Pro in EDA Causes Severe Hypohidrotic Ectodermal Dysplasia in a Chinese Family. Gene 491, 246–250. doi:10.1016/j.gene.2011.10.009
Ma, D., Tan, J., Zhou, J., Zhang, J., Cheng, J., Luo, C., et al. (2019). A Novel Splice Site Mutation in the UBE2A Gene Leads to Aberrant mRNA Splicing in a Chinese Patient with X-Linked Intellectual Disability Type Nascimento. Mol. Genet. Genomic Med. 7, e976. doi:10.1002/mgg3.976
Moura, E., Rotenberg, I. S., and Pimpão, C. T. (2019). X-linked Hypohidrotic Ectodermal Dysplasia-General Features and Dental Abnormalities in Affected Dogs Compared with Human Dental Abnormalities. Top. companion animal Med. 35, 11–17. doi:10.1053/j.tcam.2019.03.002
Okita, T., Asano, N., Yasuno, S., and Shimomura, Y. (2019). Functional Studies for a Dominant Mutation in the EDAR Gene Responsible for Hypohidrotic Ectodermal Dysplasia. J. Dermatol 46, 710–715. doi:10.1111/1346-8138.14983
Pääkkönen, K., Cambiaghi, S., Novelli, G., Ouzts, L. V., Penttinen, M., Kere, J., et al. (2001). The Mutation Spectrum of the EDA Gene in X-Linked Anhidrotic Ectodermal Dysplasia. Hum. Mutat. 17, 349.
Pigno, M. A., Blackman, R. B., Cronin, R. J., and Cavazos, E. (1996). Prosthodontic Management of Ectodermal Dysplasia: a Review of the Literature. J. Prosthet. Dent. 76, 541–545. doi:10.1016/s0022-3913(96)90015-3
Schneider, P., Street, S. L., Gaide, O., Hertig, S., Tardivel, A., Tschopp, J., et al. (2001). Mutations Leading to X-Linked Hypohidrotic Ectodermal Dysplasia Affect Three Major Functional Domains in the Tumor Necrosis Factor Family Member Ectodysplasin-A. J. Biol. Chem. 276, 18819–18827. doi:10.1074/jbc.m101280200
Trzeciak, W. H., and Koczorowski, R. (2016). Molecular Basis of Hypohidrotic Ectodermal Dysplasia: an Update. J. Appl. Genet. 57, 51–61. doi:10.1007/s13353-015-0307-4
Vaidya, S., Risbud, M., Kshar, A., and Ramdurg, P. (2013). Hereditary Ectodermal Dysplasia: Report of 11 Patients from a Family. Indian J. Dent. Res. 24, 502–506. official publication of Indian Society for Dental Research. doi:10.4103/0970-9290.118373
Wang, X., Zhang, Z., Yuan, S., Ren, J., Qu, H., Zhang, G., et al. (2020). A Novel EDA1 Missense Mutation in X-Linked Hypohidrotic Ectodermal Dysplasia. Med. Baltim. 99, e19244. doi:10.1097/md.0000000000019244
Wimmer, K., Roca, X., Beiglböck, H., Callens, T., Etzler, J., Rao, A. R., et al. (2007). Extensive In Silico Analysis of NF1 Splicing Defects Uncovers Determinants for Splicing Outcome upon 5′ Splice-Site Disruption. Hum. Mutat. 28, 599–612. doi:10.1002/humu.20493
Wright, J. T., Fete, M., Schneider, H., Zinser, M., Koster, M. I., Clarke, A. J., et al. (2019). Ectodermal Dysplasias: Classification and Organization by Phenotype, Genotype and Molecular Pathway. Am. J. Med. Genet. 179, 442–447. doi:10.1002/ajmg.a.61045
Keywords: hypohidrotic ectodermal dysplasia, EDA, whole-exome sequencing, splicing variant, cryptic splice site, HED, ectodysplasin A
Citation: Wang Y, Zhang C, Zhou B, Hui L, Zheng L, Chen X, Wang S, Yang L, Hao S and Zhang Q (2022) Three Variants Affecting Exon 1 of Ectodysplasin A Cause X-Linked Hypohidrotic Ectodermal Dysplasia: Clinical and Molecular Characteristics. Front. Genet. 13:916340. doi: 10.3389/fgene.2022.916340
Received: 09 April 2022; Accepted: 23 May 2022;
Published: 06 July 2022.
Edited by:
Ming Li, Shanghai Jiao Tong University, ChinaReviewed by:
Yanshan Liu, Wuxi Children’s Hospital, ChinaCopyright © 2022 Wang, Zhang, Zhou, Hui, Zheng, Chen, Wang, Yang, Hao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinghua Zhang, emhhbmdxaW5naHVhX2dtY2hAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.