 Paolo Enrico Maltese1*†
Paolo Enrico Maltese1*† Leonardo Colombo2†
Leonardo Colombo2† Salvatore Martella2
Salvatore Martella2 Luca Rossetti2
Luca Rossetti2 Said El Shamieh3
Said El Shamieh3 Lorenzo Sinibaldi4,5Chiara Passarelli4Andrea Maria Coppè6Luca Buzzonetti6
Lorenzo Sinibaldi4,5Chiara Passarelli4Andrea Maria Coppè6Luca Buzzonetti6 Benedetto Falsini6,7
Benedetto Falsini6,7 Pietro Chiurazzi8Giorgio Placidi7Benedetta Tanzi1Matteo Bertelli1,9‡
Pietro Chiurazzi8Giorgio Placidi7Benedetta Tanzi1Matteo Bertelli1,9‡ Giancarlo Iarossi6‡
Giancarlo Iarossi6‡- 1Magi’s Lab S.R.L., Rovereto, Italy
- 2Department of Ophthalmology, ASST Santi Paolo e Carlo Hospital, University of Milan, Milan, Italy
- 3Department of Medical Laboratory Technology, Faculty of Health Sciences, Beirut Arab University, Beirut, Lebanon
- 4Translational Cytogenomics Research Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 5Rare Disease and Medical Genetics, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 6Department of Ophthalmology, Bambino Gesù Children’s Hospital, Rome, Italy
- 7Fondazione Policlinico Universitario “A. Gemelli” IRCCS/Universita’ Cattolica del S. Cuore, Ophthalmology Unit, Rome, Italy
- 8UOC Genetica Medica, Fondazione Policlinico Universitario “A. Gemelli” IRCCS & Istituto di Medicina Genomica, Universita’ Cattolica del S. Cuore, Rome, Italy
- 9MAGI Euregio s.c.s., Bolzano, Italy
Purpose: Describing the clinical and genetic features of an ethnically heterogeneous group of (inherited retinal diseases) IRD patients from different underrepresented countries, referring to specialized Italian Hospitals, and expanding the epidemiological spectrum of the IRD in understudied populations.
Methods: The patients’ phenotypes underwent were characterized by exhaustive ophthalmological examinations, including morpho-functional testing. Genetic testing was performed using next-generation sequencing (NGS) and gene sequencing panels targeting a specific set of genes, Sanger sequencing and—when necessary—multiplex ligation-dependent probe amplification (MLPA) to better identify the genotype. When possible, segregation analysis was performed in order to confirm unsolved cases.
Results: The article reports the results of the phenotypes and genotypes of 123 IRD probands, 69 males and 54 females, mean age 41 (IQR, 54–30) years, disease onset at 13 (IQR, 27.25–5) years. Thirty-three patients out of 123 (26.8%) were Africans (North/Northwest Africa), 21 (17.1%) Asians, 19 (15.4%) Americans (South/Central America) and 50 (40.7%) Europeans (Eastern Europe). Retinitis pigmentosa was the most represented phenotype (56%), followed by cone dystrophy (11%) and Leber congenital amaurosis (7%), while ABCA4 was the most frequently mutated gene (18%), followed by USH2A (9%) and RPGR (5%). About ABCA4 variants found in Stargardt disease, macular and cone dystrophies were predominant in Asian (42%) and European (21%) patients. The most represented inheritance pattern was autosomal recessive, while a higher frequency of homozygous patients versus compound heterozygotes as compared to previous studies on Italian IRD patients was evidenced, reflecting a possible higher frequency of inbreeding marriages.
Conclusion: Though limited by the relatively low number of patients, the present paper paints a picture of the clinical and genetic features of IRD patients from understudied ethnic groups referred to Italian specialized hospitals and extended the epidemiological studies on underrepresented world regional areas.
