 Ling Sun
Ling Sun Yu-mei Xie
Yu-mei Xie Shu-shui Wang
Shu-shui Wang Zhi-wei Zhang*
Zhi-wei Zhang*- Department of Pediatric Cardiology, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
Background: Common cardiac abnormalities in Noonan syndrome (NS) include congenital heart diseases (CHD), pulmonary valve stenosis and hypertrophic cardiomyopathy (HCM). Molecular diagnoses are enabling earlier and more precise diagnosis of patients who have a subtle or atypical presentation. The aims of this study were to investigate genotype-phenotype associations with respect to Noonan syndrome (NS)-associated cardiac abnormalities and catheter or surgery-based interventions conditions.
Methods: From January 2019 to December 2021, 22 children with a confirmed molecular diagnosis of NS combined with cardiovascular abnormalities were consecutively enrolled into the current study. A comprehensive review was carried out of echocardiography and electrocardiogram results, second-generation whole-exome sequencing results and catheter or surgery-based interventions conditions.
Results: The main manifestations of electrocardiogram abnormalities were QTc prolongation, abnormal Q wave in the precordial lead and limb lead, right ventricular hypertrophy and left or right deviation of the electrical axis. The most commonly detected abnormality was pulmonary valve dysplasia with stenosis, seen in 15 (68.2%) patients, followed by atrial septal defect in 11 (50%) patients. Seven genes (RAF1, RIT1, SOS1, PTPN11, BRAF, SOS2, and LZTR1) were found to contain disease-associated variants. The most commonly observed genetic mutations were PTPN11 (27%) and RAF1 (27%). Each genotype was associated with specific phenotypic findings. RIT1, SOS1, PTPN11, and SOS2 had common echocardiography features characterized by pulmonary valve stenosis, while RAF1 was characterized by HCM. Interestingly, patients with BRAF mutations were not only characterized by HCM, but also by pulmonary valve stenosis. In the cohort there was only one patient carrying a LZTR1 mutation characterized by left ventricle globose dilation. Ten cases underwent catheter or surgery-based interventions. All the operations had immediate results and high success rates. However, some of the cases had adverse outcomes during extended follow-up. Based on the genotype-phenotype associations observed during follow-up, BRAF and RAF1 genotypes seem to be poor prognostic factors, and multiple interventions may be required for NS patients with severe pulmonary stenosis or myectomy for HCM.
Conclusions: The identification of causal genes in NS patients has enabled the evaluation of genotype-cardiac phenotype relationships and prognosis of the disease. This may be beneficial for the development of therapeutic approaches.
Introduction
Noonan syndrome (NS) is an autosomal dominant disorder that is characterized by facial dysmorphism (hypertelorism, ptosis and low-set ears), short stature, congenital heart defects and other additional extracardiac features (Romano et al., 2010). Although Noonan syndrome is mainly transmitted as a dominant trait, an autosomal recessive form of this condition has recently been associated with biallelic LZTR1 variants (Johnston et al., 2018). The incidence of NS is estimated to be 1 in 1000–2500 live births (Allanson et al., 1985). One of the principal features of NS, which is also the cause of seeking early treatment, is cardiac involvement, most frequently including congenital heart diseases, pulmonary valve stenosis and hypertrophic cardiomyopathy (HCM) (Romano et al., 2010). With the elucidation of the underlying genetic variants causing NS, numerous studies have revealed that NS and NS-like disorders are ‘RASopathies’ or ‘RAS/mitogen-activated protein kinase (MAPK) syndromes’ because they share aberrant signaling through the RAS/MAPK pathway that controls cell proliferation, differentiation and survival (Aoki et al., 2008; Tidyman and Rauen, 2009). Recently, some studies have discussed the genotype-phenotype associations together with the prognostic assessment in these patients. A large study was published by Calcagni et al. in 2020. The authors reported the cardiac features in 440 molecularly characterized patients with RASopathies enrolled from seven centers participating in the CArdiac Rasopathy NETwork—CARNET study (Calcagni et al., 2020). Among them, 312 subjects presented a common congenital heart defect or HCM, whereas atypical findings were reported in 45 patients. Leoni et al. assessed genotype-cardiac phenotype correlations in a single-center cohort of 116 patients with a molecularly confirmed RASopathy (Leoni et al., 2022). Yi et al. assessed the therapeutic landscape for the treatment of heart disease in RASopathies (Yi et al., 2022). In addition, the clinical presentation and natural history of RASopathy-related HCM was extensively described in two recent reviews (Calcagni et al., 2018; Lioncino et al., 2022). To evaluate genotype-cardiac phenotype relationships and improve prognoses for patients with NS, it is necessary to look for genotype-phenotype associations with respect to NS-associated congenital heart diseases (CHD), and a personalized targeted therapy approach should be developed according to genotype-phenotype correlations.
In this study, we reviewed the cardiac manifestations of patients with a confirmed molecular diagnosis of NS by evaluating clinical echocardiography and electrocardiogram features and performing molecular analyses of the known causative genes of NS. We also evaluated genotype-cardiac phenotype associations.
Methods
Study Participants
This retrospective study was approved by the Clinical Research Ethic Committee of Guangdong Provincial People’s Hospital (ethical approval number KY-Z-2021-191-01 and KY2020-033-01). From January 2019 to December 2021, 22 children with a confirmed molecular diagnosis of NS combined with cardiovascular abnormalities were consecutively enrolled into the current study after parental written informed consent was obtained. Some of the children underwent corrective cardiac surgery or catheter-based interventions at Guangdong Provincial People’s Hospital, the largest tertiary cardiac center in China. We reviewed the clinical phenotypes of cardiac structural anomalies evaluated by electrocardiogram, echocardiography findings and follow-up data.
Data Collection
Transthoracic echocardiographic examination was carried out preoperatively using IE33, IE Epic-7C (Philips, Andover, MA, United States) and Vivid E 9 (GE Healthcare, Horten, Norway) ultrasound systems with a transducer of 5–8 Mhz. The preoperative and postoperative TTE images and electrocardiogram were reviewed by two independent experienced pediatric cardiologists. If there were discrepancies between these two observers, another independent experienced pediatric cardiologist was invited to review the images.