Introduction
Inherited retinal diseases (IRDs) represent a heterogeneous group of disorders, characterized by different retinal cells primarily affected, age of onset, disease progression, and mode of inheritance, with a syndromic or non-syndromic clinical presentation. Depending on the retinal cells primarily involved in the degenerative process, they are classified in retinitis pigmentosa (RP, also known as rod-cone), cone-rod, and macular dystrophies. Most of these forms are degenerative, but some phenotypes, such as congenital stationary night blindness (Zeitz et al., 2015) and achromatopsia (Tsang and Sharma, 2018), are generally considered to be not progressive or slowly progressive. However, clinical presentations of IRDs can be heterogeneous and sometimes the diagnostic boundaries are not distinct. Although in most cases of IRDs only ophthalmic manifestations are present (nonsyndromic), over 70 forms of syndromic IRDs have been reported (Werdich et al., 2014; OMIM, https://www.ncbi.nlm.nih.gov/omim). The most common one is Usher syndrome (USH), characterized by RP and hearing loss (Tsang et al., 2018). Inheritance can be autosomal recessive (AR), autosomal dominant (AD), or X-linked (XL). Mitochondrial and digenic patterns of inheritance have also been described. To date, over 260 genes have been implicated in the etiology of IRD (RetNet, Retinal Information Network, https://sph.uth.edu/Retnet/). However, the contribution of each gene to the overall prevalence of the disease is relatively small and for many of them only a few reported cases of pathogenic mutations have been reported worldwide. Moreover, in approximately 30% of IRD patients, the underlying genes are still to be discovered. These factors make the genetics of IRDs very challenging (Duncan et al., 2018). To date, many reports have described the clinical and epidemiological characteristics of the various forms of inherited retinal dystrophies in the western world and in countries with specialized hospitals. However, IRD patients coming from regions with limited access to specialized structures to diagnose retinal diseases are strongly understudied or underrepresented in current literature. These patients often receive their first diagnosis of IRD abroad or remain misdiagnosed because of the scarce knowledge in the field of retinal diseases in their countries of origin, thus representing a significant lack of information for the epidemiological and clinical studies worldwide. Here, we report on an ethnically heterogeneous group of IRD patients from different underrepresented countries, referring to Italian Hospitals, describing their clinical and genetic features and expanding the epidemiological spectrum of IRDs in understudied populations.
Materials and Methods
Selection of Subjects
The cohort presented in this manuscript was recruited at the IRDs Units of the following Italian hospitals, all members of ERN-EYE: ASST Santi Paolo e Carlo Hospital—University of Milan, Bambino Gesù IRCCS Children’s Hospital—Rome, and Agostino Gemelli hospital/Università Cattolica del Sacro Cuore—Rome. All patients underwent detailed clinical examinations to assess their clinical diagnosis. Ophthalmic examination included best-corrected visual acuity (BCVA), color-vision test, slit-lamp examination, visual field analysis, spectral-domain optical coherence tomography (SD-OCT), fundus autofluorescence (FAF), full-field electroretinogram (ERG), and multifocal electroretinogram (MfERG) recorded according to the ISCEV standards.
All patients received genetic counseling to explain the risks and benefits of genetic testing and gave their informed consent.
The study conformed to the Helsinki declaration of 1984 and subsequent amendments. Ethics Committee approval was obtained (Ethical Committee of Azienda Sanitaria dell’Alto Adige, Italy - Approval No. 132-2020).
Genetic Testing
Genetic testing was performed at MAGI’s laboratories (MAGI’s Lab s.r.l., Rovereto, TN, Italy, and MAGI Euregio s.c.s., Bolzano, Italy) and at the Genetic Unit of the Bambino Gesù children’s hospital. All protocols and their technical aspects have been published elsewhere (Marceddu et al., 2019; Iarossi et al., 2021).
Briefly, DNA was extracted from blood or saliva using a commercial kit (E.Z.N.A. Blood DNA kit Omega Bio-Tek; Norcross, GA, United States) or from blood with Qiagen columns (QIAamp DNA minikit; Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA was analyzed via next-generation sequencing (NGS) on an Illumina MiSeq personal sequencer (Illumina, San Diego, CA) using an in-house bioinformatics pipeline or using the Twist Custom Panel kit (Twist Bioscience, South San Francisco, CA, United States) according to the manufacturer’s protocol on a NovaSeq6000 platform (Illumina, San Diego, CA, United States).
Sanger sequencing, using a Beckman Coulter CEQ 8000 sequencer (Beckmann Coulter, Milan, Italy), was performed to validate all genetic variants identified in the probands and for family segregation studies.
The pathogenicity of all identified variants was evaluated according to American College of Medical Genetics and Genomics guidelines (ACMG) (Richards et al., 2015), with the help of VarSome (https://varsome.com/) (Kopanos et al., 2019), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and gnomAD (https://gnomad.broadinstitute.org/) databases. Only variants of unknown significance (VUS), likely pathogenic (LP) variants and pathogenic (P) variants were considered in our analysis and the test was defined as “positive”.
A positive genetic test was considered solved when patients had: pathogenic and/or likely pathogenic variants in dominant and X-linked (XL) genes, even in the absence of family history; VUS variants on dominant and X-linked genes, with the support of family history or of the family segregation study; at least two variants in recessive genes, with a segregation study to verify biallelism. Without a family segregation study, probands carrying ≥2 potentially disease-causing variants were considered unsolved.
Statistical Analysis
Mann–Whitney test for independent samples was used to compare pairs of medians (MedCalc Sofware, Mariakerke, Belgium). Quantitative data are reported as the median and interquartile range (IQR, QR3-QR1).
Results
We report the results of genetic analyses on 123 IRD probands, 69 males and 54 females, mean age 41 (IQR, 54–30) years, disease onset 13 (IQR, 27.25–5) years. Pediatric patients (disease onset 0-14 years) represent 52.3% of the whole population. 36.6% of probands had a family history of IRDs, while for 63.4% of probands the family history was absent or unknown and they were defined sporadic cases.
Genetic testing was extended in 28 families, considering 52 probands’ relatives (24 males and 28 females, of which six were affected and 46 were healthy), thus reaching a total of 175 tested individuals. A total of 79 probands (64.2%) carried potentially disease-causing genetic variants, 10 (12.7%) carried variants in AD genes, 61 (77.2%; 30 homozygotes and 31 compound heterozygotes) carried variants in AR genes and 8 (10.1%) carried variants in XL genes.
A summary of the phenotypes tested in the 123 probands and the phenotypes and genes with variants found in the 79 patients who tested positive to the genetic test are reported in Figures 1A–C.
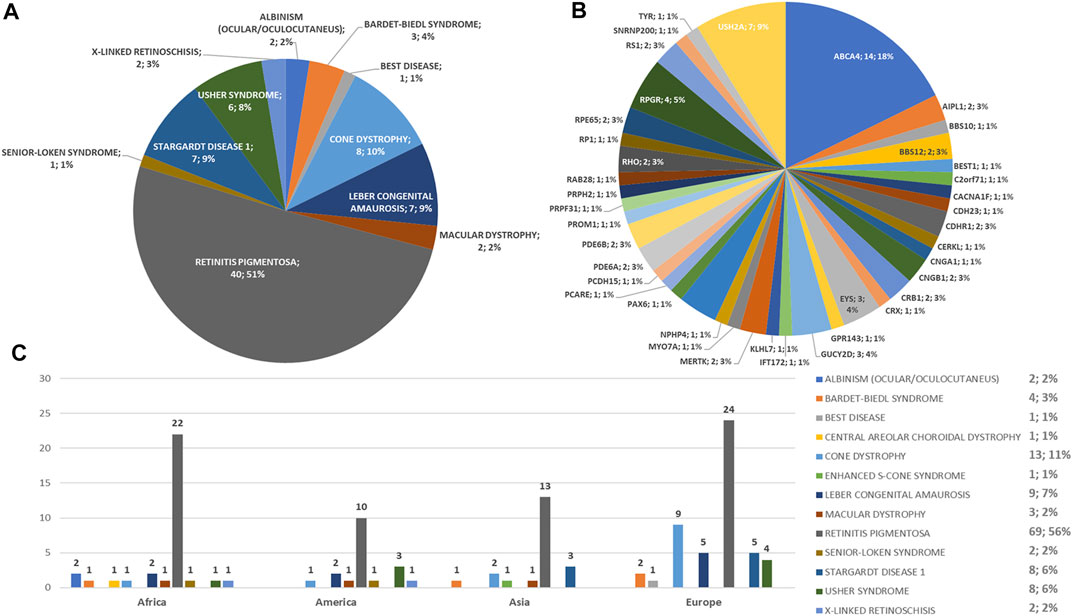
FIGURE 1. Phenotype and genotype distributions of understudied ethnic groups. Pie charts show (A) the phenotypes and (B) genotypes of positive cases. (C) Bar chart shows the number of examined patients, divided per phenotype.
Regarding the origins of the patients, 33 out of 123 (26.8%) were Africans (North/Northwest Africa), 21 (17.1%) Asians, 19 (15.4%) Americans (South/Central America) and 50 (40.7%) Europeans (Eastern Europe). The diseases’ distribution across the four continents is shown in Figure 1C. The countries of origin of the probands and the respective distribution of genes and phenotypes are shown in Figures 2–5. Detailed descriptions of probands, phenotypes, and genotypes are listed in Supplementary Table S1-S4. RP was the most represented phenotype (56%), followed by cone dystrophy (11%) and Leber congenital amaurosis (7%), while ABCA4 was the most frequently mutated gene (18%), followed by USH2A (9%) and RPGR (5%) (Figure 1). ABCA4 variants (found in Stargardt disease, macular, and cone dystrophies) were predominant in Asian (42%) and European (21%) probands (Figures 3, 5, respectively). Demographic data, diagnostic yields and inheritance patterns in the different populations are shown in Table 1. The most represented inheritance pattern was the autosomal recessive one for all the four populations, about 50% caused by homozygous variants in all populations except Asians (Table 1).