Molecular Genetic Analyses
Molecular diagnosis was performed using second-generation whole-exome sequencing. Genomic DNA was extracted from peripheral blood using the Solpure Blood DNA kit (Magen), then fragmented by Q800R Sonicator (Qsonica) to generate 200–500 bp insert fragments. Custom designed NimbleGen SeqCap probes (Roche NimbleGen, Madison, Wis) were used for in-solution hybridization to enrich target sequences. Enriched DNA samples were indexed and sequenced on a NextSeq500 sequencer (Illumina, San Diego, Calif) with 100 cycles of single end reads, according to the manufacturer’s protocols. The target gene capture/NGS assay provides an average read depth of approximately 200×. The insufficiently covered (<20×) target exons were analyzed by using gene-specific amplicon-based Sanger sequencing. Deleterious mutations and novel variants detected by using NGS were confirmed by means of Sanger sequencing. Variants interpretation was manipulated according to the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015). When an NS molecular diagnosis report was suspected, a clinical geneticist was invited to evaluate the diagnosis. The NS patients and their molecular diagnoses are shown in Table 1.
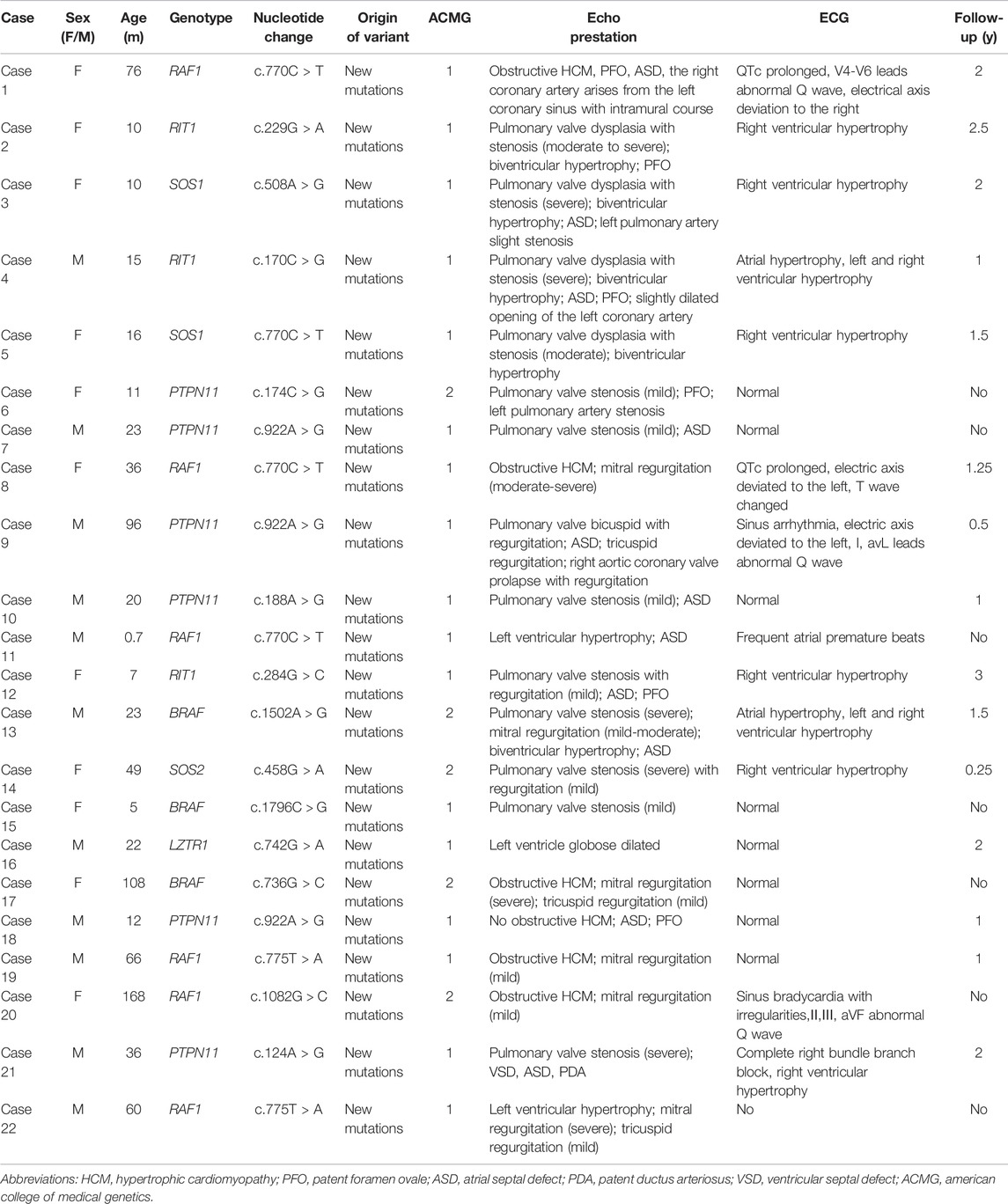
TABLE 1. NS patients and their clinical characteristics.
Statistical Analysis
Continuous variables are presented as mean ± standard deviation if normally distributed, otherwise they are presented as the median (interquartile range). Categorial variables are presented as number and proportion. All analyses were performed using SPSS Statistics 23 software (IBM, Chicago, IL, United States).
Results
Electrocardiographic Findings
A total of 22 children with NS were included in the current analysis. There were 11 female patients and 10 male patients. Their ages ranged from 0.7 to 168 months old (mean age: 39.5 ± 41.6 months). All patients had confirmed diagnoses by molecular analysis. The preoperative echocardiography and electrocardiogram features and genotypes are listed in Table 1. Follow-up ranged from 0.5 to 3 years (mean follow-up: 1.02 ± 0.94 years). Case 22 had no follow-up and no electrocardiogram. Thirteen cases (62%) had electrocardiographic abnormalities. The main manifestations of electrocardiogram abnormalities were QTc prolongation, abnormal Q wave in the precordial lead and limb lead, right ventricular hypertrophy and left or right deviation of the electrical axis.