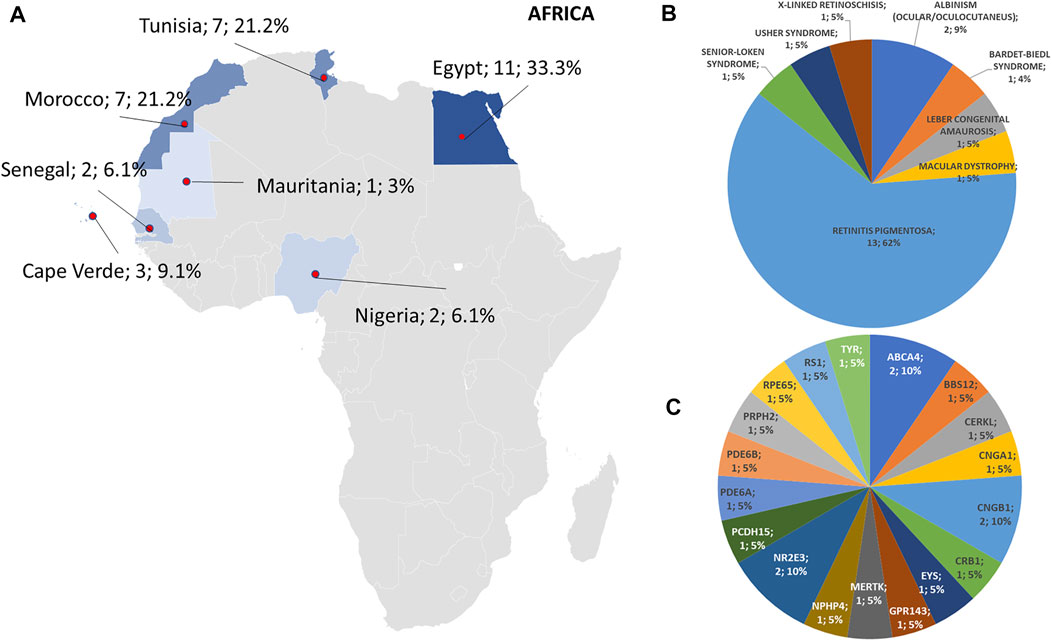
FIGURE 2. Phenotype and genotype distributions in African countries. (A) The map shows the numbers and percentages of examined patients. Pie charts show the (B) phenotypes and (C) genotypes of positive cases.
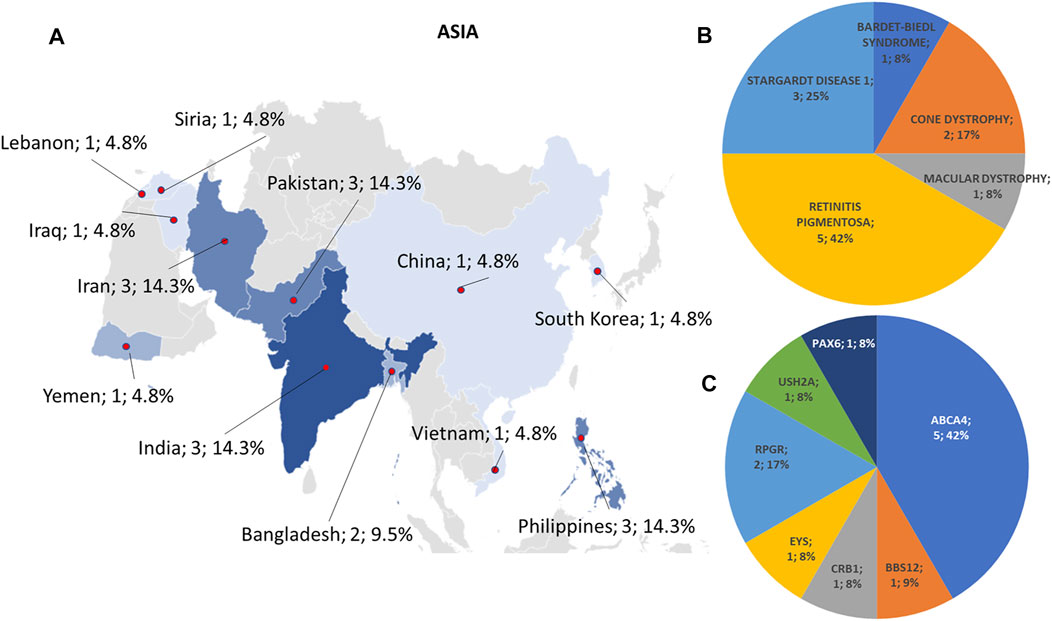
FIGURE 3. Phenotype and genotype distributions in Asian countries. (A) The map shows the numbers and percentages of examined patients. Pie charts show (B) the phenotypes and (C) genotypes of positive cases.
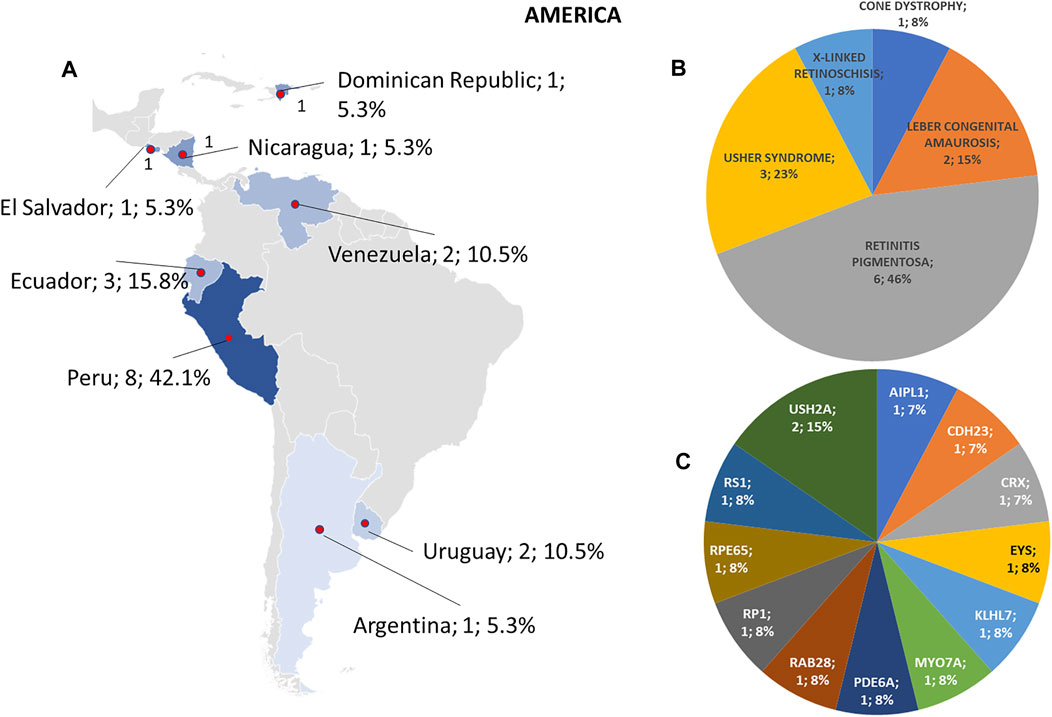
FIGURE 4. Phenotype and genotype distributions in American countries. (A) The map shows the numbers and percentages of examined patients. Pie charts show (B) the phenotypes and (C) genotypes of positive cases.
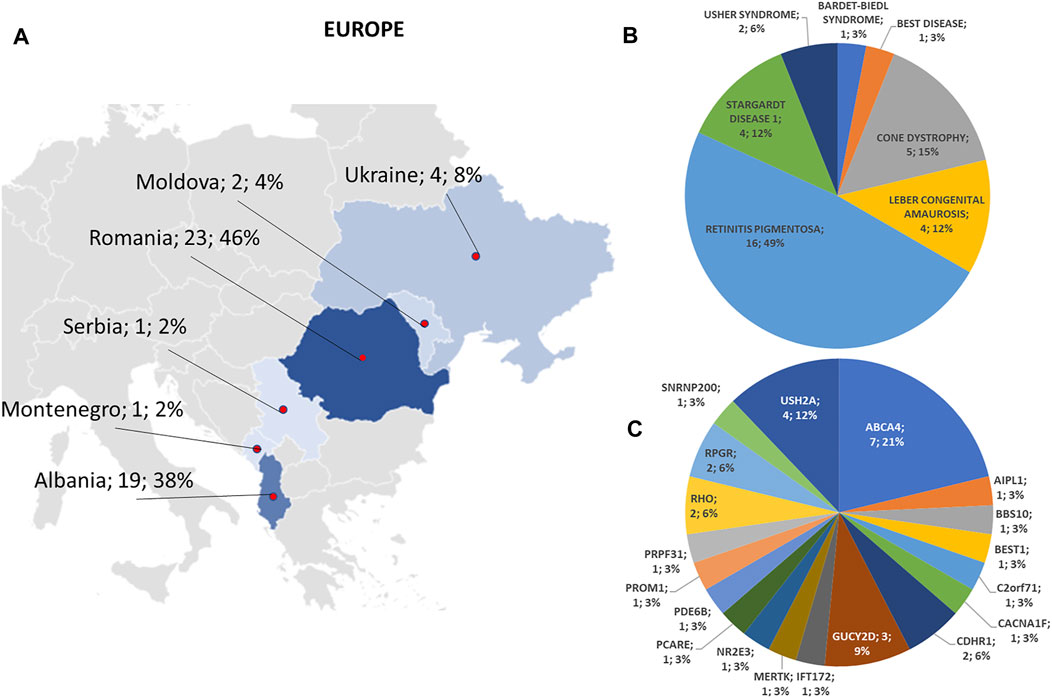
FIGURE 5. Phenotype and genotype distributions in European countries. (A) The map shows the numbers and percentages of examined patients. Pie charts show (B) the phenotypes and (C) genotypes of positive cases.