Cardiac Abnormalities
All 22 NS patients had cardiac abnormalities. The most commonly detected abnormality was pulmonary valve dysplasia with stenosis, seen in 15 (68.2%) patients, followed by atrial septal defect (ASD) in 11 (50%) patients, biventricular hypertrophy in seven (31.8%) patients, left HCM in six (27.3%) patients, mitral regurgitation in six (27.3%) patients, patent foramen ovale (PFO) in six (27.3%) patients, both coronary artery anomalies and pulmonary branches stenosis in two (9.1%) patients and left ventricle globose dilated in one (4.5%) patient (Figure 1).
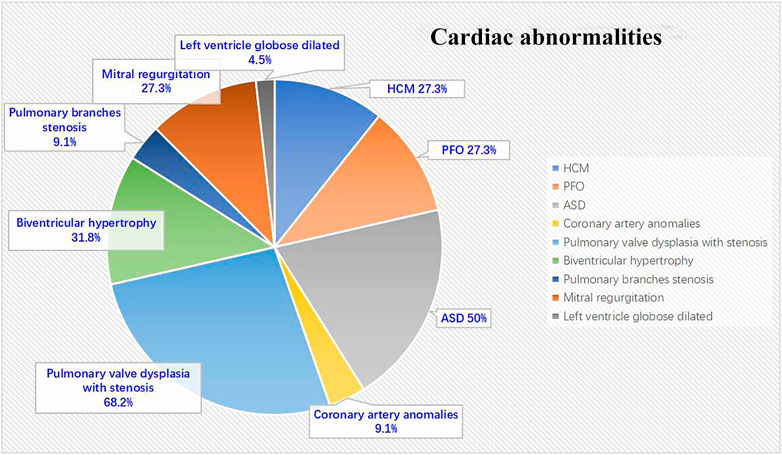
FIGURE 1. Cardiac abnormalities in NS patients (N = 22).
Genotype–Cardiac Phenotype Abnormalities
The genotype distribution in the 22 NS patients is listed in Figure 2, including mutation of RAF1 in six patients (27%), RIT1 in three patients (14%), SOS1 in two patients (9%), PTPN11 in six patients (27%), BRAF in three patients (14%), SOS2 in one patient (4.5%) and LZTR1 in one patient (4.5%). The most commonly observed genetic mutations in patients were in PTPN11 and RAF1. Each genotype had specific echocardiography findings. Patients with RAF1 mutations were characterized by left ventricular hypertrophy (obstructive or not obstructive), ASD and mitral regurgitation. Patients with RIT1 mutation were characterized by pulmonary valve dysplasia with stenosis and ASD/PFO. Patients with mutations in SOS1 had pulmonary valve dysplasia with stenosis, biventricular hypertrophy and ASD. PTPN11-mutation carriers had pulmonary valve stenosis and ASD. BRAF carriers had pulmonary valve stenosis, obstructive HCM, ASD, and mitral regurgitation. SOS2-mutation carriers had pulmonary valve stenosis with regurgitation. Only one patient carrying a LZTR1 mutation had left ventricle globose dilation (Table 2). Among all the pathogenic variants, we found that patients with mutations in RIT1, SOS1, PTPN11, and SOS2 had common echocardiography figures characterized by pulmonary valve stenosis, while RAF1 patients had HCM. Interestingly, BRAF patients were characterized not only by HCM but also by pulmonary valve stenosis, meaning poor prognoses.
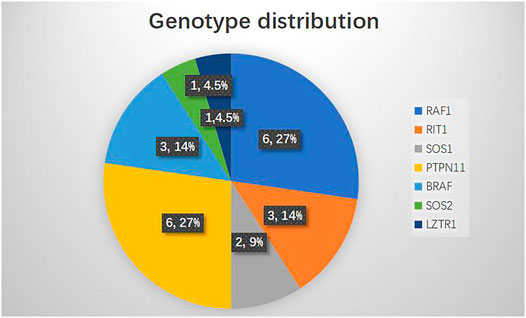
FIGURE 2. Genotype distribution in NS patients (N = 22).

TABLE 2. Genotype–cardiac phenotype abnormalities in NS patients (N = 22).
Catheter or Surgery-Based Interventions and Follow-Up Data
A total of ten cases underwent catheter or surgery-based interventions (Table 3). Percutaneous balloon pulmonary valve dilation (PBPV) was seen in two (20%) patients, percutaneous closure of ASD in two (20%) patients and complex surgery in three (30%) patients. Another three cases underwent not only PBPV, but also complex surgery because of poor outcomes (Figure 3). The genotype distribution in 10 NS patients is listed in Figure 4, including SOS1 in two patients (20%), RIT1 in one patient (10%), RAF1 in one patient (10%), PTPN11 in three patients (30%), BRAF in two patients (20%) and SOS2 in one patient (10%). All surgeries had immediate results and high success rates. However, some of the cases had adverse outcomes during extended follow-up. According to the follow-up results of the last echocardiography, the degree of pulmonary valve stenosis and the left ventricular outflow tract severity were determined from measurements of mean and peak gradients across the obstruction, and were quantitatively divided into mild, medium, and severe grades, following the recommendations of ESC guidelines (Baumgartner et al., 20202021) and EAE/ASE (Baumgartner et al., 2009). SOS1, RIT1, PTPN11, and SOS2 mutations seemed to have good prognosis, while BRAF mutations and RAF1 mutations seemed to have poor prognosis because of medium or severe mean and peak gradients across the obstruction of pulmonary valve or the left ventricular outflow tract.