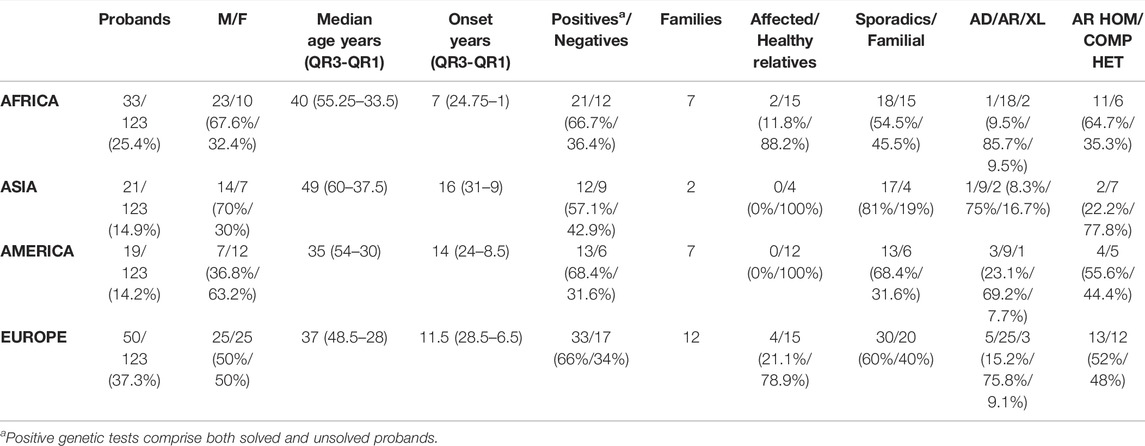
TABLE 1. Characteristics of the studied populations. By comparing the median age at onset of the whole population (13 years, IQR 27.25–5) with those derived from each of the four continents, there was not statistical differences (p = 0.29 Africa Vs. All; p = 0.26 Asia Vs. All; p = 0.89 America Vs. All; p = 0.87 Europe Vs. All). Legend: M, male; F, female; AD, autosomal dominant; AR, autosomal recessive; XL, X-linked; HEM, COMP HET, compound heterozygous; HOM, homozygous.
A total of 113 different variants were identified (Supplementary Table S5), 33 of which were new (Supplementary Table S6).
Discussion
Epidemiological and clinical studies on patients affected by IRDs from countries with poorly specialized health systems are largely underrepresented in literature. Only a few reports have been published so far, revealing interesting aspects of the disease manifestations due to the geographic and physical connectivity within families with a higher prevalence of recessive expression caused by consanguinity. Such a situation is particularly advantageous for genetic studies of retinal dystrophies, as it may offer an opportunity to find more prevalent genes in these extended families due to common ancestry. Moreover, the analysis of IRD clinical and genetic expression in understudied populations may improve the epidemiological knowledge of these diseases worldwide. Our previous studies represented the genetics of nonsyndromic RP and USH (Colombo et al., 2021) and macular and cone/cone-rod dystrophy (Falsini et al., 2022) in large Italian series. In this study, we present a similar approach but considering non-Italian patients and families, selected among the understudied ethnic groups referred to Italian specialized hospitals, depicting a picture, though limited in number, of the different expressions of the disease in different world areas. As expected (O'Neal and Luther, 2021), and in line with other reports on understudied ethnic groups, the prevalent disease in our patients is represented by RP, affecting 56% of the patients and reaching a prevalence of 63% with the inclusion Leber congenital amaurosis. Nevertheless, only 37.7% of patients presenting a RP phenotype had a proper genotypic correlation, consistent with the results reported by our group in a cohort of RP and USH patients (Figure 6). As reported in our previous work, also in this case the criteria to deem a case “solved” were very stringent: recessive forms were considered unsolved even if they presented two pathogenic variants but lacked a family segregation study to determine whether the alleles were in cis- or trans-configuration. Indeed, 50% of the 60 AR patients carried heterozygous variants, but only for 9 out of 30 of them it was possible to confirm the biallelic phase of the variants by family segregation studies (Supplementary Tables S1-S4). Moreover, from a raw analysis of our group of patients across the different regions, RP is more prevalent in patients from Africa than the other continents, whereas cone dystrophy is more represented in patients from Eastern Europe. Among maculopathies, Stargardt disease is the most common one in our group of patients, being more widespread in Asian and Eastern European patients in our cohort.
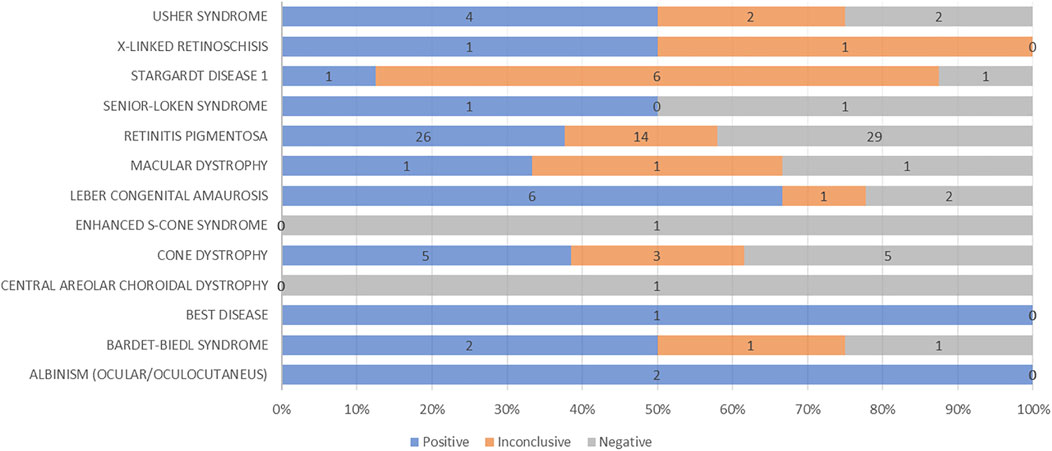
FIGURE 6. Graphical representation of diagnostic yield for each phenotype analyzed by genetic testing.
Although our cohort is not too numerous, it allowed us to make some comparisons with the previously published series regarding the genotypic distribution of IRDs. Considering the whole cohort, our data are in line with the literature: the greatest prevalence is of the ABCA4 and USH2A genes for the AR IRDs, RHO and PRPH2 for the dominant forms and RPGR for the x-linked forms (Carrigan et al., 2016; Stone et al., 2017; Hanany et al., 2020; Pontikos et al., 2020; Colombo et al., 2021; Perea-Romero et al., 2021; Falsini et al., 2022). Interestingly, no cases of USH2A-related dystrophy were found among Africans, similarly to what was reported by Habibi et al. in a cohort of patients from Tunisia and by Roberts et al. in a small cohort of African patients (Roberts et al., 2016; Habibi et al., 2020). Moreover, no ABCA4 cases were detected in the South American cohort, in contrast with Motta et al., in which ABCA4 was the most frequent disease-causing gene among the Brazilian population (Motta et al., 2018).
Comparing the allelic state of the recessive phenotypes, a higher frequency of homozygous patients versus compound heterozygotes emerges (50%/50% in this study; 33.2%/66.8% in nonsyndromic AR-RP and USH; 13.4%/86.6% in AR macular and cone/cone-rod dystrophy) (Colombo et al., 2021; Falsini et al., 2022), reflecting a possible higher frequency of consanguine marriages. This type of marriage is indeed very frequent in the cultures of some of the populations considered here.
This study has some limitations. Although the total number of patients considered is not too small, and the literature associated with the ethnic groups here reported is very scarce, this number is not sufficient to represent realistically the geographic distribution of phenotypes and genotypes and, unfortunately, it does not allow any statistically relevant comparison. Furthermore, in many of the countries reported here, genetic testing is not considered a health priority and the genetics of IRDs remain undetermined, thus limiting the general knowledge on the spread of these diseases. This is why epidemiological studies, such as the one presented here, aim to fill, at least partially, the resulting gaps in this field.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethical Committee of Azienda Sanitaria dell’Alto Adige, Italy (Approval No. 132-2020). Informed consent was obtained from all subjects for the use of their anonymized genetic results for research.
Author Contributions
PM, LC, GI and MB contributed to conception and design of the study. PM and LC organized the database. GI, PM and LC wrote the first draft of the manuscript. SEL, LS, PC and BF wrote sections of the manuscript. PM performed the statistical analysis. SM, LS, AMC, LB, BF, PC, GP, LR and GI contributed to patients’ clinical evaluation. BT and CP performed the genetic tests. PM, GI, MB and LC supervised the manuscript. LR, LB, GI, PM and MB administered the project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
IG, LB, LC, LR, PM, MB and BF are members of the European Reference Network for Rare Eye Diseases (ERN-EYE)—Project ID No 739534. Authors wish to thank the European Reference Network dedicated to Rare Eye Diseases (ERN-EYE) for their support in encouraging collaboration among institutions that specialize in Inherited Retinal Dystrophies.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.914345/full#supplementary-material
References
Carrigan, M., Duignan, E., Malone, C. P. G., Stephenson, K., Saad, T., McDermott, C., et al. (2016). Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 6, 33248. doi:10.1038/srep33248
Colombo, L., Maltese, P. E., Castori, M., El Shamieh, S., Zeitz, C., Audo, I., et al. (2021). Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Invest. Ophthalmol. Vis. Sci. 62 (2), 13. doi:10.1167/iovs.62.2.13
Duncan, J. L., Pierce, E. A., Laster, A. M., Daiger, S. P., Birch, D. G., Ash, J. D., et al. (2018). Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Trans. Vis. Sci. Tech. 7 (4), 6. doi:10.1167/tvst.7.4.6