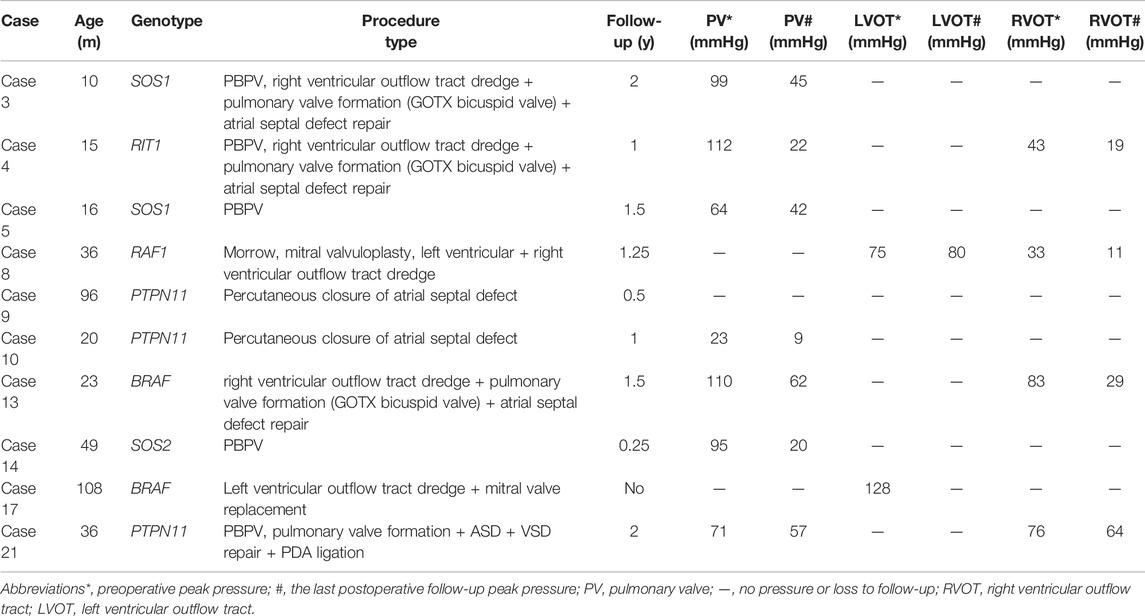
TABLE 3. Catheter or surgery-based interventions and follow-up clinical data.

FIGURE 3. Operation distributions in interventions patients.
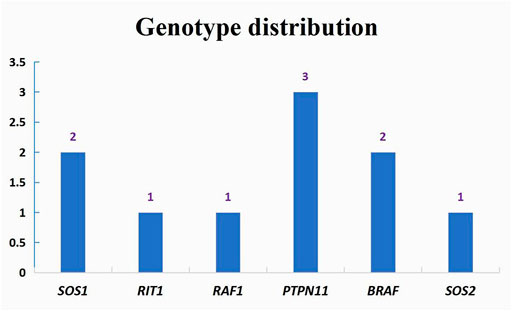
FIGURE 4. Genotype distribution in interventions patients.
Among ten NS patients, cases 3, 4, and 21 had common features of pulmonary valve dysplasia with stenosis, and each experienced interventions twice. The first interventions for cases 3 and 4 were PBPV, but afterward, the pulmonary valve peak differential pressures increased gradually at 1 year of follow-up, and both patients then experienced a second intervention [right ventricular outflow tract dredge + pulmonary valve (GOTX bicuspid valve) formation]. After 1 year of follow-up, the peak differential pressure of the pulmonary valve increased gradually, but more slowly than PBPV (Figure 5). The first intervention of case 21 was pulmonary valve formation, ASD and VSD repair and PDA ligation in another hospital, but 2 years later, the pulmonary valve peak differential pressure was 71 mmHg, so PBPV was performed in our hospital. After 2 years of follow-up, the peak differential pressure of the pulmonary valve was 57 mmHg. From the clinical data, we speculate that PBPV was not a good choice for NS patients. Three patients had only complex surgery. Case 13 and 17 with BRAF mutation showed pulmonary valve stenosis or HCM. Case 13 underwent right ventricular outflow tract dredge, pulmonary valve formation (GOTX bicuspid valve) and ASD repair. The pulmonary valve peak differential pressure decreased from 110 to 62 mmHg after 1 and a half years. Case 17 was lost to follow-up. Before the operation, the patient had severe left ventricular outflow tract stenosis and mitral regurgitation. Case 8, with a RAF1 mutation, showed severe obstructive HCM and mitral regurgitation, then underwent Morrow and mitral valvuloplasty procedures. However, the left ventricular outflow tract peak differential pressure still increased to 80 mmHg after 15 months, which was not a good outcome. Thus, for HCM in NS patients, surgery appears to lead to poor outcomes.
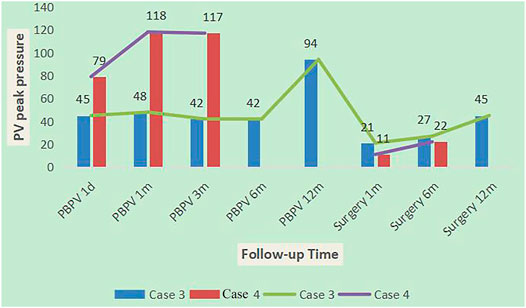
FIGURE 5. Follow-up results in case 3 and case 4.
Discussion
NS is the second most common syndromic cause of congenital heart disease, exceeded in prevalence only by trisomy 21 (Marino et al., 1999). Cardiac defects problems are the most common reasons that prompted these patients to see a doctor. There are several cardiovascular phenotypes abnormalities in NS. The most common phenotypes are pulmonary valve dysplasia with stenosis (50–62%), HCM (20%), ASDs (6–10%) and unusual electrocardiographic patterns (50%). Other cardiac abnormalities were also noted, like peripheral pulmonary stenosis, ventricular septal defect, mitral valve abnormalities, atrioventricular canal, aortic valve stenosis, aortic coarctation and coronary artery anomalies (Sanchez-Cascos, 1983; Patton, 1994; Shaw et al., 2007). Rarely, tetralogy of Fallot or patent ductus arteriosus are observed in NS. Often, patients display complex cardiac phenotypes with multiple defects such as pulmonary valve stenosis and aortic valve stenosis or CHD and HCM (Prendiville et al., 2014). This result is not entirely consistent with our clinical data from a single center. In this study, we found that the most commonly detected abnormality was pulmonary valve dysplasia with stenosis (68.2%), followed by ASD (50%), biventricular hypertrophy (31.8%), left HCM (27.3%), mitral regurgitation (27.3%), PFO (27.3%), both coronary artery anomalies and pulmonary branches stenosis (9.1%) and left ventricle globose dilation (4.5%). In particular, the incidence of ASD was significantly higher than that reported in literature, due to the small cohort. Also, we observed left ventricle globose dilation, which has not been reported in previous literature. The presentation was similar to dilated cardiomyopathy.
With the development of whole-exome sequencing, genetic testing is very useful for diagnosis of NS or NS-like disorders. If a patient has a mild or atypical presentation, genotyping could establish the diagnosis. To establish better treatment and management, it is necessary to look for genotype-phenotype associations with respect to NS-associated CHD phenotypes. There are eight known genotypes that cause NS, in the genes PTPN11, SOS1, RAF1, KRAS, NRAS, BRAF, SHOC2, and CBL (Romano et al., 2010). Recently, however, studies have found several novel genes associated with NS, like RIT1, RASA2, A2ML1, SOS2 and LZTR1 (Aoki et al., 2016). In our study, we detected variants in seven genes associated with NS. Mutations in four known genes (PTPN11, RAF1, SOS1, BRAF) and three novel genes (RIT1, SOS2, LZTR1) and, overall, PTPN11 (27%) and RAF1 (27%) were the most common.
PTPN11 encodes the Src homology-2 domain containing protein tyrosine phosphatase (Tartaglia et al., 2001; Tartaglia et al., 2002; Tartaglia et al., 2010). Studies suggest that 50% of cases of NS are caused by missense, gain-of-function mutations in PTPN11 (Tartaglia et al., 2001; Maheshwari et al., 2002; Tartaglia et al., 2002). NS in patients with PTPN11 mutations is more likely be associated with pulmonary valve stenosis or ASD (ostium secundum type) (Yoshida et al., 2004; Sznajer et al., 2007). This is consistent with the findings of our study. Six patients had pulmonary valve stenosis or ASD with PTPN11 mutations. RAF1 was another commonly mutated gene, often associated with HCM and negatively associated with pulmonary valve stenosis (Pandit et al., 2007). NS cases with RAF1 mutations were estimated to comprise 5–15% of the population (Pandit et al., 2007; Razzaque et al., 2007). RAF1 mutations prevent the phosphorylation of Ser259, causing a gain of function of the kinase, contributing to the autoinhibition of RAF1, affecting the activation loop and reducing the activity of the kinase (Pandit et al., 2007; Tidyman and Rauen, 2009). Our study found that six patients had RAF1 mutations, and the cardiac abnormalities included not only HCM (obstructive or non-obstructive), but also ASD and mitral regurgitation. SOS1 mutations most often result in pulmonary valve stenosis, much like PTPN11 mutations (Lepri et al., 2011). Missense mutations in SOS1 account for roughly 10% of cases of NS (Tidyman and Rauen, 2009; Tartaglia et al., 2010), close to our study results (9%). The protein product of SOS1 is a guanine nucleotide exchange factor that activates RAS proteins by displacing GDP, enabling GTP to bind through mass action (Egan and Weinberg, 1993; Takai et al., 2001). Other than pulmonary valve stenosis, SOS1 was associated with pulmonary valve dysplasia, biventricular hypertrophy and ASD in our study. BRAF mutations were rare, but have also been reported in individuals meeting clinical diagnostic criteria for NS. BRAF mutation-associated NS is characterized by HCM (Razzaque et al., 2007; Nyström et al., 2008; Sarkozy et al., 2009). However, BRAF predisposed patients to both HCM and valve abnormalities and accounted for roughly 14% of cases of NS in our study.
Three novel genotypes were also related to cardiac abnormalities. The mutations in RIT1 in NS patients are located in its G1 domain (p.S35T) and in the switch I region that is included in its G2 domain (p.A57G) (Aoki et al., 2016). Chen et al. (Chen et al., 2014) identified RIT1 mutations in five out of 27 (18.5%) individuals with NS. Bertola et al. (Bertola et al., 2014) and Gos et al. (Gos et al., 2014) identified RIT1 mutations in six out of 70 (8.6%) individuals and four out of 106 individuals (3.8%), respectively. Aoki et al. (Aoki et al., 2016) suggested that the frequency of RIT1 mutations can be estimated at ∼5% in patients with NS. Our study found that 3/22 (14%) NS patients had RIT1 mutations, significantly higher than the data reported in the literature. This may be related to the ethnicity of the population and the small number of patients, which requires further study. RIT1 mutations predispose patients to both HCM and valve abnormalities (Gelb et al., 2015), while our findings related to cardiac malformation were pulmonary valve dysplasia with stenosis, ASD and PFO. To date, few studies have reported cardiac abnormalities related to SOS2 and LZTR1. SOS2 is homologous to SOS1, and the identified variants, p.M267K and p.T376S, were located in the DH domain of SOS2 (Aoki et al., 2016). In our study, we found that pulmonary valve stenosis with regurgitation was related to cardiac malformation, which was similar to SOS1. Another novel genotype in LZTR1 had a unique cardiac phenotype, left ventricle globose dilation, with a presentation similar to dilated cardiomyopathy. LZTR1 is located within the 3-Mb-long region that is often deleted in 22q11 deletion syndrome patients (Kurahashi et al., 1995). Some studies have found that somatic and germline mutations in LZTR1 were associated with glioblastoma multiforme (Frattini et al., 2013) and multiple schwannomas (Piotrowski et al., 2014). Our study first reported this unique cardiac malformation related to LZTR1. Additional studies should be performed on mutational and cardiac malformation analyses.
A total of ten cases underwent catheter or surgery-based interventions in our study. Major interventions were PBPV, percutaneous closure of ASD and complex surgery. Cardiac malformations that require management were severe pulmonary valve stenosis, obstructive HCM and congenital heart disease of hemodynamic significance. In this group NS patients, we tried to describe the trend of prognosis for the correlations between genotypes and cardiac phenotypes according to the follow-up results of the last echocardiogram after catheter or surgery-based interventions. SOS1, RIT1, PTPN11, and SOS2 mutations showed common cardiac abnormalities such as pulmonary valve stenosis. For mild pulmonary valve stenosis in NS, stenosis tends to be nonprogressive and unlikely to require intervention. For moderate or severe obstruction, like BRAF mutations and RAF1 mutations, the rates of intervention are higher (∼50 and 100%, respectively) (Linglart and Gelb, 2020). McCrindle reported that the standard intervention using PBPV was often not as successful as was typical for pulmonary valve stenosis because of frequent dysplasia with commissural fusion and thickened pulmonary valve leaflets (McCrindle, 1994). We found that some of the cases had good results via PBPV, such as case 5 and case 14 with mutations in SOS1 and SOS2, respectively, but this may be related to our short follow-up period. Cases like case 3 and 4, with mutations in SOS1 and RIT1, respectively, did not show good results after the first PBPV, so they underwent surgical valvotomy again, which appeared to be highly successful. However, case 21 with a mutation in PTPN11 first underwent surgical valvotomy with poor results at 2 years of follow-up, then underwent PBPV again, which appeared to be successful. Holzmann et al. (Holzmann et al., 2018) reported that the pulmonary gradient remained unfavorably elevated in 80% of patients with NS after the initial PBPV and reintervention was necessary in up to 65% of cases (Prendiville et al., 2014). The clinical data from our center suggests that multiple interventions may be required for NS patients with severe pulmonary stenosis, whether PBPV or valvulotomy. Additionally, when the first intervention fails, the second intervention appears to be highly successful. Additional follow-up data are needed.
.Obstructive HCM in NS is another complex problem in the clinic. It can lead to severe heart failure and arrhythmias with progression of HCM. Most patients prefer to choose surgery. In our study, two genotypes in RAF1 and BRAF resulted in a common cardiac abnormality, obstructive HCM. These two genotypes seemed to have poor prognosis. While myectomy for HCM in a patient with NS is initially useful, with time, the left ventricular outflow tract may become obstructed again. Wilkinson et al. (Wilkinson et al., 2012) reported that patients with NS with HCM have a worse risk profile at presentation compared with other children with HCM, resulting in significant early mortality (22% at 1 year). Thus, heart transplantation may be a preferrable approach. It is encouraging that much progress has been made in our understanding of the molecular genetic causes of NS, and novel therapeutic drugs have been given to patients. To date, there are some publications describing the use of RAS pathway inhibition as useful for NS (Andelfinger et al., 2019; Mek Inhibitor Reverses Hypertrophic Cardiomyopathy, 2019; Dori et al., 2020; Meisner et al., 2021; Mussa et al., 2021; Kontaridis et al., 2022). Trametinib, a MEK-inhibitor approved for treatment of RAS/MAPK-mutated cancers, is an emerging treatment option for HCM in NS. Trametinib was given to two infants with the severe, rapidly progressive form of HCM with RIT1 mutations and no adverse effects have been observed as of yet (Andelfinger et al., 2019). However, further study of the effects of trametinib and other pathway inhibitors for pulmonary valve stenosis or HCM in NS is necessary.
Study Limitations
The retrospective design of the study had some limitations. We tried to grade the correlations between genotypes and cardiac phenotypes according to the follow-up results of the last echocardiogram after catheter or surgery-based interventions. However, the relatively small size of the cohort may have biased the results. Since each genotype group consisted of only a few individuals, we limited our analysis to describing prognostic trends for cardiac phenotypes within each group.
Conclusion
Molecular diagnoses are enabling earlier and more precise identification of patients with a subtle or atypical presentation of NS. The identification of causal genes in NS patients has enabled the evaluation of genotype-cardiac phenotype relationships and may be beneficial to the development of therapeutic approaches. Although there are many extracardiac malformations, in some cases, progressive cardiac malformations still need to be managed, which will lead to better prognoses for patients with this disorder. In addition, NS-specific therapeutic medications require further research.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Clinical Research Ethic Committee of Guangdong Provincial People’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual (s), and minor (s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LS contributed to conception and manuscript writing. Y-mX contributed to design and formal analysis. S-sW and Zw-Z contributed to editing, supervision and funding acquisition. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 82070321).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allanson, J. E., Hall, J. G., Hughes, H. E., Preus, M., Witt, R. D., Opitz, J. M., et al. (1985). Noonan Syndrome: the Changing Phenotype. Am. J. Med. Genet. 21 (3), 507–514. doi:10.1002/ajmg.1320210313
Andelfinger, G., Marquis, C., Raboisson, M.-J., Théoret, Y., Waldmüller, S., Wiegand, G., et al. (2019). Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll. Cardiol. 73 (17), 2237–2239. doi:10.1016/j.jacc.2019.01.066
Aoki, Y., Niihori, T., Inoue, S.-i., and Matsubara, Y. (2016). Recent Advances in RASopathies. J. Hum. Genet. 61 (1), 33–39. doi:10.1038/jhg.2015.114
Aoki, Y., Niihori, T., Narumi, Y., Kure, S., and Matsubara, Y. (2008). The RAS/MAPK Syndromes: Novel Roles of the RAS Pathway in Human Genetic Disorders. Hum. Mutat. 29 (8), 992–1006. doi:10.1002/humu.20748
Baumgartner, H., De Backer, J., Babu-Narayan, S. V., Budts, W., Chessa, M., Diller, G. P., et al. (20202021). 2020 ESC Guidelines for the Management of Adult Congenital Heart Disease. Eur. Heart J. 42 (6), 563–645. doi:10.1093/eurheartj/ehaa554
Baumgartner, H., Hung, J., Bermejo, J., Chambers, J. B., Evangelista, A., Griffin, B. P., et al. (2009). Echocardiographic Assessment of Valve Stenosis: EAE/ASE Recommendations for Clinical Practice. Eur. J. Echocardiogr. 10 (1), 1–25. quiz 101-2. doi:10.1093/ejechocard/jen303
Bertola, D. R., Yamamoto, G. L., Almeida, T. F., Buscarilli, M., Jorge, A. A. L., Malaquias, A. C., et al. (2014). Further Evidence of the Importance ofRIT1in Noonan Syndrome. Am. J. Med. Genet. 164 (11), 2952–2957. doi:10.1002/ajmg.a.36722
Calcagni, G., Adorisio, R., Martinelli, S., Grutter, G., Baban, A., Versacci, P., et al. (2018). Clinical Presentation and Natural History of Hypertrophic Cardiomyopathy in RASopathies. Heart Fail. Clin. 14 (2), 225–235. doi:10.1016/j.hfc.2017.12.005
Calcagni, G., Gagliostro, G., Limongelli, G., Unolt, M., De Luca, E., Digilio, M. C., et al. (2020). Atypical Cardiac Defects in Patients with RASopathies: Updated Data on CARNET Study. Birth Defects Res. 112 (10), 725–731. doi:10.1002/bdr2.1670
Chen, P.-C., Yin, J., Yu, H.-W., Yuan, T., Fernandez, M., Yung, C. K., et al. (2014). Next-generation Sequencing Identifies Rare Variants Associated with Noonan Syndrome. Proc. Natl. Acad. Sci. U.S.A. 111 (31), 11473–11478. doi:10.1073/pnas.1324128111
Dori, Y., Smith, C., Pinto, E., Snyder, K., March, M. E., Hakonarson, H., et al. (2020). Severe Lymphatic Disorder Resolved with MEK Inhibition in a Patient with Noonan Syndrome and SOS1 Mutation. Pediatrics 146 (6). doi:10.1542/peds.2020-0167
Egan, S. E., and Weinberg, R. A. (1993). The Pathway to Signal Achievement. Nature 365 (6449), 781–783. doi:10.1038/365781a0
Frattini, V., Trifonov, V., Chan, J. M., Castano, A., Lia, M., Abate, F., et al. (2013). The Integrated Landscape of Driver Genomic Alterations in Glioblastoma. Nat. Genet. 45 (10), 1141–1149. doi:10.1038/ng.2734
Gelb, B. D., Roberts, A. E., and Tartaglia, M. (2015). Cardiomyopathies in Noonan Syndrome and the Other RASopathies. Prog. Pediatr. Cardiol. 39 (1), 13–19. doi:10.1016/j.ppedcard.2015.01.002
Gos, M., Fahiminiya, S., Poznański, J., Klapecki, J., Obersztyn, E., Piotrowicz, M., et al. (2014). Contribution ofRIT1mutations to the Pathogenesis of Noonan Syndrome: Four New Cases and Further Evidence of Heterogeneity. Am. J. Med. Genet. 164 (9), 2310–2316. doi:10.1002/ajmg.a.36646
Holzmann, J., Tibby, S. M., Rosenthal, E., Qureshi, S., Morgan, G., and Krasemann, T. (2018). Results of Balloon Pulmonary Valvoplasty in Children with Noonan's Syndrome. Cardiol. Young 28 (5), 647–652. doi:10.1017/s1047951117002827
Johnston, J. J., van der Smagt, J. J., Rosenfeld, J. A., Pagnamenta, A. T., Alswaid, A., Baker, E. H., et al. (2018). Autosomal Recessive Noonan Syndrome Associated with Biallelic LZTR1 Variants. Genet. Med. 20 (10), 1175–1185. doi:10.1038/gim.2017.249
Kontaridis, M. I., Roberts, A. E., Schill, L., Schoyer, L., Stronach, B., Andelfinger, G., et al. (2022). The Seventh International RASopathies Symposium: Pathways to a Cure-Expanding Knowledge, Enhancing Research, and Therapeutic Discovery. Am. J. Med. Genet. A 188 (6), 1915–1927. doi:10.1002/ajmg.a.62716
Kurahashi, H., Akagi, K., Inazawa, J., Ohta, T., Niikawa, N., Kayatani, F., et al. (1995). Isolation and Characterization of a Novel Gene Deleted in DiGeorge Syndrome. Hum. Mol. Genet. 4 (4), 541–549. doi:10.1093/hmg/4.4.541
Leoni, C., Blandino, R., Delogu, A. B., De Rosa, G., Onesimo, R., Verusio, V., et al. (2022). Genotype‐cardiac Phenotype Correlations in a Large Single‐center Cohort of Patients Affected by RASopathies: Clinical Implications and Literature Review. Am. J Med Genet. Pt A 188 (2), 431–445. doi:10.1002/ajmg.a.62529
Lepri, F., De Luca, A., Stella, L., Rossi, C., Baldassarre, G., Pantaleoni, F., et al. (2011). SOS1 Mutations in Noonan Syndrome: Molecular Spectrum, Structural Insights on Pathogenic Effects, and Genotype-Phenotype Correlations. Hum. Mutat. 32 (7), 760–772. doi:10.1002/humu.21492
Linglart, L., and Gelb, B. D. (2020). Congenital Heart Defects in Noonan Syndrome: Diagnosis, Management, and Treatment. Am. J. Med. Genet. 184 (1), 73–80. doi:10.1002/ajmg.c.31765
Lioncino, M., Monda, E., Verrillo, F., Moscarella, E., Calcagni, G., Drago, F., et al. (2022). Hypertrophic Cardiomyopathy in RASopathies. Heart Fail. Clin. 18 (1), 19–29. doi:10.1016/j.hfc.2021.07.004
Maheshwari, M., Belmont, J., Fernbach, S., Ho, T., Molinari, L., Yakub, I., et al. (2002). PTPN11 Mutations in Noonan Syndrome Type I: Detection of Recurrent Mutations in Exons 3 and 13. Hum. Mutat. 20 (4), 298–304. doi:10.1002/humu.10129
Marino, B., Digilio, M. C., Toscano, A., Giannotti, A., and Dallapiccola, B. (1999). Congenital Heart Diseases in Children with Noonan Syndrome: An Expanded Cardiac Spectrum with High Prevalence of Atrioventricular Canal. J. Pediatr. 135 (6), 703–706. doi:10.1016/s0022-3476(99)70088-0
McCrindle, B. W. (1994). Independent Predictors of Long-Term Results after Balloon Pulmonary Valvuloplasty. Valvuloplasty and Angioplasty of Congenital Anomalies (VACA) Registry Investigators. Circulation 89 (4), 1751–1759. doi:10.1161/01.cir.89.4.1751
Meisner, J. K., Bradley, D. J., and Russell, M. W. (2021). Molecular Management of Multifocal Atrial Tachycardia in Noonan's Syndrome with MEK1/2 Inhibitor Trametinib. Circ. Genom Precis. Med. 14 (5), e003327. doi:10.1161/CIRCGEN.121.003327
Mek Inhibitor Reverses Hypertrophic Cardiomyopathy in RIT1 Mutated Noonan Syndrome: For the First Time, Hypertrophic Cardiomyopathy Was Reversed in Noonan Syndrome Associated with a RIT1 Mutation. Am. J. Med. Genet. A. 2019;179(8):1408–1409.doi:10.1002/ajmg.a.40456
Mussa, A., Carli, D., Giorgio, E., Villar, A. M., Cardaropoli, S., Carbonara, C., et al. (2021). MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease. Genes. (Basel) 13 (1). doi:10.3390/genes13010006
Nyström, A.-M., Ekvall, S., Berglund, E., Björkqvist, M., Braathen, G., Duchen, K., et al. (2008). Noonan and Cardio-Facio-Cutaneous Syndromes: Two Clinically and Genetically Overlapping Disorders. J. Med. Genet. 45 (8), 500–506. doi:10.1136/jmg.2008.057653
Pandit, B., Sarkozy, A., Pennacchio, L. A., Carta, C., Oishi, K., Martinelli, S., et al. (2007). Gain-of-function RAF1 Mutations Cause Noonan and LEOPARD Syndromes with Hypertrophic Cardiomyopathy. Nat. Genet. 39 (8), 1007–1012. doi:10.1038/ng2073
Piotrowski, A., Xie, J., Liu, Y. F., Poplawski, A. B., Gomes, A. R., Madanecki, P., et al. (2014). Germline Loss-Of-Function Mutations in LZTR1 Predispose to an Inherited Disorder of Multiple Schwannomas. Nat. Genet. 46 (2), 182–187. doi:10.1038/ng.2855
Prendiville, T. W., Gauvreau, K., Tworog-Dube, E., Patkin, L., Kucherlapati, R. S., Roberts, A. E., et al. (2014). Cardiovascular Disease in Noonan Syndrome. Archives Dis. Child. 99 (7), 629–634. doi:10.1136/archdischild-2013-305047
Razzaque, M. A., Nishizawa, T., Komoike, Y., Yagi, H., Furutani, M., Amo, R., et al. (2007). Germline Gain-Of-Function Mutations in RAF1 Cause Noonan Syndrome. Nat. Genet. 39 (8), 1013–1017. doi:10.1038/ng2078
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Romano, A. A., Allanson, J. E., Dahlgren, J., Gelb, B. D., Hall, B., Pierpont, M. E., et al. (2010). Noonan Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Pediatrics 126 (4), 746–759. doi:10.1542/peds.2009-3207
Sanchez-Cascos, A. (1983). The Noonan Syndrome. Eur. Heart J. 4 (4), 223–229. doi:10.1093/oxfordjournals.eurheartj.a061452
Sarkozy, A., Carta, C., Moretti, S., Zampino, G., Digilio, M. C., Pantaleoni, F., et al. (2009). GermlineBRAFmutations in Noonan, LEOPARD, and Cardiofaciocutaneous Syndromes: Molecular Diversity and Associated Phenotypic Spectrum. Hum. Mutat. 30 (4), 695–702. doi:10.1002/humu.20955
Shaw, A. C., Kalidas, K., Crosby, A. H., Jeffery, S., and Patton, M. A. (2007). The Natural History of Noonan Syndrome: a Long-Term Follow-Up Study. Arch. Dis. Child. 92 (2), 128–132. doi:10.1136/adc.2006.104547
Sznajer, Y., Keren, B., Baumann, C., Pereira, S., Alberti, C., Elion, J., et al. (2007). The Spectrum of Cardiac Anomalies in Noonan Syndrome as a Result of Mutations in the PTPN11 Gene. Pediatrics 119 (6), e1325–e1331. doi:10.1542/peds.2006-0211
Takai, Y., Sasaki, T., and Matozaki, T. (2001). Small GTP-Binding Proteins. Physiol. Rev. 81 (1), 153–208. doi:10.1152/physrev.2001.81.1.153
Tartaglia, M., Kalidas, K., Shaw, A., Song, X., Musat, D. L., van der Burgt, I., et al. (2002). PTPN11 Mutations in Noonan Syndrome: Molecular Spectrum, Genotype-Phenotype Correlation, and Phenotypic Heterogeneity. Am. J. Hum. Genet. 70 (6), 1555–1563. doi:10.1086/340847
Tartaglia, M., Mehler, E. L., Goldberg, R., Zampino, G., Brunner, H. G., Kremer, H., et al. (2001). Mutations in PTPN11, Encoding the Protein Tyrosine Phosphatase SHP-2, Cause Noonan Syndrome. Nat. Genet. 29 (4), 465–468. doi:10.1038/ng772
Tartaglia, M., Zampino, G., and Gelb, B. D. (2010). Noonan Syndrome: Clinical Aspects and Molecular Pathogenesis. Mol. Syndromol. 1 (1), 2–26. doi:10.1159/000276766
Tidyman, W. E., and Rauen, K. A. (2009). The RASopathies: Developmental Syndromes of Ras/MAPK Pathway Dysregulation. Curr. Opin. Genet. Dev. 19 (3), 230–236. doi:10.1016/j.gde.2009.04.001
Wilkinson, J. D., Lowe, A. M., Salbert, B. A., Sleeper, L. A., Colan, S. D., Cox, G. F., et al. (2012). Outcomes in Children with Noonan Syndrome and Hypertrophic Cardiomyopathy: a Study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 164 (3), 442–448. doi:10.1016/j.ahj.2012.04.018
Yi, J.-S., Perla, S., and Bennett, A. M. (2022). An Assessment of the Therapeutic Landscape for the Treatment of Heart Disease in the RASopathies. Cardiovasc Drugs Ther. doi:10.1007/s10557-022-07324-0
Keywords: cardiovascular abnormalities, echocardiography, genotype-phenotype, noonan syndrome, pulmonary valve stenosis
Citation: Sun L, Xie Y-m, Wang S-s and Zhang Z-w (2022) Cardiovascular Abnormalities and Gene Mutations in Children With Noonan Syndrome. Front. Genet. 13:915129. doi: 10.3389/fgene.2022.915129
Received: 07 April 2022; Accepted: 23 May 2022;
Published: 13 June 2022.
Edited by:
Tieliu Shi, Hunan University of Arts and Science, ChinaReviewed by:
Simone Martinelli, National Institute of Health (ISS), ItalySina Zoghi, Shiraz University of Medical Sciences, Iran
Copyright © 2022 Sun, Xie, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-shui Wang, d3NzY29tZUAxMjYuY29t; Zhi-wei Zhang, ZHJ6aGFuZ3poaXdlaUBzaW5hLmNvbQ==