Falsini, B., Placidi, G., De Siena, E., Chiurazzi, P., Minnella, A. M., Savastano, M. C., et al. (2022). Genetic Characteristics of 234 Italian Patients with Macular and Cone/Cone-Rod Dystrophy. Sci. Rep. 12 (1), 3774. doi:10.1038/s41598-022-07618-1
Habibi, I., Falfoul, Y., Turki, A., Hassairi, A., El Matri, K., Chebil, A., et al. (2020). Genetic Spectrum of Retinal Dystrophies in Tunisia. Sci. Rep. 10 (1), 11199. doi:10.1038/s41598-020-67792-y
Hanany, M., Rivolta, C., and Sharon, D. (2020). Worldwide Carrier Frequency and Genetic Prevalence of Autosomal Recessive Inherited Retinal Diseases. Proc. Natl. Acad. Sci. U. S. A. 117 (5), 2710–2716. doi:10.1073/pnas.1913179117
Iarossi, G., Coppè, A. M., Passarelli, C., Maltese, P. E., Sinibaldi, L., Cappelli, A., et al. (2021). Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. Int. J. Mol. Sci. 22 (16), 8617. doi:10.3390/ijms22168617
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: The Human Genomic Variant Search Engine. Bioinforma. Oxf. Engl. 35 (11), 1978–1980. doi:10.1093/bioinformatics/bty897
Marceddu, G., Dallavilla, T., Guerri, G., Manara, E., Chiurazzi, P., and Bertelli, M. (2019). PipeMAGI: An Integrated and Validated Workflow for Analysis of NGS Data for Clinical Diagnostics. Eur. Rev. Med. Pharmacol. Sci. 23 (15), 6753–6765. doi:10.26355/eurrev_201908_18566
Motta, F. L., Martin, R. P., Filippelli-Silva, R., Salles, M. V., and Sallum, J. M. F. (2018). Relative Frequency of Inherited Retinal Dystrophies in Brazil. Sci. Rep. 8 (1), 15939. doi:10.1038/s41598-018-34380-0
O'Neal, T. B., and Luther, E. E. (2021). Retinitis Pigmentosa. Treasure Island (FL): StatPearls Publishing.
Perea-Romero, I., Gordo, G., Iancu, I. F., Del Pozo-Valero, M., Almoguera, B., Blanco-Kelly, F., et al. (2021). Genetic Landscape of 6089 Inherited Retinal Dystrophies Affected Cases in Spain and Their Therapeutic and Extended Epidemiological Implications. Sci. Rep. 11 (1), 1526. doi:10.1038/s41598-021-81093-y
Pontikos, N., Arno, G., Jurkute, N., Schiff, E., Ba-Abbad, R., Malka, S., et al. (2020). Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 127 (10), 1384–1394. doi:10.1016/j.ophtha.2020.04.008
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roberts, L., Ratnapriya, R., du Plessis, M., Chaitankar, V., Ramesar, R. S., and Swaroop, A. (2016). Molecular Diagnosis of Inherited Retinal Diseases in Indigenous African Populations by Whole-Exome Sequencing. Invest. Ophthalmol. Vis. Sci. 57 (14), 6374–6381. doi:10.1167/iovs.16-19785
Stone, E. M., Andorf, J. L., Whitmore, S. S., DeLuca, A. P., Giacalone, J. C., Streb, L. M., et al. (2017). Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 124 (9), 1314–1331. doi:10.1016/j.ophtha.2017.04.008
Tsang, S. H., Aycinena, A. R. P., and Sharma, T. (2018). Ciliopathy: Usher Syndrome. Adv. Exp. Med. Biol. 1085, 167–170. doi:10.1007/978-3-319-95046-4_32
Tsang, S. H., and Sharma, T. (2018). Rod Monochromatism (Achromatopsia). Adv. Exp. Med. Biol. 1085, 119–123. doi:10.1007/978-3-319-95046-4_24
Werdich, X. Q., Place, E. M., and Pierce, E. A. (2014). Systemic Diseases Associated with Retinal Dystrophies. Semin. Ophthalmol. 29 (5-6), 319–328. doi:10.3109/08820538.2014.959202
Keywords: inherited retinal diseases, understudied ethnic groups, genetic epidemiology, next generation sequencing, sanger sequencing
Citation: Maltese PE, Colombo L, Martella S, Rossetti L, El Shamieh S, Sinibaldi L, Passarelli C, Coppè AM, Buzzonetti L, Falsini B, Chiurazzi P, Placidi G, Tanzi B, Bertelli M and Iarossi G (2022) Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals. Front. Genet. 13:914345. doi: 10.3389/fgene.2022.914345
Received: 06 April 2022; Accepted: 10 June 2022;
Published: 28 June 2022.
Edited by:
Sander W. Van Der Laan, University Medical Center Utrecht, NetherlandsReviewed by:
Dusanka Savic Pavicevic, University of Belgrade, SerbiaJing Yu, University of Oxford, United Kingdom
Copyright © 2022 Maltese, Colombo, Martella, Rossetti, El Shamieh, Sinibaldi, Passarelli, Coppè, Buzzonetti, Falsini, Chiurazzi, Placidi, Tanzi, Bertelli and Iarossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Enrico Maltese, cGFvbG8ubWFsdGVzZUBhc3NvbWFnaS5vcmc=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship