 Longbo Li
Longbo Li Jia Liu
Jia Liu Zhenzhen Chen
Zhenzhen Chen Junnan Wang
Junnan Wang- Department of Cardiology, Second Hospital of Jilin University, Changchun, China
Cardiovascular disease (CVD) is a leading cause of morbidity and mortality worldwide. Recent studies have shown that n6-methyladenosine (m6A) plays a major role in cardiovascular homeostasis and pathophysiology. These studies have confirmed that m6A methylation affects the pathophysiology of cardiovascular diseases by regulating cellular processes such as differentiation, proliferation, inflammation, autophagy, and apoptosis. Moreover, plenty of research has confirmed that m6A modification can delay the progression of CVD via the post-transcriptional regulation of RNA. However, there are few available summaries of m6A modification regarding CVD. In this review, we highlight advances in CVD-specific research concerning m6A modification, summarize the mechanisms underlying the involvement of m6A modification during the development of CVD, and discuss the potential of m6A modification as a therapeutic target of CVD.
1 Introduction
Cardiovascular disease (CVD), including cardiac diseases and vascular diseases, is a leading cause of morbidity and mortality worldwide and regarded as a significant public health problem (Leong et al., 2017; Andersson and Vasan, 2018). Over the past decade, there is a tremendous development in the diagnosis and treatment of CVD, however, it is still challenging to approach the rising morbidity and mortality rates in CVD patients. A big problem is that the mechanisms regulating CVD onset and progression are still not entirely clear. It has been realized that genetics and environments play great role in the onset and progression of CVD, and with the help of molecular biology technique, we could explore the role of genetics in the pathophysiology of CVD in the cellular and molecular level and develop genetic therapy agents for CVD (Ylä-Herttuala and Baker, 2017). However, neither the genetics nor the environments are sufficient to fully explain the onset and progression of CVD. Recent discoveries revealed that environments could alter gene expression without genetic changes, indicating that there is a link between genetics and environments (Prasher et al., 2020). For example, hypoxia could change the expression of hypoxia-related genes without changing DNA sequence of these genes; this regulation of gene expression without the change of DNA sequences in response to environmental factors is called epigenetics, through which we can explore the link between genetics and environments (Jones et al., 2016).
Epigenetics, including histone modification, DNA methylation, RNA methylation and non-coding RNA molecules, has attracted tremendous research interests due to its special regulation of gene expression. Latest research revealed that epigenetic modifications is heritable, reversible, and regulated by a serious of proteins, such as writers (that deposit them), readers (to interpret them) and erasers (to remove them) (Cavalli and Heard, 2019). Additionally, more and more researches confirmed that epigenetics plays a great role in the onset and development of CVD through regulating the expression of CVD-related genes, which further affect cell function and contribute to the progression of CVD (Prasher et al., 2020). The role of histone modification, DNA methylation and non-coding RNA in the onset and development of CVD have been well studied, however, m6A modification, the most common and abundant epigenetic modification in eukaryotic RNA (Chen Y.-S. et al., 2021; Shen Z.-J. et al., 2021), has attracted tremendous research interests just in recent years. Emerging studies confirmed that m6A modification play significant role in the pathophysiology of CVD, such as the proliferation, autophagy, apoptosis, and inflammation of cardiovascular system cells, through post-transcriptional regulation of CVD-related RNA. Regulation of m6A methylation has been proven to reverse the progression of CVDs and may be a therapeutic target for the treatment of CVDs (Zhao K. et al., 2020). However, given this large number of researches, few reviews have been published regarding the relationship between m6A modification and CVD, despite the ability of the latter to cause major acute cardiovascular events. To achieve an in-depth understanding of the function and regulatory mechanism of m6A modification regarding CVD, it is necessary to summarize existing knowledge of m6A modification in relation to CVD. Such a summary will contribute to the prevention, diagnosis, and treatment of CVD.
To this end, herein, we review the role of m6A modification in CVD. First, we introduce the molecular mechanism, regulation, and biological function of m6A modification. Second, we summarize the mechanisms of m6A modification that are involved in the cardiovascular pathophysiology. Third, we discuss the potential role of m6A modification in CVD.
2 m6A Methylation
2.1 Molecular Mechanism of m6A Methylation
Methylation of adenine at the 6N position, commonly called m6A methylation (Wu et al., 2021), was first discovered in 1974 in the messenger RNA (mRNA) of mammalian cells (Desrosiers et al., 1974). Since its discovery, only few functional studies have been conducted on m6A modification because of a lack of reliable detection technologies. In 2012, however, a breakthrough came in the form of m6A sequencing, which can identify target transcriptions that have undergone m6A modification (Dominissini et al., 2012; Meyer et al., 2012).
m6A modification is characterized by its dynamic reversibility, wide distribution, and highly conservative nature. As the most abundant RNA modification in eukaryotes, m6A modification is widely found in almost all types of RNA (Wu S. et al., 2020), such as mRNA, transfer RNA (tRNA), circular RNA (circRNA), long non-coding RNA (lncRNA) RNA, and ribosomal RNA (rRNA), and more than 25% of human transcripts are modified by m6A (Wu et al., 2021). m6A modification is not static but is instead dynamically regulated by m6A methyltransferases and demethylases (Liu et al., 2014; Zhang W. et al., 2021). Additionally, this type of epigenetic modification is enriched near stop codons and in 3’ untranslated regions (UTRs). Moreover, it is distributed across the highly conserved RRACH sequence (R = A or G and H = A, U, or C) (Xia et al., 2021), where adenosine is modified to m6A.
2.2 Regulation of m6A Methylation Modification
m6A methylation is a dynamic and reversible process that is primarily regulated by three proteases: m6A methyltransferase (writer), m6A demethylase (eraser), and m6A RNA-binding proteins (reader; Figure 1). The m6A methyltransferase complex catalyzes m6A modification, m6A demethylase removes the methyl group and modifies m6A to A, and m6A RNA-binding proteins recognize and combine m6A-methylation sites to regulate RNA mentalism. The combined action of m6A methyltransferase and m6A demethylase ensures that m6A RNA methylation remains balanced in cells. Therefore, many functional studies on m6A modification have been performed by either knocking down or overexpressing m6A methyltransferase or demethylase (Shen et al., 2015; Dorn et al., 2019; Ma et al., 2020; Wang et al., 2020; Wang P. et al., 2021; Takemoto et al., 2021; Xia et al., 2021).
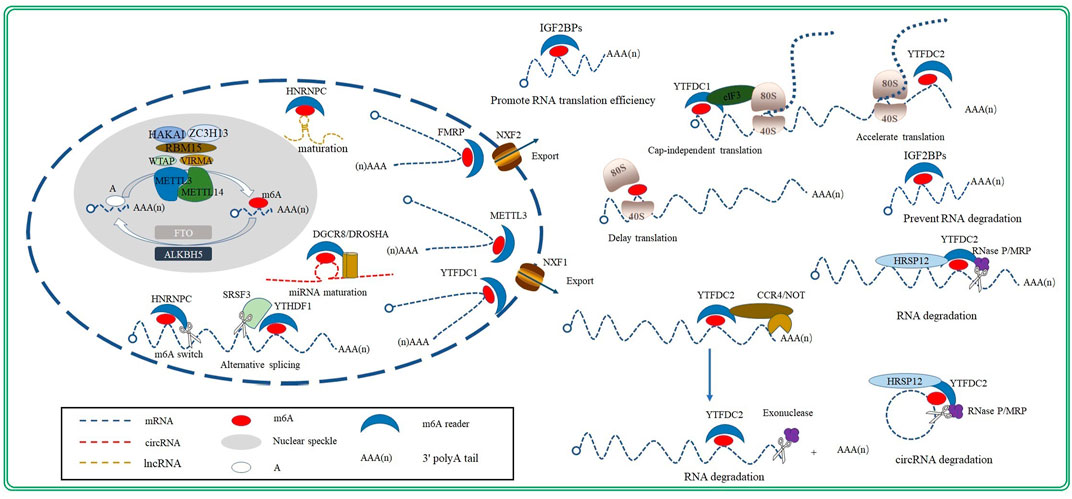
FIGURE 1. Regulation and biological function of m6A modification.
2.2.1 m6A Methyltransferase (Writer)
The m6A methyltransferase complex, consisting of methyltransferase-like3 (METTL3), methyltransferase-like14 (METTL14), and Wilms tumor 1associated protein (WTAP), specifically recognizes the RRACH sequence in RNA and catalyzes the modification of adenosine to m6A (Liu et al., 2014; Figure 1). METTL3 is a key enzyme within this complex; it possesses an S-adenosylmethionine (SAM)-binding domain that can transfer methyl groups from the m6A-methylation substrate to the sixth N of adenosine. Moreover, METTL3 is the only catalytic subunit in the m6A methyltransferase complex; therefore, its deletion inactivates the complex (Geula et al., 2015). The sequence homology between METTL14 and METTL3 can reach as high as 43%. However, unlike METTL3, METTL14 has no catalytic activity. METTL14 has an arginine-glycine-glycine domain at its C-terminus, which provides a platform for an RNA and greatly improves the complex’s stability and catalytic ability (Liu et al., 2014; Schöller et al., 2018; Chen Y.-S. et al., 2021). Moreover, METTL3 and METTL14 form the core of the m6A methyltransferase complex (Liu et al., 2014) and localizes these complex to the nuclear speckle, which is enriched in splicing factors (Liu et al., 2014; Alarcón et al., 2015a; Liu et al., 2015; Wang et al., 2016).
Similar to METTL14, WTAP does not exhibit methyltransferase activity; however, its knockdown significantly reduces the level of m6A modification in RNA, to an even more severe degree than METTL14 knockdown. This suggests that WTAP is essential to m6A modification (Duan et al., 2019). Furthermore, studies have confirmed that WTAP helps localize the m6A methyltransferase complex to the nuclear speckle and recruits it to the substrate RNA (Ping et al., 2014; Zhang W. et al., 2021; Wu et al., 2021).
Studies have also identified other protein components, such as Vir-like m6A methyltransferase-associated protein (VIRMA), RNA binding motif protein15 (RBM15), Cbl photo oncogene like1 (HAKAI), and zinc finger CCCH-type containing 13 (ZC3H13), in the m6A methyltransferase complex by co-immunoprecipitation (Liu et al., 2014; Yue et al., 2018; Zhu et al., 2020; Wang P. et al., 2021, Wang et al., 2021 J.; Zhang et al., 2021b; Figure 1). Moreover, studies have discovered that the ablation of VIRMA, RBM15, HAKAI or ZC3H13decreased the degree of m6A methylation in RNAs, indicating that these proteins are subunits of the m6A methyltransferase complex. However, their roles in the catalytic process remain to be studied (Patil et al., 2016; Růžička et al., 2017; Wen et al., 2018).
2.2.2 m6A Demethylase (Eraser)
Two m6A demethylases have been identified in eukaryotes: FAT mass and obesity-associated protein (FTO) and ALKB homologue5 protein (ALKBH5) (Zheng et al., 2013). They are both Fe(II)/α-ketoglutarate-dependent dioxygenases, and both are members of the ALKBH family (Zaccara et al., 2019). Additionally, FTO and ALKBH5 are both localized to the nucleus, and their knockdown significantly increases m6A-methylation levels (Shen et al., 2015; Shen W. et al., 2021; Zhao Y. et al., 2021). The discovery of m6A demethylases revealed the reversibility of m6A modification, indicating that m6A modification is a dynamically regulated process (Wu et al., 2021).
FTO (also referred to as ALKBH9) was first identified as an obesity-related gene; it exhibits dioxygenase activity that oxidizes m6A to form an N6-hydroxymethyladenosine intermediate, which is further oxidized to N6-formyladenosine. Both N6-hydroxymethyladenosine and N6-formyladenosine remove formaldehyde or formic acid molecules and form adenylate to complete RNA demethylation (Fu et al., 2013). Unlike FTO, ALKBH5 directly demethylates the m6A site; it skips the production of intermediates (Zheng et al., 2013; Figure 1). FTO and ALKBH5 both are essential to cardiac homeostasis. Studies have confirmed that FTO and ALKBH5 play a significant role in the development of embryo heart and cardiovascular disease, such as atherosclerosis, coronary heart disease (CHD) and heart failure (Mathiyalagan et al., 2019; Song et al., 2019; Shen W. et al., 2021).
2.2.3 m6A RNA-Binding Proteins (Reader)
The m6A RNA-binding proteins, called readers, recognize and bind to the m6A methylation sites to regulate RNA mentalism (Wu et al., 2021). The most well-known m6A RNA-binding protein is a member of the large YTH family, which comprises the YTH domain-containing family protein 1–3 (YTHDF1/2/3) and YTH domain-containing protein 1–2 (YTHDC1/2) subfamilies. Members of the YTH family possess a YTH domain, which constitutes an aromatic pocket structure that can recognize and bind to m6A-methylation sites. Within the YTHDF subfamily, YTHDF1 can increase the ribosome-occupancy rate and improve translational efficiency by directly interacting with translation-initiation factors, while YTHDF2 facilitate the degradation of mRNA modified by m6A (Sheth and Parker, 2003). Studies have demonstrated that YTHDF2 knockout increases the stability of target mRNAs, which can prolong their lifespan by ∼30% (Wang X. et al., 2014; Schwartz et al., 2014; Wang et al., 2015). Furthermore, YTHDF3 acts as a “buffer” for YTHDF1 and YTHDF2, as it binds to m6A-methylated RNA that has entered the cytoplasm. It can promote the combination of YTHDF1/YTHDF2 and target mRNA, thus accelerating the translation or degradation of the target transcript induced by YTHDF1 or YTHDF2 (Shi et al., 2017).
YTHDC1, as a member of the YTHDC subfamily, recognizes m6A-methylation sites and enhances the binding of target transcripts to serine- and arginine-rich splicing factor 3 (SRSF3) and nuclear RNA-export factor 1 (NXF1). In this way, it promotes the nuclear export of mRNA in an m6A-dependent manner (Roundtree et al., 2017). Additionally, YTHDC2 regulates RNA translation and decay by recognizing m6A-methylation sites on target RNA (Mao et al., 2019).
Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), including IGF2BP1, IGF2BP2, and IGF2BP3, is another important family of m6A RNA-binding proteins. Functionally, IGF2BPs promote the stability and the translation efficiency of target m6A-modified mRNA (Zhao Y. et al., 2020). Advances in research have uncovered new m6A RNA-binding proteins, such as heterogeneous nuclear ribonucleoprotein C (HNRNPC) and heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) (Wu et al., 2021). Interestingly, the m6A methyltransferase METTL3 also functions as an m6A RNA-binding protein under certain circumstances (Alarcón et al., 2015a; Liu et al., 2015; Zhou et al., 2019).
2.3 m6A Methylation and Post-Transcriptional Regulation
m6A modification, as well as other types of epigenetic, regulate gene expression by affecting the physiological properties of RNA transcripts, including their charge, base pairing, secondary structure, and protein–RNA interactions (Yang et al., 2018). Furthermore, m6A modification participates in almost every stage of the RNA life cycle, and it determines the fate of m6A-methylated RNA (Kmietczyk et al., 2019; Zhao K. et al., 2020).
2.3.1 Post-Transcriptional Regulation of mRNAs by m6A Methylation
2.3.1.1 mRNA Maturation
The level of m6A modification is proportional to the amount of mature mRNA in the cytoplasm. In fact, the down-regulation of m6A modification has been shown to significantly decrease the amount of mature RNA in the cytoplasm, suggesting that m6A modification participates in the process of mRNA maturation. Splicing is a key process during mRNA maturation in eukaryotic cells. Both m6A methyltransferases and demethylases are located in nuclear speckles, where splicing occurs. This suggests that m6A modification and mRNA splicing may be connected in some way. In fact, m6A modification was initially recognized as a splicing regulator (Zhao et al., 2017); and its interaction with splicing factors may represent one of the mechanisms through which m6A modification influences mRNA splicing. Studies have confirmed that m6A modification in alternatively spliced exons promotes alternatively spliced retention by recruiting SRSF3 and blocking the binding of the exon-skipping factor SRSF10, after being recognized by YTHDC1 (Roignant and Soller, 2017; Figure 1). The down-regulation of m6A modification by the knockout of m6A methyltransferases has been shown to reduce the splicing efficiency rate (Wei et al., 1975; Csepany et al., 1990; Gerken et al., 2007). Interaction with RNA-binding proteins is another mechanism of m6A modification during the regulation of mRNA splicing. HNRNPC is an RNA-binding protein that promotes mRNA processing and maturation by binding to a single-stranded RNA-binding motif hidden in pre-mRNA molecules. m6A modification make this single-stranded RNA-binding motif exposed to HNRNPC by weakening the base pairing. This allows HNRNPC to bind to target mRNA while promoting pre-mRNA processing and maturation (Figure 1); this process is called “m6A switch” (Liu et al., 2015).
Mature mRNAs have a 5′ cap and a 3′ tail to initiate translation and avoid nuclease-mediated degradation. Methylphosphate capping enzyme (MEPCE) is a crucial enzyme required for 5′ capping. Warda et al. (2017) performed co-immunoprecipitation and discovered that MEPCE is a part of the m6A methyltransferase complex. This implies that m6A methylation may be involved in 5′ capping of mRNAs (Warda et al., 2017). Furthermore, more than 70% of m6A methylations are located in the last exons of mRNAs, which affects the position of the 3′ polyA tail. This is evidenced by the knockout of m6A methyltransferases or m6A RNA-binding proteins, altering the position of the 3′ polyA mRNA tail and modifying the 3′-UTR (Beal et al., 2007; Warda et al., 2017).
2.3.1.2 Nuclear Export
Mature mRNA must translocate from the nucleus to the cytoplasm to perform its myriad functions; however, the knockout of m6A methyltransferases can hinder the nuclear export of mRNA (Fustin et al., 2013; Lesbirel et al., 2018). It has been confirmed that m6A modification facilitate the nuclear export of mRNA through the nuclear export complex, or via export complex factors. Specifically, m6A modification recruits the transcription–export (TREX) complex through METTL3; TREX then recruits NXF1, thus promoting the nuclear export of mRNA (Zheng et al., 2013; Lesbirel et al., 2018; Figure 1). Similarly, the reader proteins YTHDC1 (Roundtree et al., 2017) and FMRP (Lai et al., 2006; Hsu et al., 2019) promote mRNA export by transporting m6A-methylated mRNAs to the nuclear export receptors NXF1 and NXF2 (Figure 1), respectively.
2.3.1.3 RNA Translation
Eukaryotic initiation factor (eIF) 4E, which is a 7-methylguanosine-containing mRNA cap-binding protein, can recruit eIF3 and initiate cap-dependent translation. However, m6A located in the 5′ UTR of mRNA can be combined by ribosomes as a substitute for the 5′ cap. In this way, it can promote the initiation of cap-independent translation. Specifically, in the absence of eIF4E, the 5’ UTR of m6A binds to eIF3 through YTHDF1 (Wang et al., 2015; Lin et al., 2016), thus enabling mRNA to bind to ribosomes. This allows the consequent initiation of cap-independent translation (Berulava et al., 2020; Figure 1).
m6A modification exert a dual effect on the translation elongation of mRNA. On the one hand, m6A modification slows the binding of the ternary complex to the ribosomal A site and then delay translation elongation (Choi et al., 2016; Figure 1). On the other hand, the m6A RNA-binding protein YTHDC2 functions as an RNA helicase; it recognizes an m6A-methylation site and then accelerates translation elongation by unwinding the secondary structure of mRNA (Liu and Zhou, 2021; Figure 1). T Therefore, a balanced m6A-methylation process forms the basis of translation elongation, whereas an imbalance can influence the translation process.
Approximately >25% of m6A modifications in the entire transcriptome occur in the 3′ UTR, near the mRNA stop codon (Meyer et al., 2012), suggesting that m6A modifications may also participate in the termination of mRNA translation. However, the exact mechanism of the m6A modification’s role in translation termination requires further study.
2.3.1.4 mRNA Degradation
The function that m6A modification plays in the regulation of mRNA degradation was identified in 1978. The half-lives of m6A methylated mRNA were found to be significantly shorter than those of mRNA without m6A modification. Following studies confirmed that the up-regulation of m6A modification could facilitate mRNA degradation. Moreover, further studies have since revealed that YTHDF2 induced m6A-mediated mRNA degradation. YTHDF2 binds to the m6A-modification sites of mRNA with its C-terminus, whereas its N-terminus localizes mRNA to P body where RNA is degraded (Sheth and Parker, 2003). Currently, there are two known pathways for m6A-methylation-mediated mRNA degradation: the YTHDF2–carbon catabolite-repression 4 (CCR4)/negative on TATA-less (NOT) complex pathway (Figure 1) and the YTHDF2–heat-responsive protein 12 (HRSP12)–ribonuclease (RNase) P/mitochondrial RNA-processing (MRP) endonuclease complex pathway (Figure 1). YTHDF2 facilitates the deadenylation of mRNA by recruiting the CCR4/NOT complex, thereby promoting rapid, exonuclease-mediated mRNA degradation (Wang X. et al., 2014). On the other hand, m6A-methylated target mRNA containing HRSP12-binding sites have been found to be preferentially bound by HRSP12. This consequently promotes the binding of YTHDF2 to target mRNA. Thereafter, YTHDF2 accelerates mRNA degradation by recruiting the RNase P/MRP complex, which mediates mRNA degradation via its endonuclease activity (Park et al., 2019; Figure 1).
2.3.2 Non-Coding RNA
In addition to directly acting upon mRNA, m6A modification also regulates gene expression by acting on non-coding RNA. The DiGeorge syndrome critical region 8 (DGCR8)/Drosha ribonuclease III (DROSHA) complex acts as a crucial cleavage enzyme that participates in the processing and maturation of microRNA (miRNA). Specifically, m6A modification promotes the binding of primary-miRNA (pri-miRNA) with DGCR8, which further recruits DROSHA to promote maturation of pri-miRNA (Alarcón et al., 2015b; Figure 1). Similarly, m6A modification in the hairpin region of lncRNA also facilitates their processing and maturation by loosening the hairpin structure and facilitating their binding to HNRNPC via the m6A switch mechanism mentioned above (Figure 1). The closed-loop structure of circular RNA (circRNA) offers protection from exonuclease degradation; nevertheless, m6A can accelerate its degradation through the YTHDF2–HRSP12–RNase P/MRP pathway (Figure 1). Notably, m6A modification also affects the function of non-coding RNA. For example, miRNA can direct the silencing complex to degrade target mRNA. However, IGF2BPs can block target mRNA from binding to miRNA by recruiting human antigen R, thereby improving mRNA stability (Fan and Steitz, 1998; Peng et al., 1998).
As mentioned above, m6A modification participates in almost every stage of RNA metabolism, including RNA maturation, nuclear export, translation, and degradation 48. This indicates that m6A modification has the potential to prevent and treat diseases by regulating the fate and function of RNA.
3 m6A Methylation and Cardiovascular Pathophysiology
The process of m6A methylation is critical for the development, differentiation, homeostatic maintenance, and stress response of the cardiovascular system. Many m6A modifications are present in the heart (Dorn et al., 2019), comprising approximately a quarter of the total transcripts in healthy mouse and human hearts (Berulava et al., 2020). A high-throughput sequencing study discovered that m6A-methylated RNAs are widely involved in myocardial development, energy metabolism, stress response, and myocardial remodeling in human and mouse hearts (Berulava et al., 2020). Normal m6A modification levels maintain cardiac homeostasis, but METTL3 overexpression can disturb cardiac homeostasis and induce cardiomyocyte hypertrophy (Dorn et al., 2019). Therefore, regulating m6A methylation may be a novel approach for reversal of CVD progression.
3.1 Regulation of Cell Proliferation
Studies have confirmed that m6A methylation promotes tumorigenesis and tumor progression by regulating cell proliferation through the AKT, p38/ERK, and Wnt/β-catenin pathways (Liu et al., 2018; Deng et al., 2019; Li Y. et al., 2021). And latest research suggest that m6A methylation also plays great roles in cellular proliferation of cardiovascular system (Liu et al., 2018; Deng et al., 2019; Li Y. et al., 2021). One study found that ALKBH5 knockdown inhibits cardiomyocyte proliferation in neonatal mice, whereas overexpression promotes cardiomyocyte proliferation following myocardial infarction. Furthermore, ALKBH5 promotes YAP translation by enhancing the stability of YTHDF1 mRNA in an m6A dependent manner (Han et al., 2021b), which enables cardiomyocytes to re-enter the cell cycle and proliferate (Table 1). Upregulation of WTAP expression in vascular smooth muscle cells (VSMCs) promotes p16 expression by increasing m6A methylation level of p16 mRNA and inhibits cell proliferation of VSMCs (Zhu et al., 2020; Table 1). Similarly, FTO knockdown increases m6A methylation of SM22α; then the m6A RNA-binding protein IGF2BPs recognizes m6A-methylated SM22α mRNA and enhances its stability and expression, thus inhibiting the proliferation and migration of VSMCs in type 2 diabetes patients, while also improving intimal hyperplasia (Zhang et al., 2022; Table 1). Of note, cell proliferation plays different roles in various CVDs. For example, cell proliferation reduces myocardial infarct size and improves cardiac function; however, hyperproliferation of VSMCs can cause atherosclerosis. Therefore, it may be inefficient to regulate cell proliferation by altering m6A methylase or demethylase expression. Alternatively, precise regulation of m6A methylation levels using gene editing technologies may be a good approach to the regulation of cell proliferation of cardiovascular system (Liu X.-M. et al., 2019; Rau et al., 2019).
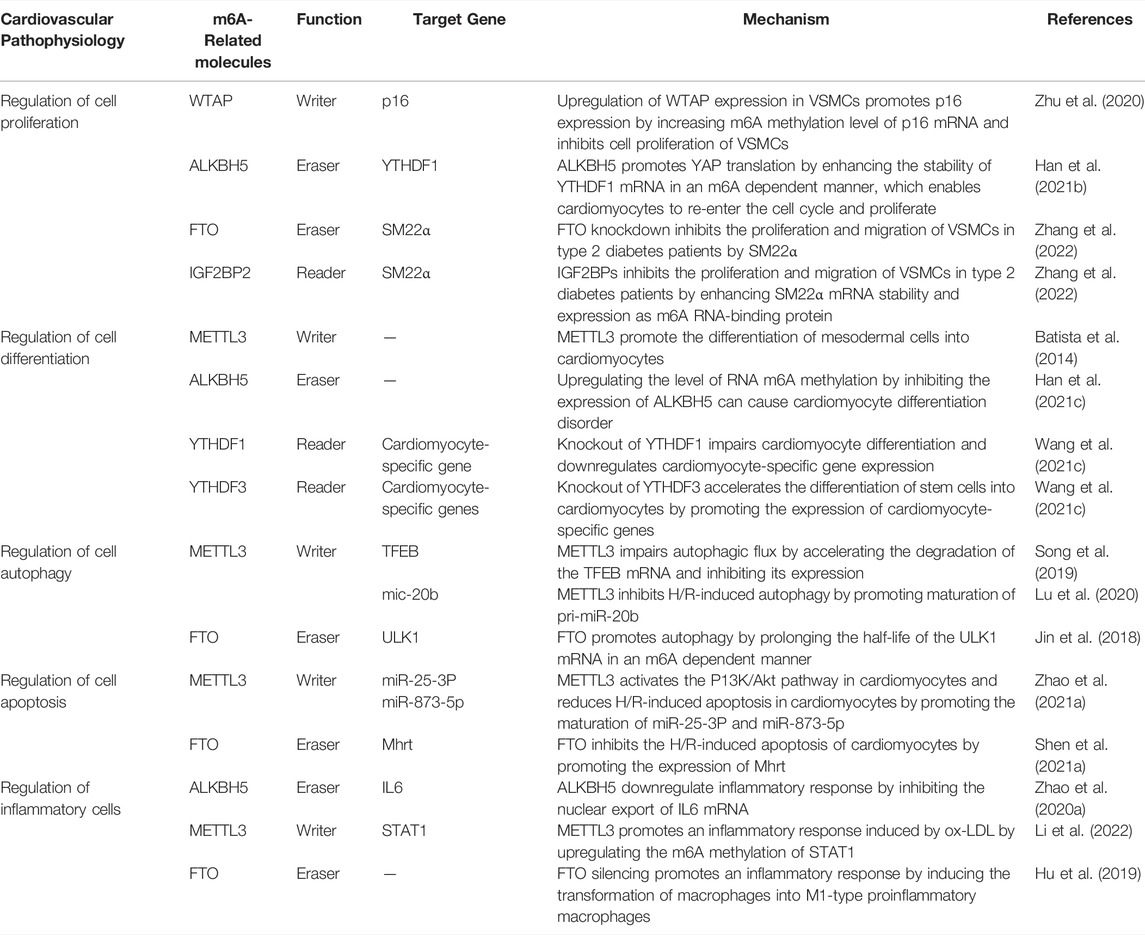
TABLE 1. The role of m6A methylation in cardiovascular pathophysiology.
3.2 Regulation of Cell Differentiation
Presently, studies have confirmed that m6A methylation plays several roles in post-transcriptional modification during stem cell differentiation, and it determines the fate of embryonic stem cells (Batista et al., 2014; Wang Y. et al., 2014; Slobodin et al., 2017; Wen et al., 2018; Kwon et al., 2019; Han et al., 2021c; Wang S. et al., 2021). In fact, complete knockout of the m6A methyltransferases METTL3 (Geula et al., 2015) and METTL14 (Yoon et al., 2017) leads to embryonic death, suggesting that m6A methylation is critical for embryonic development and organ differentiation. During embryonic development, the heart develops from mesodermal cells. In this regard, a study has found that when mesodermal cells differentiate into cardiomyocytes, the level of RNA m6A methylation is significantly upregulated. Furthermore, an in vitro differentiation experiment on mouse embryonic stem cells (ESCs) revealed that only 3% of METTL3-knockout ESCs generated beating cardiomyocytes, whereas 50% of the control cells generated beating cardiomyocytes (Batista et al., 2014; Table 1). The m6A demethylase ALKBH5 is also involved in ESC-directed differentiation of cardiomyocytes. Thus, upregulating the level of RNA m6A methylation by inhibiting the expression of ALKBH5 can cause cardiomyocyte differentiation disorder. In contrast, overexpression of ALKBH5 inhibits this differentiation (Han et al., 2021c; Table 1). m6A RNA-binding proteins are also crucial in the differentiation of stem cells into cardiomyocytes. For example, knockout of YTHDF1 in mouse embryos severely impairs cardiomyocyte differentiation and downregulates cardiomyocyte-specific gene expression (Wang S. et al., 2021; Table 1). In contrast, knockout of YTHDF3 accelerates the differentiation of stem cells into cardiomyocytes by promoting the expression of cardiomyocyte-specific genes (Wang S. et al., 2021; Table 1). Thus, YTHDF1 and YTHDF3 play opposing roles in the differentiation of stem cells into cardiomyocytes, wherein YTHDF3 regulates cell differentiation by partially inhibiting the action of YTHDF1 (Wang S. et al., 2021). Therefore, m6A methylation regulates cell differentiation in the cardiovascular system, and its absence can lead to severe differentiation disorders.
3.3 Regulation of Cell Autophagy
Autophagy, in which m6A methylation modification plays a role, protects the heart from damage by inhibiting cardiomyocyte apoptosis (Saito et al., 2019). Ischemia-reperfusion (I/R) increases the level of m6A methylation in the mRNA of the transcription factor EB (TFEB) by inducing the expression of METTL3. This accelerates the degradation of the TFEB mRNA and inhibits TFEB expression, impairs autophagic flux, and promotes apoptosis (Song et al., 2019). In addition, m6A methylation regulates autophagy via miRNAs. In H/R-treated endothelial cells, METTL3 recruits DGCR8 to bind to pri-miR-20b in an m6A-dependent manner, promoting maturation of pri-miR-20b. The mature mic-20b consequently inhibits H/R-induced autophagy by inhibiting unc-51-like kinase 1 (ULK1) expression (Lu et al., 2020; Table 1). In contrast, FTO can directly reverses m6A methylation on ULK1, thereby prolonging the half-life of the ULK1 mRNA and promoting autophagy (Jin et al., 2018; Table 1). Notably, regulation of autophagy by m6A methylation is tissue- and disease-specific. For example, m6A methylation in CVDs inhibits H/R-induced autophagic flux, but in other diseases, m6A methylation promotes autophagy by accelerating its initiation (Chen X. et al., 2021). Indeed, the effect that m6A methylation has on autophagy depends on the target mRNA it modifies. Therefore, mapping the landscape of m6A-methylated target mRNAs can help to further explore the mechanism of m6A methylation in autophagy.
3.4 Regulation of Cell Apoptosis
Currently, studies investigating the role of m6A methylation in the regulation of apoptosis have presented contradictory observations. For instance, METTL3 enhances the binding of miR-25-3P and miR-873-5p to DGCR8 in an m6A-dependent manner to promote their maturation, which in turn activates the P13K/Akt pathway in cardiomyocytes and reduces H/R-induced apoptosis in cardiomyocytes (Zhao X. et al., 2021; Table 1). Interestingly, overexpression of FTO promotes the expression of Mhrt by downregulating m6A modification of Mhrt and inhibits the H/R-induced apoptosis of cardiomyocytes (Shen W. et al., 2021; Table 1). METTL3 and FTO have opposing effects on cardiomyocyte apoptosis, likely because of differential regulation of m6A methylation in different target mRNAs. Furthermore, studies have confirmed that targeting point mutations in the m6A modification sites of target transcripts using gene editing technologies can regulate apoptosis (Hao et al., 2020); thus, m6A methylation is a potentially effective means of regulating apoptosis. However, target transcripts that affect apoptosis first need to be identified.
3.5 Regulation of Inflammatory Cells
m6A methylation affects the autoimmunity and inflammatory response by regulating inflammatory cells, such as macrophages (Bechara and Gaffen, 2021). For instance, ALKBH5 inhibits the nuclear export of interleukin-6 (IL6) mRNA by demethylation, thus downregulating an inflammatory response (Zhao J. et al., 2020; Table 1). On the contrary, METTL3 promotes an inflammatory response induced by oxidized low-density lipoprotein (ox-LDL) by upregulating the m6A methylation of STAT1 (Li et al., 2022; Table 1). Similar to METTL3 overexpression, FTO silencing induces the transformation of macrophages into M1-type proinflammatory macrophages (Hu et al., 2019), thereby promoting an inflammatory response (Table 1). At present, the role and mechanisms of m6A methylation in regulating inflammation are still being studied. What we know is that MAPK and NF-κB inflammatory signaling pathways are targets of m6A methylation (Yu et al., 2019); With the progress of research, many other inflammatory signaling pathways regulated by m6A methylation will be found.
4 m6A Methylation and CVDs
CVDs, including cardiac and vascular diseases, are the leading cause of death i humans. As mentioned above, m6A affects the pathophysiology of CVDs by regulating cell proliferation, differentiation, autophagy, apoptosis, and inflammation (Song et al., 2019; Wang Y.-J. et al., 2021). Therefore, regulation of m6A methylation may possibly reverse the progression and provide new treatment strategies for CVDs. m6A sequencing data is crucial to screen key m6A target genes and we have summarize the publicly available m6A sequencing data in Gene Expression Omnibus (GEO) related to CVD in Table 2.
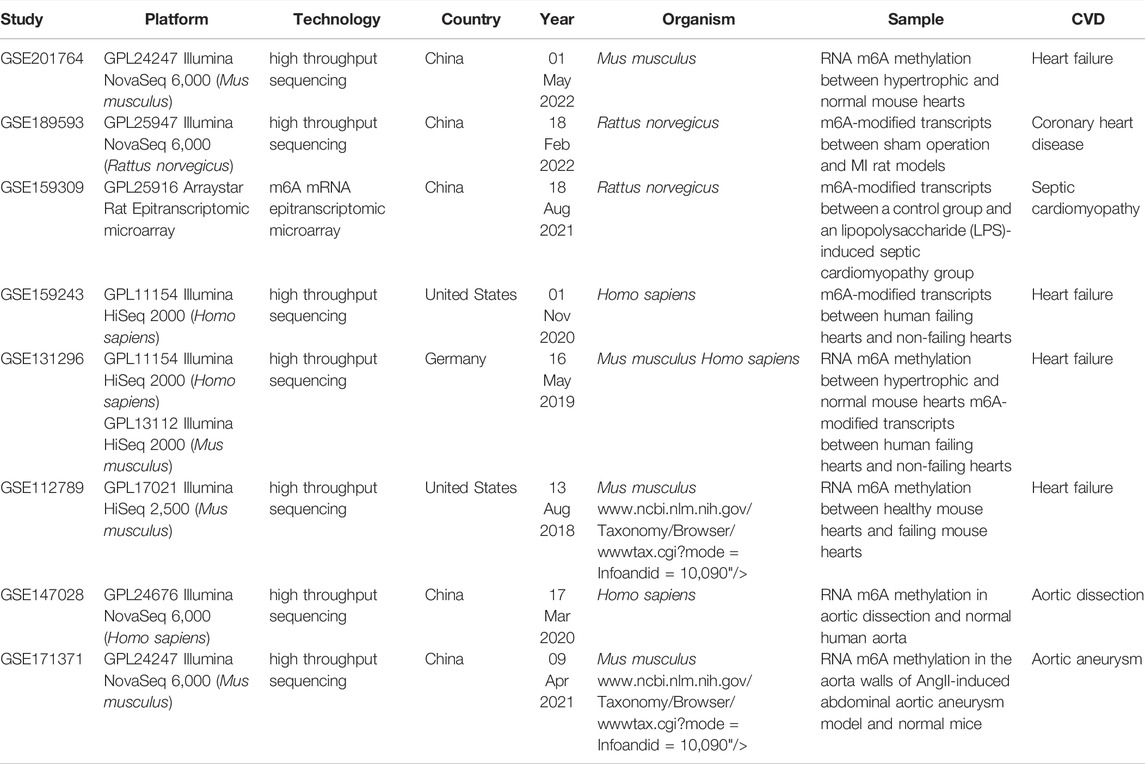
TABLE 2. m6A sequencing and microarray data in GEO related to CVD.
4.1 m6A Methylation and Vascular Diseases
Vascular diseases include atherosclerosis, pulmonary hypertension, and aortic aneurysm/dissection. As mentioned above, m6A methylation plays great role in the pathophysiology of vascular diseases, such as regulating endothelial cell proliferation, inflammation, and VSMC proliferation. While research on m6A methylation in vascular diseases is still in its infancy, m6A methylation seems to be a potential treatment target for vascular diseases.
4.1.1 m6A Methylation and Atherosclerosis
Atherosclerosis, the primary culprit of cardiovascular disease, is characterized by the formation of atherosclerotic or fibrous plaques in the arterial intima of primarily large and medium-sized arteries. And for now, the mechanism of arteriosclerosis is still unclear. Recently, many studies have confirmed that the level of m6A methylation were significantly changed and it can delay the progression of atherosclerosis through the post-transcriptional regulation of RNA.(Zhang BY. et al., 2020; Guo et al., 2020; Jian et al., 2020; Chien et al., 2021; Gong et al., 2021).
The inflammation of endothelial cell is an initial factor in the development of coronary atherosclerosis. Jian et al. found significantly increased RNA m6A modification levels in a tumor necrosis factor (TNF)-α-induced inflammation model of endothelial cell through up-regulation of METTL14. Moreover, they identified that the ablation of METTL14 significantly inhibited the expression of TNF-α-induced inflammatory factors, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cellular adhesion molecule-1 (VCAM-1) (Jian et al., 2020; Table 3 and Figure 2). Zhang et al., meanwhile, found that METTL3 aggravated the inflammatory response of monocytes and promoted monocyte–endothelial cell adhesion by inducing the degradation of peroxisome proliferators-activated receptor γ coactivator l alpha (PGC-1α) mRNA in an m6A dependent way (Zhang X. et al., 2021; Table 3 and Figure 2). Chien et al. discovered that METTL3 up-regulated nucleotide-binding domain leucine-rich repeat pyrin domain containing 1 (NLRP1) and down-regulated Kruppel-like factor 4 (KLF4) through m6A modification of NLRP1 and KLF4 mRNA, and effectively promoted TNF-α-mediated inflammation in endothelial cells (Chien et al., 2021; Table 3 and Figure 2). Moreover, Li et al. and Liu et al. both reported that METTL3 modified signal transducer and activator of transcription 1 (STAT1) coding sequence and the 3’ UTR via m6A modification, thereby improving STAT1 mRNA stability, upregulating STAT1 protein levels, and promoting the M1 polarization of macrophages and inflammatory response (Liu Y. et al., 2019; Li et al., 2022; Table 3 and Figure 2).
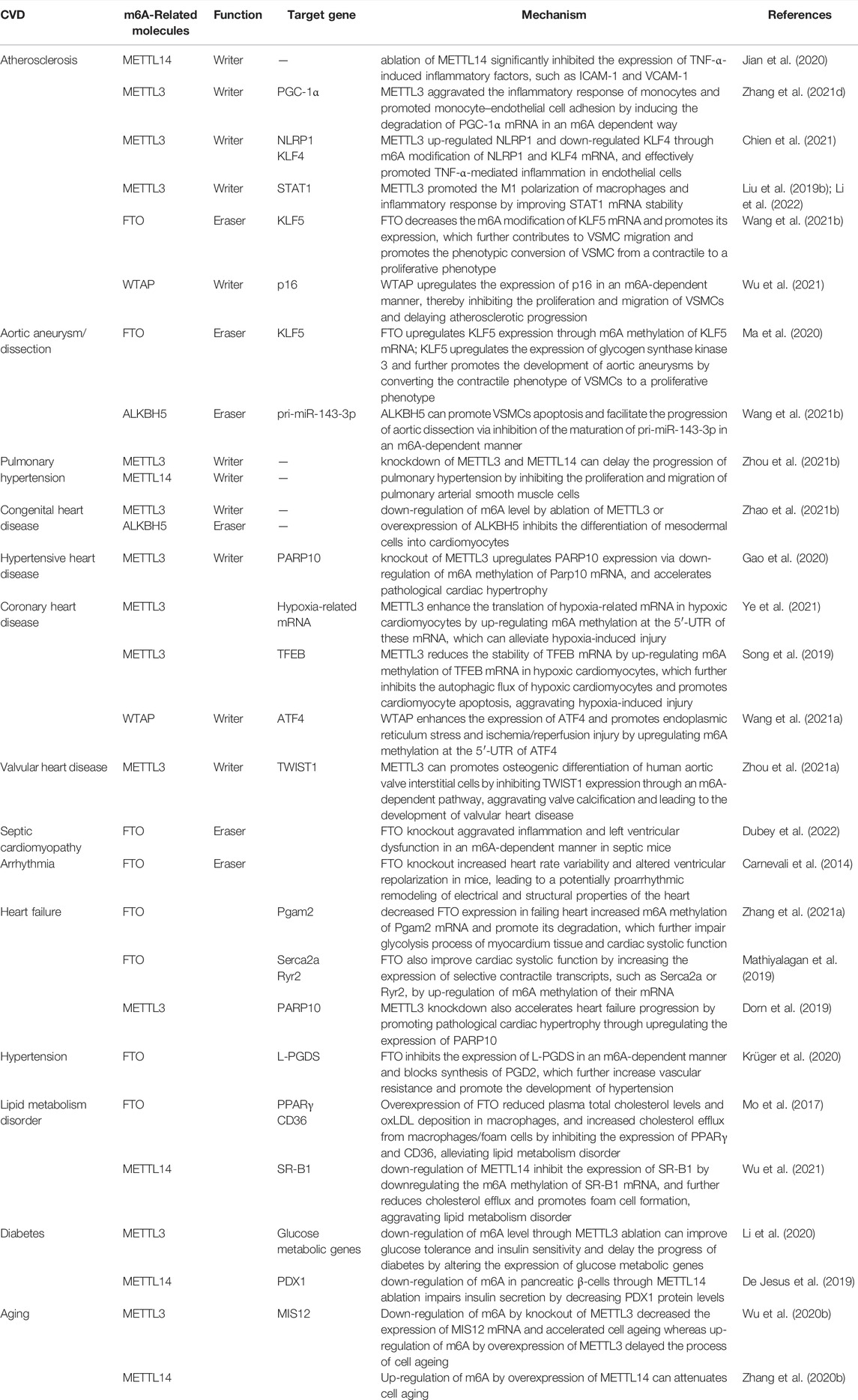
TABLE 3. The role of m6A methylation in CVD.
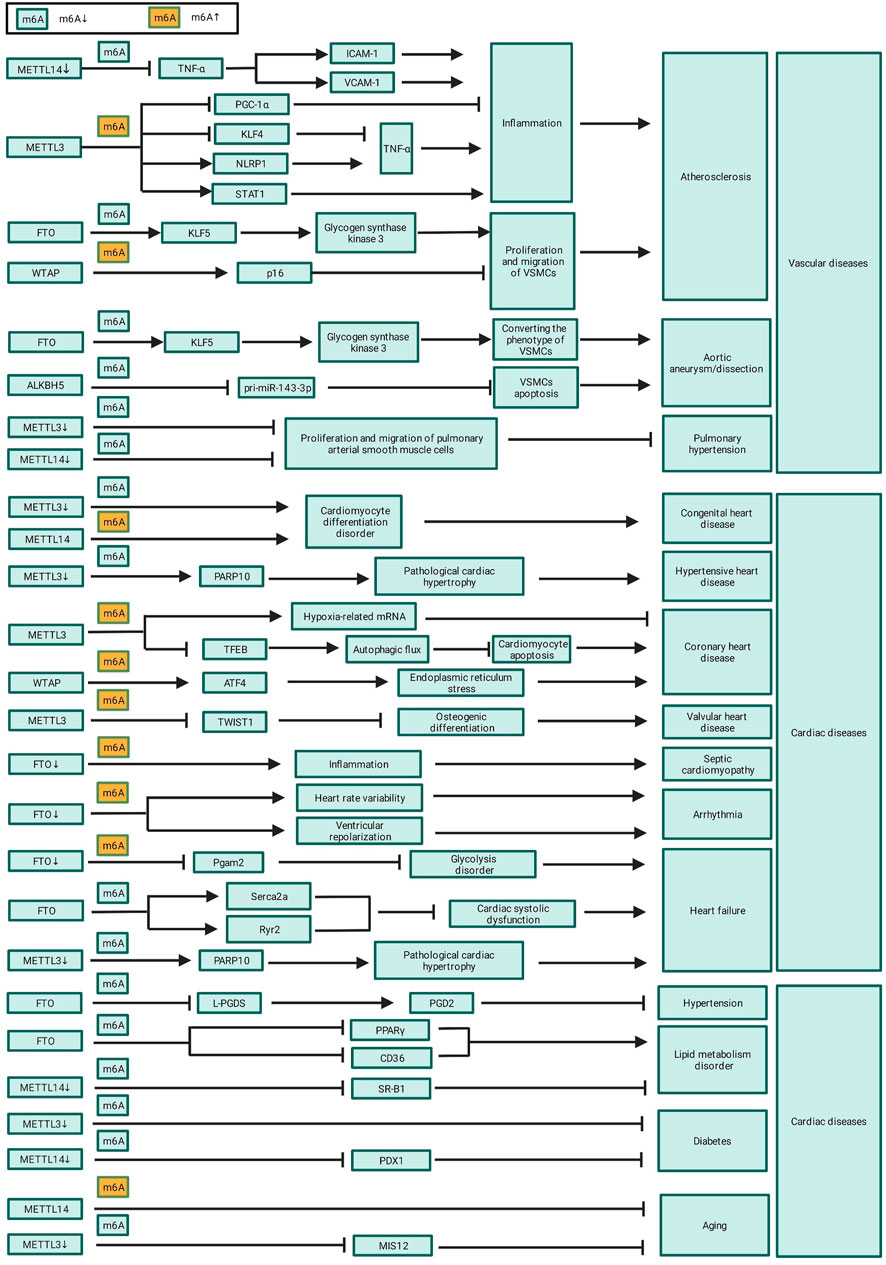
FIGURE 2. The role of m6A in CVD.
m6A modification also participates in the progress of atherosclerosis through regulating the proliferation, migration, and phenotypic transformation of VSMCs 12. Specifically, FTO promotes the expression of Krueppel-like factor 5 (KLF5) by decreasing the m6A modification of KLF5 mRNA; this consequently upregulates the downstream expression of glycogen synthase kinase 3. This, in turn, contributes to VSMC migration and promotes the phenotypic conversion of VSMCs from a contractile to a proliferative phenotype, thereby promoting the development of atherosclerosis (Wang P. et al., 2021; Table 3 and Figure 2). Conversely, WTAP upregulates the expression of p16 in an m6A-dependent manner, thereby inhibiting the proliferation and migration of VSMCs and delaying atherosclerotic progression (Wu et al., 2021; Table 3 and Figure 2).
4.1.2 m6A Methylation and Aortic Aneurysm/Dissection
Compared to normal human, the level of m6A methylation in aortic tissues of aortic aneurysm patients is significantly upregulated. This high level of m6A methylation is associated with an increased risk of aortic aneurysm rupture, indicating that m6A methylation is involved in the development and prognosis of aortic aneurysm and dissection (He et al., 2019). m6A methylation is possibly involved in the progression of aortic dissection via regulation of smooth muscle cell (Li T. et al., 2021). Ma et al. found that FTO upregulates KLF5 expression through m6A methylation of KLF5 mRNA; KLF5 upregulates the expression of glycogen synthase kinase 3 and further promotes the development of aortic aneurysms by converting the contractile phenotype of VSMCs to a proliferative phenotype (Ma et al., 2020; Table 3 and Figure 2). Wang J. et al. (2021) found that ALKBH5 can promote VSMCs apoptosis and facilitate the progression of aortic dissection via inhibition of the maturation of pri-miR-143-3p in an m6A-dependent manner (Wang P. et al., 2021; Table 3 and Figure 2).
4.1.3 m6A Methylation and Pulmonary Hypertension
Several studies have reported that m6A methylation levels are significantly upregulated in a rat model of pulmonary hypertension, and these differentially m6A methylated genes proved to be involved in inflammation, glycolysis, endothelial cell receptor activation, and lung development, which can accelerate the progress of pulmonary hypertension (Xu et al., 2021; Zeng et al., 2021). Down-regulation of m6A modification by knockdown of METTL3 and METTL14 can delay the progression of pulmonary hypertension by inhibiting the proliferation and migration of pulmonary arterial smooth muscle cells (Zhou X.-L. et al., 2021; Table 3 and Figure 2). In addition, m6A methylation was also found to affect the progression of pulmonary hypertension through the circRNA-miRNA-mRNA network, and two pulmonary hypertension-related circRNAs, circXpo6 and circTmtc3, were identified (Su et al., 2020). Of note, m6A methylation is a biomarker of epigenetic modifications in key genes that regulate pulmonary arterial pressure and lung development; however, further study is needed to characterize the specific regulatory mechanisms underlying the involvement of m6A methylation in pulmonary hypertension.
4.2 m6A Methylation and Cardiac Diseases
Cardiac diseases include congenital, hypertensive, atherosclerotic, and valvular heart diseases as well as cardiomyopathy, arrhythmia, and heart failure. As mentioned above, m6A methylation play a crucial role in the pathogenesis of cardiac diseases. Currently, the landscape of m6A modifications in hypertensive heart disease and heart failure patients has been mapped; thus, future research needs to focus on mapping the landscape and modification patterns in other cardiac diseases, in order to identify key transcripts that regulate the progression of cardiac diseases.
4.2.1 m6A Methylation and Congenital Heart Disease
The heart is developed from mesodermal cells during embryonic development; abnormal differentiation of embryonic stem cells lead to congenital heart disease (Kattman et al., 2007; Bu et al., 2009). m6A methylation has been proved to be a critical role in embryonic stem cell differentiation and cardiomyocyte proliferation. Additionally, m6A methylation is a key regulator of maintaining the pluripotency of embryonic stem cells, reprogramming of somatic cells, and differentiation and proliferation of stem and progenitor cells (Malla et al., 2019). As previously mentioned, down-regulation of m6A level by ablation of METTL3 or overexpression of ALKBH5 inhibits the differentiation of mesodermal cells into cardiomyocytes (Table 3; Figure 2). Therefore, there is a link between m6A methylation and congenital heart diseases. Exploring the specific role and therapeutic potential of m6A methylation in the progression of congenital heart disease will help to the prevention and treatment of congenital heart disease.
4.2.2 m6A and Hypertensive Heart Disease
Hypertensive heart disease is charactered by left ventricular hypertrophy and heart failure caused by uncontrolled hypertension. Cardiomyocyte hypertrophy is an adaptive response to afterloads caused by uncontrolled hypertension; however, persistent cardiomyocyte hypertrophy induces cardiomyocyte apoptosis, necrosis, and fibrotic proliferation, ultimately causing heart failure (Krüger et al., 2020).
Some studies have shown that the percentage of m6A-methylated RNA was significantly increased in hypertension-induced hypertrophic cardiomyocytes compared to that of healthy cardiomyocytes (Dorn et al., 2019; Berulava et al., 2020) and that the degree of cardiac hypertrophy is positively correlated with the level of m6A methylation (Dorn et al., 2019; Chen Y.-S. et al., 2021). Overexpression of METTL3 can induce compensatory cardiomyocyte hypertrophy without deteriorating cardiac function (Dorn et al., 2019), whereas knockout of METTL3 upregulates PARP10 expression via down-regulation of m6A methylation of Parp10 mRNA, and accelerates pathological cardiac hypertrophy (Gao et al., 2020; Table 3 and Figure 2). Down-regulation of m6A might be one of the mechanisms by which hypertension-induced pathological cardiac hypertrophy and heart failure.
4.2.3 m6A Methylation and CHD
CHD, also called ischemia heart disease, is characterized by a reduction of oxygen supply due to coronary atherosclerosis. Shen et al. found that their is substantial difference between the transcriptomes and proteomes of hypoxic cardiomyocytes, implying epigenetic modifications of RNA occurred in hypoxic cardiomyocytes (Shen et al., 2020).
Rapid gene expression after hypoxia stress is required to prevent cardiomyocytes from the hypoxia-induced injury. As the most abundant RNA modification, m6A methylation is dramatically up-regulated in cardiomyocytes after hypoxia exposure (Fry et al., 2017; Chokkalla et al., 2019; Ye et al., 2021). Ye et al. reported that m6A methylation at the 5′-UTR of hypoxia-related mRNA is upregulated via overexpression of METTL3 induced by hypoxia; and this promote the localization of eIF4A2 to target genes and enhance the translation of these genes in hypoxic cardiomyocytes, which can alleviate hypoxia-induced injury (Ye et al., 2021; Table 3 and Figure 2). However, up-regulation of m6A methylation maybe a double-edged sword after hypoxia exposure. Song et al. found that the METTL3 reduces the stability of TFEB mRNA by up-regulating m6A methylation of TFEB mRNA in hypoxic cardiomyocytes, which further inhibits the autophagic flux of hypoxic cardiomyocytes and promotes cardiomyocyte apoptosis, aggravating hypoxia-induced injury (Song et al., 2019). Activation of transcription factor 4 (ATF4) enhances the expression of endoplasmic reticulum stress-related genes and contributes to myocardial ischemia/reperfusion injury by promoting endoplasmic reticulum stress. The m6A methylation at the 5′-UTR of ATF4 is up-regulated through increased WTAP expression induced by ischemia/reperfusion injury and this promote the binding of eIF3A to ATF4, which further enhances the expression of ATF4 and promotes endoplasmic reticulum stress and ischemia/reperfusion injury (Wang J. et al., 2021).
4.2.4 m6A Methylation and Valvular Heart Disease
Redifferentiation of interstitial cells in the human aortic valve into osteoblast-like cells is an important mechanism of aortic valve calcification, which is a primary cause of heart valve disease. METTL3 can promotes osteogenic differentiation of human aortic valve interstitial cells by inhibiting twist-related protein 1 (TWIST1) expression through an m6A-dependent pathway, aggravating valve calcification and leading to the development of valvular heart disease (Zhou T. et al., 2021). At present, there are only a few studies on the role of m6A methylation in valvular heart disease. Thus, further research is needed to decipher the exact mechanism and therapeutic value of m6A methylation in this disease.
4.2.5 m6A Methylation and Septic Cardiomyopathy
Approximately, septic cardiomyopathy occurs in 50% of septic shock patients. Moreover, cardiac dysfunction caused by septic cardiomyopathy is a key cause of high mortality in these patients (Zaky et al., 2014). m6A methylation pattern in cardiac tissue of septic shock rat model was found to be significantly different from control rat. Additionally, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses showed that differentially m6A-methylated mRNAs are mainly involved in interleukin-17 (IL17) signaling pathways, TNF signaling pathway, and cytokine receptor interaction pathway, suggesting that m6A methylation possibly participate in the progression of septic cardiomyopathy through immune and inflammatory response (Han Y.-C. et al., 2021; Shen Z.-J. et al., 2021). FTO was proved to be a significant regulator of the m6A methylation level in the myocardial tissue of septic mice. FTO knockout aggravated inflammation and left ventricular dysfunction in an m6A-dependent manner in septic mice (Dubey et al., 2022). Therefore, m6A methylation may be a potential target for alleviating cardiac dysfunction in septic shock patients.
4.2.6 m6A Methylation and Arrhythmia
To date, only a few studies explored the relationship between m6A modifications and arrhythmias. In one study, m6A demethylase FTO knockout increased heart rate variability and altered ventricular repolarization in mice, leading to a potentially proarrhythmic remodeling of electrical and structural properties of the heart (Table 3; Figure 2). It is suggested that m6A methylation can make the mice more prone to stress-induced tachyarrhythmias (Carnevali et al., 2014). However, further research is required to identify the m6A-regulated target genes associated with arrhythmia and to assess the involvement of other m6A methyltransferases and demethylases.
4.2.7 m6A Methylation and Heart Failure
Extensive studies have confirmed that the level of m6A methylation is significantly upregulated. METTL3 and FTO were proved to be the key regulators of m6A methylation in failure heart. The expression of METTL3 is increased while that of FTO is reduced; the synergistic function of METTL3 and FTO up-regulate the level of m6A methylation in failing hearts (Hinger et al., 2021). GO and KEGG pathway analysis show that these differential m6A methylation modifications are not randomly distributed but actually enriched in certain mRNA subsets, including mRNAs involved in glycolytic metabolism, mitochondrial function, myocardial fibrosis and cardiac contractility (Berulava et al., 2020; Zhang et al., 2021a; Hinger et al., 2021).
Glycolysis is essential to myocardial energy metabolism; myocardial energy metabolic disorders would directly affect cardiac systolic function. In a transverse aortic constriction (TAC)-induced heart failure model, Zhang et al. found that decreased FTO expression in failing heart increased m6A methylation of Pgam2 mRNA and promote its degradation, which further impair glycolysis process of myocardium tissue and cardiac systolic function (Zhang et al., 2021a). FTO also improve cardiac systolic function by increasing the expression of selective contractile transcripts, such as Serca2a or Ryr2, by up-regulation of m6A methylation of their mRNA (Mathiyalagan et al., 2019). These suggest FTO is a potential therapeutic in the treatment of heart failure.
However, METTL3 knockdown also accelerates heart failure progression by promoting pathological cardiac hypertrophy through upregulating the expression of PARP10 as mentioned above (Dorn et al., 2019). This may be explained by the fact that METTL3 and FTO participate in the progression of heart failure by regulating m6A methylation of target transcriptions rather regulating than the global level of m6A methylation. The target transcriptions of METTL3 or FTO determine their roles of in the progression of heart failure.
4.3 M6A Methylation and CVD Risk Factors
4.3.1 m6A Methylation and Hypertension
A preliminary understanding on how m6A methylation affects the development of hypertension is primarily based on the high-throughput sequencing data of hypertension patients. Mo et al. identified more m6A-single nucleotide polymorphism (m6A-SNP) loci in hypertension patients than in healthy individuals. Remarkably, they identified 1,236 hypertension-specific m6A-SNPs (Mo et al., 2019). Wu et al. found that coding regions of mRNA were enriched with m6A modifications in hypertensive rats. Additionally, compared to those of control mice, hypertensive mice exhibited significant differences in the number of m6A modifications. Furthermore, GO and KEGG pathway analyses revealed that m6A-modified genes are involved in inflammation, proximal tubule development, RNA methyltransferase activity, water channel activity, actin cytoskeleton pathway regulation, and neuroligand receptor activity.
Krüger et al. (2020) revealed that FTO-mediated m6A demethylation plays a key role in regulating arterial myogenic contraction and vascular resistance. Prostaglandin D2 (PGD2) activates G protein-coupled receptors expressed on vascular smooth muscle membranes to promote the relaxation of VSMCs, delaying the progression of hypertension. Lipocalin-type prostaglandin D synthase (L-PGDS) is the main enzyme to synthesize PGD2. In human and mouse blood vessels, FTO inhibits the expression of L-PGDS in an m6A-dependent manner and blocks synthesis of PGD2, which further increase vascular resistance and promote the development of hypertension (Krüger et al., 2020).
4.3.2 m6A Methylation and Lipid Metabolism Disorder
Disorders of lipid metabolism are common drivers of atherosclerosis. Notably, m6A methylation affects blood lipid levels and elevates blood lipid deposition, aggravating atherosclerotic progression (Davis et al., 2014; Chedraui et al., 2016). Overexpression of FTO reduced plasma total cholesterol levels and ox-LDL deposition in macrophages, and increased cholesterol efflux from macrophages/foam cells by inhibiting the expression of peroxisome proliferator-activated receptor γ (PPARγ) and cluster of differentiation 36 (CD36), alleviating lipid metabolism disorder (Mo et al., 2017; Table 3 and Figure 2). Wu et al. found that down-regulation of METTL14 inhibit the expression of scavenger receptor B-type 1 (SR-B1) by downregulating the m6A methylation of SR-B1 mRNA, and further reduces cholesterol efflux and promotes foam cell formation, aggravating lipid metabolism disorder (Wu et al., 2021; Table 3 and Figure 2).
4.3.3 m6A Methylation and Diabetes
Li et al. found that m6A level is increased in diabetes mice model due to upregulation of METTL3 induced by diabetes. To verify the role of m6A in diabetes, they constructed METTL3 knockout mice and confirmed that down-regulation of m6A level through METTL3 ablation can improve glucose tolerance and insulin sensitivity and delay the progress of diabetes by altering the expression of glucose metabolic genes (Li et al., 2020). However, different from the findings of Li et al., Jesus et al. verified that m6A level and METTL14 expression in pancreatic β-cells of diabetes patients is decreased, and down-regulation of m6A in pancreatic β-cells through METTL14 ablation impairs insulin secretion by decreasing pancreatic and duodenal homeobox 1 (PDX1) protein levels (De Jesus et al., 2019). This difference can be interpreted with the poor conservatism of m6A modification between different organs. But they both confirmed that m6A modification is involved in the progress of diabetes. Further studies are needed to explore the mechanism of m6A modification involved in the progress of diabetes.
4.3.4 m6A Methylation and Aging
Aging is an important risk factor of CVD. Numerous reports have explored the relationship of m6A modification and aging, and found that m6A modification participated in the ageing process. Wu et al. found that the level of m6A and METTL3 is reduced in aged cells. Down-regulation of m6A by knockout of METTL3 decreased the expression of MIS12 mRNA and accelerated cell ageing whereas up-regulation of m6A by overexpression of METTL3 delayed the process of cell ageing (Wu Z. et al., 2020). Up-regulation of m6A by overexpression of METTL14 can also attenuates cell aging (Zhang J. et al., 2020). These studies show that m6A modification has the potential as a therapeutic target in the treatment of aging-related disease.
5 Prospects and Summary
Over the past 10 years, researchers have mapped the m6A methylation landscape in CVDs, such as heart failure and coronary heart disease, confirmed that regulation of m6A methylation can reverse pathophysiological processes. Additionally, there is a complicated interrelations between m6A and other epigenetics regulation layers like histone modifications and non-coding RNAs. For example, m6A demethylase ALKBH5 as mentioned above facilitate the progression of aortic dissection via inhibition of the maturation of pri-miR-143-3p in an m6A-dependent manner. The intricate crosstalk between m6A and other epigenetics regulation layers triggers epigenetic remodeling, further affecting the progression of CVDs. So, it is a promising direction to explore the relationship of m6A and other epigenetics regulation layers.
Another challenge is that m6A methylases and demethylases regulate m6A methylation of RNA without high-specificity. They also regulate the m6A methylation levels of other genes when they were used to regulate the metabolism of target transcriptions, which bring obstacles in the application of these m6A methylation regulators. However, studies using gene editing technologies to regulate the m6A modifications of a single target transcript have achieved remarkable results (Liu X.-M. et al., 2019; Rau et al., 2019), indicating that gene editing may be a more precise tool to regulate m6A methylation.
In all, we discussed the biological role of m6A methylation and its potential therapeutic role in CVDs. At present, research investigating the role of m6A methylation in the occurrence and development of CVDs is still in its infancy; however, future research may identify additional functions and underlying mechanisms of m6A methylation in CVDs, which will be a major advance in the field of epigenetics.
Author Contributions
LL wrote and edited the manuscript; ZC, JL, and XL collated the literature; NX edited and revised the manuscript; JW designed the study.
Funding
This work was supported by Development and Reform Commission of Jilin Province (2020C036-3), Jilin Province Department of Finance (2020SCZT003), Scientific Research Project of Education Department of Jilin Province (Grant No. JJKH20211167KJ), and the Science and Technology Development of Jilin Province (20200301003RQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alarcón, C. R., Goodarzi, H., Lee, H., Liu, X., Tavazoie, S., and Tavazoie, S. F. (2015a). HNRNPA2B1 Is a Mediator of m6A-dependent Nuclear RNA Processing Events. Cell 162, 1299–1308. doi:10.1016/j.cell.2015.08.011
Alarcón, C. R., Lee, H., Goodarzi, H., Halberg, N., and Tavazoie, S. F. (2015b). N6-methyladenosine Marks Primary microRNAs for Processing. Nature 519, 482–485. doi:10.1038/nature14281
Andersson, C., and Vasan, R. S. (2018). Epidemiology of Cardiovascular Disease in Young Individuals. Nat. Rev. Cardiol. 15, 230–240. doi:10.1038/nrcardio.2017.154
Batista, P. J., Molinie, B., Wang, J., Qu, K., Zhang, J., Li, L., et al. (2014). m6A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell 15, 707–719. doi:10.1016/j.stem.2014.09.019
Beal, P. A., Maydanovych, O., and Pokharel, S. (2007). The Chemistry and Biology of RNA Editing by Adenosine Deaminases. Nucleic Acids Symp. Ser. 51, 83–84. doi:10.1093/nass/nrm042
Bechara, R., and Gaffen, S. L. (2021). '(m6)A' Stands for 'autoimmunity': Reading, Writing, and Erasing RNA Modifications during Inflammation. Trends Immunol. 42, 1073–1076. doi:10.1016/j.it.2021.10.002
Berulava, T., Buchholz, E., Elerdashvili, V., Pena, T., Islam, M. R., Lbik, D., et al. (2020). Changes in m6A RNA Methylation Contribute to Heart Failure Progression by Modulating Translation. Eur. J. Heart Fail. 22, 54–66. doi:10.1002/ejhf.1672
Bu, L., Jiang, X., Martin-Puig, S., Caron, L., Zhu, S., Shao, Y., et al. (2009). Human ISL1 Heart Progenitors Generate Diverse Multipotent Cardiovascular Cell Lineages. Nature 460, 113–117. doi:10.1038/nature08191
Carnevali, L., Graiani, G., Rossi, S., Al Banchaabouchi, M., Macchi, E., Quaini, F., et al. (2014). Signs of Cardiac Autonomic Imbalance and Proarrhythmic Remodeling in FTO Deficient Mice. PloS One 9, e95499. doi:10.1371/journal.pone.0095499
Cavalli, G., and Heard, E. (2019). Advances in Epigenetics Link Genetics to the Environment and Disease. Nature 571, 489–499. doi:10.1038/s41586-019-1411-0
Chedraui, P., Pérez-López, F. R., Pérez-López, F. R., Escobar, G. S., Espinoza-Caicedo, J. A., Montt-Guevara, M., et al. (2016). Polymorphisms of the FTO and MTHFR Genes and Vascular, Inflammatory and Metabolic Marker Levels in Postmenopausal Women. J. Endocrinol. Invest. 39, 885–890. doi:10.1007/s40618-016-0443-7
Chen, X., Wang, J., Tahir, M., Zhang, F., Ran, Y., Liu, Z., et al. (2021a). Current Insights into the Implications of m6A RNA Methylation and Autophagy Interaction in Human Diseases. Cell Biosci. 11, 147. doi:10.1186/s13578-021-00661-x
Chen, Y.-S., Ouyang, X.-P., Yu, X.-H., Novák, P., Zhou, L., He, P.-P., et al. (2021b). N6-Adenosine Methylation (m6A) RNA Modification: an Emerging Role in Cardiovascular Diseases. J. Cardiovasc. Trans. Res. 14, 857–872. doi:10.1007/s12265-021-10108-w
Chien, C.-S., Li, J. Y.-S., Chien, Y., Wang, M.-L., Yarmishyn, A. A., Tsai, P.-H., et al. (2021). METTL3-dependent N 6 -methyladenosine RNA Modification Mediates the Atherogenic Inflammatory Cascades in Vascular Endothelium. Proc. Natl. Acad. Sci. U.S.A. 118, e2025070118. doi:10.1073/pnas.2025070118
Choi, J., Ieong, K.-W., Demirci, H., Chen, J., Petrov, A., Prabhakar, A., et al. (2016). N6-methyladenosine in mRNA Disrupts tRNA Selection and Translation-Elongation Dynamics. Nat. Struct. Mol. Biol. 23, 110–115. doi:10.1038/nsmb.3148
Chokkalla, A. K., Mehta, S. L., Kim, T., Chelluboina, B., Kim, J., and Vemuganti, R. (2019). Transient Focal Ischemia Significantly Alters the M 6 A Epitranscriptomic Tagging of RNAs in the Brain. Stroke 50, 2912–2921. doi:10.1161/STROKEAHA.119.026433
Csepany, T., Lin, A., Baldick, C. J., and Beemon, K. (1990). Sequence Specificity of mRNA N6-Adenosine Methyltransferase. J. Biol. Chem. 265, 20117–20122. doi:10.1016/s0021-9258(17)30477-5
Davis, W., van Rensburg, S. J., Cronje, F. J., Whati, L., Fisher, L. R., van der Merwe, L., et al. (2014). The Fat Mass and Obesity-Associated FTO Rs9939609 Polymorphism Is Associated with Elevated Homocysteine Levels in Patients with Multiple Sclerosis Screened for Vascular Risk Factors. Metab. Brain Dis. 29, 409–419. doi:10.1007/s11011-014-9486-7
De Jesus, D. F., Zhang, Z., Kahraman, S., Brown, N. K., Chen, M., Hu, J., et al. (2019). m6A mRNA Methylation Regulates Human β-cell Biology in Physiological States and in Type 2 Diabetes. Nat. Metab. 1, 765–774. doi:10.1038/s42255-019-0089-9
Deng, R., Cheng, Y., Ye, S., Zhang, J., Huang, R., Li, P., et al. (2019). m6A Methyltransferase METTL3 Suppresses Colorectal Cancer Proliferation and Migration through P38/ERK Pathways. OncoTargets Ther. 12, 4391–4402. doi:10.2147/OTT.S201052
Desrosiers, R., Friderici, K., and Rottman, F. (1974). Identification of Methylated Nucleosides in Messenger RNA from Novikoff Hepatoma Cells. Proc. Natl. Acad. Sci. U.S.A. 71, 3971–3975. doi:10.1073/pnas.71.10.3971
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 485, 201–206. doi:10.1038/nature11112
Dorn, L. E., Lasman, L., Chen, J., Xu, X., Hund, T. J., Medvedovic, M., et al. (2019). The N 6 -Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 139, 533–545. doi:10.1161/CIRCULATIONAHA.118.036146
Duan, H. C., Wang, Y., and Jia, G. (2019). Dynamic and Reversible RNA N 6 - Methyladenosine Methylation. WIREs RNA 10, e1507. doi:10.1002/wrna.1507
Dubey, P. K., Patil, M., Singh, S., Dubey, S., Ahuja, P., Verma, S. K., et al. (2022). Increased m6A-RNA Methylation and FTO Suppression Is Associated with Myocardial Inflammation and Dysfunction during Endotoxemia in Mice. Mol. Cell. Biochem. 477, 129–141. doi:10.1007/s11010-021-04267-2
Fan, X. C., and Steitz, J. A. (1998). Overexpression of HuR, a Nuclear-Cytoplasmic Shuttling Protein, Increases the Invivo Stability of ARE-Containing mRNAs. EMBO J. 17, 3448–3460. doi:10.1093/emboj/17.12.3448
Fry, N. J., Law, B. A., Ilkayeva, O. R., Holley, C. L., and Mansfield, K. D. (2017). N6-methyladenosine Is Required for the Hypoxic Stabilization of Specific mRNAs. Rna 23, 1444–1455. doi:10.1261/rna.061044.117
Fu, Y., Jia, G., Pang, X., Wang, R. N., Wang, X., Li, C. J., et al. (2013). FTO-mediated Formation of N6-Hydroxymethyladenosine and N6-Formyladenosine in Mammalian RNA. Nat. Commun. 4, 1798. doi:10.1038/ncomms2822
Fustin, J.-M., Doi, M., Yamaguchi, Y., Hida, H., Nishimura, S., Yoshida, M., et al. (2013). RNA-methylation-dependent RNA Processing Controls the Speed of the Circadian Clock. Cell 155, 793–806. doi:10.1016/j.cell.2013.10.026
Gao, X.-Q., Zhang, Y.-H., Liu, F., Ponnusamy, M., Zhao, X.-M., Zhou, L.-Y., et al. (2020). The piRNA CHAPIR Regulates Cardiac Hypertrophy by Controlling METTL3-dependent N6-Methyladenosine Methylation of Parp10 mRNA. Nat. Cell Biol. 22, 1319–1331. doi:10.1038/s41556-020-0576-y
Gerken, T., Girard, C. A., Tung, Y.-C. L., Webby, C. J., Saudek, V., Hewitson, K. S., et al. (2007). The Obesity-Associated FTO Gene Encodes a 2-oxoglutarate-dependent Nucleic Acid Demethylase. Science 318, 1469–1472. doi:10.1126/science.1151710
Geula, S., Moshitch-Moshkovitz, S., Dominissini, D., Mansour, A. A., Kol, N., Salmon-Divon, M., et al. (2015). m 6 A mRNA Methylation Facilitates Resolution of Naïve Pluripotency toward Differentiation. Science 347, 1002–1006. doi:10.1126/science.1261417
Gong, C., Fan, Y., and Liu, J. (2021). METTL14 Mediated m6A Modification to LncRNA ZFAS1/RAB22A: A Novel Therapeutic Target for Atherosclerosis. Int. J. Cardiol. 328, 177. doi:10.1016/j.ijcard.2020.12.002
Guo, M., Yan, R., Ji, Q., Yao, H., Sun, M., Duan, L., et al. (2020). IFN Regulatory Factor-1 Induced Macrophage Pyroptosis by Modulating m6A Modification of Circ_0029589 in Patients with Acute Coronary Syndrome. Int. Immunopharmacol. 86, 106800. doi:10.1016/j.intimp.2020.106800
Han, Y.-C., Xie, H.-Z., Lu, B., Xiang, R.-L., Zhang, H.-P., Li, J.-Y., et al. (2021a). Lipopolysaccharide Alters the m6A Epitranscriptomic Tagging of RNAs in Cardiac Tissue. Front. Mol. Biosci. 8, 670160. doi:10.3389/fmolb.2021.670160
Han, Z., Wang, X., Xu, Z., Cao, Y., Gong, R., Yu, Y., et al. (2021b). ALKBH5 Regulates Cardiomyocyte Proliferation and Heart Regeneration by Demethylating the mRNA of YTHDF1. Theranostics 11, 3000–3016. doi:10.7150/thno.47354
Han, Z., Xu, Z., Yu, Y., Cao, Y., Bao, Z., Gao, X., et al. (2021c). ALKBH5-mediated m6A mRNA Methylation Governs Human Embryonic Stem Cell Cardiac Commitment. Mol. Ther. - Nucleic Acids 26, 22–33. doi:10.1016/j.omtn.2021.05.019
Hao, J., Li, C., Lin, C., Hao, Y., Yu, X., Xia, Y., et al. (2020). Targeted Point Mutations of the m6A Modification in miR675 Using RNA-Guided Base Editing Induce Cell Apoptosis. Biosci. Rep. 40, BSR20192933. doi:10.1042/BSR20192933
He, Y., Xing, J., Wang, S., Xin, S., Han, Y., and Zhang, J. (2019). Increased m6A Methylation Level Is Associated with the Progression of Human Abdominal Aortic Aneurysm. Ann. Transl. Med. 7, 797. doi:10.21037/atm.2019.12.65
Hinger, S. A., Wei, J., Dorn, L. E., Whitson, B. A., Janssen, P. M. L., He, C., et al. (2021). Remodeling of the m6A Landscape in the Heart Reveals Few Conserved Post-transcriptional Events Underlying Cardiomyocyte Hypertrophy. J. Mol. Cell. Cardiol. 151, 46–55. doi:10.1016/j.yjmcc.2020.11.002
Hsu, P. J., Shi, H., Zhu, A. C., Lu, Z., Miller, N., Edens, B. M., et al. (2019). The RNA-Binding Protein FMRP Facilitates the Nuclear Export of N6-Methyladenosine-Containing mRNAs. J. Biol. Chem. 294, 19889–19895. doi:10.1074/jbc.AC119.010078
Hu, F., Tong, J., Deng, B., Zheng, J., and Lu, C. (2019). MiR-495 Regulates Macrophage M1/M2 Polarization and Insulin Resistance in High-Fat Diet-Fed Mice via Targeting FTO. Pflugers Arch. - Eur. J. Physiol. 471, 1529–1537. doi:10.1007/s00424-019-02316-w
Jian, D., Wang, Y., Jian, L., Tang, H., Rao, L., Chen, K., et al. (2020). METTL14 Aggravates Endothelial Inflammation and Atherosclerosis by Increasing FOXO1 N6-Methyladeosine Modifications. Theranostics 10, 8939–8956. doi:10.7150/thno.45178
Jin, S., Zhang, X., Miao, Y., Liang, P., Zhu, K., She, Y., et al. (2018). m6A RNA Modification Controls Autophagy through Upregulating ULK1 Protein Abundance. Cell Res. 28, 955–957. doi:10.1038/s41422-018-0069-8
Jones, P. A., Issa, J.-P. J., and Baylin, S. (2016). Targeting the Cancer Epigenome for Therapy. Nat. Rev. Genet. 17, 630–641. doi:10.1038/nrg.2016.93
Kattman, S. J., Adler, E. D., and Keller, G. M. (2007). Specification of Multipotential Cardiovascular Progenitor Cells during Embryonic Stem Cell Differentiation and Embryonic Development. Trends Cardiovasc. Med. 17, 240–246. doi:10.1016/j.tcm.2007.08.004
Kmietczyk, V., Riechert, E., Kalinski, L., Boileau, E., Malovrh, E., Malone, B., et al. (2019). m6A-mRNA Methylation Regulates Cardiac Gene Expression and Cellular Growth. Life Sci. Alliance 2, e201800233. doi:10.26508/lsa.201800233
Krüger, N., Biwer, L. A., Good, M. E., Ruddiman, C. A., Wolpe, A. G., DeLalio, L. J., et al. (2020). Loss of Endothelial FTO Antagonizes Obesity-Induced Metabolic and Vascular Dysfunction. Circ. Res. 126, 232–242. doi:10.1161/CIRCRESAHA.119.315531
Kwon, J., Jo, Y.-J., Namgoong, S., and Kim, N.-H. (2019). Functional Roles of hnRNPA2/B1 Regulated by METTL3 in Mammalian Embryonic Development. Sci. Rep. 9, 8640. doi:10.1038/s41598-019-44714-1
Lai, D., Sakkas, D., and Huang, Y. (2006). The Fragile X Mental Retardation Protein Interacts with a Distinct mRNA Nuclear Export Factor NXF2. Rna 12, 1446–1449. doi:10.1261/rna.94306
Leong, D. P., Joseph, P. G., McKee, M., Anand, S. S., Teo, K. K., Schwalm, J.-D., et al. (2017). Reducing the Global Burden of Cardiovascular Disease, Part 2. Circ. Res. 121, 695–710. doi:10.1161/CIRCRESAHA.117.311849
Lesbirel, S., Viphakone, N., Parker, M., Parker, J., Heath, C., Sudbery, I., et al. (2018). The m6A-Methylase Complex Recruits TREX and Regulates mRNA Export. Sci. Rep. 8, 13827. doi:10.1038/s41598-018-32310-8
Li, T., Wang, T., Jing, J., and Sun, L. (2021a). Expression Pattern and Clinical Value of Key m6A RNA Modification Regulators in Abdominal Aortic Aneurysm. J. Inflamm. Res. 14, 4245–4258. doi:10.2147/JIR.S327152
Li, Y., Sheng, H., Ma, F., Wu, Q., Huang, J., Chen, Q., et al. (2021b). RNA m6A Reader YTHDF2 Facilitates Lung Adenocarcinoma Cell Proliferation and Metastasis by Targeting the AXIN1/Wnt/β-Catenin Signaling. Cell Death Dis. 12, 479. doi:10.1038/s41419-021-03763-z
Li, Y., Zhang, Q., Cui, G., Zhao, F., Tian, X., Sun, B.-F., et al. (2020). m6A Regulates Liver Metabolic Disorders and Hepatogenous Diabetes. Genomics, Proteomics Bioinforma. 18, 371–383. doi:10.1016/j.gpb.2020.06.003
Li, Z., Xu, Q., Huangfu, N., Chen, X., and Zhu, J. (2022). Mettl3 Promotes oxLDL-Mediated Inflammation through Activating STAT1 Signaling. Clin. Lab. Anal. 36, e24019. doi:10.1002/jcla.24019
Lin, S., Choe, J., Du, P., Triboulet, R., and Gregory, R. I. (2016). The M 6 A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 62, 335–345. doi:10.1016/j.molcel.2016.03.021
Liu, J., Eckert, M. A., Harada, B. T., Liu, S.-M., Lu, Z., Yu, K., et al. (2018). m6A mRNA Methylation Regulates AKT Activity to Promote the Proliferation and Tumorigenicity of Endometrial Cancer. Nat. Cell Biol. 20, 1074–1083. doi:10.1038/s41556-018-0174-4
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 Complex Mediates Mammalian Nuclear RNA N6-Adenosine Methylation. Nat. Chem. Biol. 10, 93–95. doi:10.1038/nchembio.1432
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015). N6-methyladenosine-dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature 518, 560–564. doi:10.1038/nature14234
Liu, X.-M., Zhou, J., Mao, Y., Ji, Q., and Qian, S.-B. (2019a). Programmable RNA N6-Methyladenosine Editing by CRISPR-Cas9 Conjugates. Nat. Chem. Biol. 15, 865–871. doi:10.1038/s41589-019-0327-1
Liu, X.-M., and Zhou, J. (2021). Multifaceted Regulation of Translation by the Epitranscriptomic Modification N6-Methyladenosine. Crit. Rev. Biochem. Mol. Biol. 56, 137–148. doi:10.1080/10409238.2020.1869174
Liu, Y., Liu, Z., Tang, H., Shen, Y., Gong, Z., Xie, N., et al. (2019b). The N6-Methyladenosine (m6A)-Forming Enzyme METTL3 Facilitates M1 Macrophage Polarization through the Methylation of STAT1 mRNA. Am. J. Physiology-Cell Physiology 317, C762–C775. doi:10.1152/ajpcell.00212.2019
Lu, Y., Wang, S., Cai, S., Gu, X., Wang, J., Yang, Y., et al. (2020). Propofol-induced MiR-20b Expression Initiates Endogenous Cellular Signal Changes Mitigating Hypoxia/re-Oxygenation-Induced Endothelial Autophagy In Vitro. Cell Death Dis. 11, 681. doi:10.1038/s41419-020-02828-9
Ma, D., Liu, X., Zhang, J.-J., Zhao, J.-J., Xiong, Y.-J., Chang, Q., et al. (2020). Vascular Smooth Muscle FTO Promotes Aortic Dissecting Aneurysms via m6A Modification of Klf5. Front. Cardiovasc. Med. 7, 592550. doi:10.3389/fcvm.2020.592550
Malla, S., Melguizo-Sanchis, D., and Aguilo, F. (2019). Steering Pluripotency and Differentiation with N6-Methyladenosine RNA Modification. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1862, 394–402. doi:10.1016/j.bbagrm.2018.10.013
Mao, Y., Dong, L., Liu, X.-M., Guo, J., Ma, H., Shen, B., et al. (2019). m6A in mRNA Coding Regions Promotes Translation via the RNA Helicase-Containing YTHDC2. Nat. Commun. 10, 5332. doi:10.1038/s41467-019-13317-9
Mathiyalagan, P., Adamiak, M., Mayourian, J., Sassi, Y., Liang, Y., Agarwal, N., et al. (2019). FTO-dependent N 6 -Methyladenosine Regulates Cardiac Function during Remodeling and Repair. Circulation 139, 518–532. doi:10.1161/CIRCULATIONAHA.118.033794
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and Near Stop Codons. Cell 149, 1635–1646. doi:10.1016/j.cell.2012.05.003
Mo, C., Yang, M., Han, X., Li, J., Gao, G., Tai, H., et al. (2017). Fat Mass and Obesity-Associated Protein Attenuates Lipid Accumulation in Macrophage Foam Cells and Alleviates Atherosclerosis in Apolipoprotein E-Deficient Mice. J. Hypertens. 35, 810–821. doi:10.1097/HJH.0000000000001255
Mo, X.-B., Lei, S.-F., Zhang, Y.-H., and Zhang, H. (2019). Examination of the Associations between m6A-Associated Single-Nucleotide Polymorphisms and Blood Pressure. Hypertens. Res. 42, 1582–1589. doi:10.1038/s41440-019-0277-8
Park, O. H., Ha, H., Lee, Y., Boo, S. H., Kwon, D. H., Song, H. K., et al. (2019). Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol. Cell 74, 494–507. e8. doi:10.1016/j.molcel.2019.02.034
Patil, D. P., Chen, C.-K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m6A RNA Methylation Promotes XIST-Mediated Transcriptional Repression. Nature 537, 369–373. doi:10.1038/nature19342
Peng, S. S.-Y., Chen, C. Y., Xu, N., and Shyu, A. B. (1998). RNA Stabilization by the AU-Rich Element Binding Protein, HuR, an ELAV Protein. EMBO J. 17, 3461–3470. doi:10.1093/emboj/17.12.3461
Ping, X.-L., Sun, B.-F., Wang, L., Xiao, W., Yang, X., Wang, W.-J., et al. (2014). Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res. 24, 177–189. doi:10.1038/cr.2014.3
Prasher, D., Greenway, S. C., and Singh, R. B. (2020). The Impact of Epigenetics on Cardiovascular Disease. Biochem. Cell Biol. 98, 12–22. doi:10.1139/bcb-2019-0045
Rau, K., Rösner, L., and Rentmeister, A. (2019). Sequence-specific m6A Demethylation in RNA by FTO Fused to RCas9. Rna 25, 1311–1323. doi:10.1261/rna.070706.119
Roignant, J.-Y., and Soller, M. (2017). m 6 A in mRNA: An Ancient Mechanism for Fine-Tuning Gene Expression. Trends Genet. 33, 380–390. doi:10.1016/j.tig.2017.04.003
Roundtree, I. A., Luo, G.-Z., Zhang, Z., Wang, X., Zhou, T., Cui, Y., et al. (2017). YTHDC1 Mediates Nuclear Export of N6-Methyladenosine Methylated mRNAs. eLife 6, e31311. doi:10.7554/eLife.31311
Růžička, K., Zhang, M., Campilho, A., Bodi, Z., Kashif, M., Saleh, M., et al. (2017). Identification of Factors Required for M 6 A mRNA Methylation in Arabidopsis Reveals a Role for the Conserved E3 Ubiquitin Ligase HAKAI. New Phytol. 215, 157–172. doi:10.1111/nph.14586
Saito, T., Nah, J., Oka, S.-I., Mukai, R., Monden, Y., Maejima, Y., et al. (2019). An Alternative Mitophagy Pathway Mediated by Rab9 Protects the Heart against Ischemia. J. Clin. Invest. 129, 802–819. doi:10.1172/JCI122035
Schöller, E., Weichmann, F., Treiber, T., Ringle, S., Treiber, N., Flatley, A., et al. (2018). Interactions, Localization, and Phosphorylation of the m6A Generating METTL3-METTL14-WTAP Complex. Rna 24, 499–512. doi:10.1261/rna.064063.117
Schwartz, S., Mumbach, M. R., Jovanovic, M., Wang, T., Maciag, K., Bushkin, G. G., et al. (2014). Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 8, 284–296. doi:10.1016/j.celrep.2014.05.048
Shen, F., Huang, W., Huang, J.-T., Xiong, J., Yang, Y., Wu, K., et al. (2015). DecreasedN6-Methyladenosine in Peripheral Blood RNA from Diabetic Patients Is Associated WithFTOExpression rather ThanALKBH5. J. Clin. Endocrinol. Metabolism 100, E148–E154. doi:10.1210/jc.2014-1893
Shen, W., Li, H., Su, H., Chen, K., and Yan, J. (2021a). FTO Overexpression Inhibits Apoptosis of Hypoxia/reoxygenation-Treated Myocardial Cells by Regulating m6A Modification of Mhrt. Mol. Cell. Biochem. 476, 2171–2179. doi:10.1007/s11010-021-04069-6
Shen, Z.-J., Han, Y.-C., Nie, M.-W., Wang, Y.-N., Xiang, R.-L., and Xie, H.-Z. (2021b). Genome-wide Identification of Altered RNA m6A Profiles in Vascular Tissue of Septic Rats. Aging 13, 21610–21627. doi:10.18632/aging.203506
Shen, Z., Zeng, L., and Zhang, Z. (2020). Translatome and Transcriptome Profiling of Hypoxic-Induced Rat Cardiomyocytes. Mol. Ther. - Nucleic Acids 22, 1016–1024. doi:10.1016/j.omtn.2020.10.019
Sheth, U., and Parker, R. (2003). Decapping and Decay of Messenger RNA Occur in Cytoplasmic Processing Bodies. Science 300, 805–808. doi:10.1126/science.1082320
Shi, H., Wang, X., Lu, Z., Zhao, B. S., Ma, H., Hsu, P. J., et al. (2017). YTHDF3 Facilitates Translation and Decay of N6-Methyladenosine-Modified RNA. Cell Res. 27, 315–328. doi:10.1038/cr.2017.15
Slobodin, B., Han, R., Calderone, V., Vrielink, J. A. F. O., Loayza-Puch, F., Elkon, R., et al. (2017). Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-Adenosine Methylation. Cell 169, 326–337. e12. doi:10.1016/j.cell.2017.03.031
Song, H., Feng, X., Zhang, H., Luo, Y., Huang, J., Lin, M., et al. (2019). METTL3 and ALKBH5 Oppositely Regulate m6A Modification of TFEB mRNA, Which Dictates the Fate of Hypoxia/reoxygenation-Treated Cardiomyocytes. Autophagy 15, 1419–1437. doi:10.1080/15548627.2019.1586246
Su, H., Wang, G., Wu, L., Ma, X., Ying, K., and Zhang, R. (2020). Transcriptome-wide Map of m6A circRNAs Identified in a Rat Model of Hypoxia Mediated Pulmonary Hypertension. BMC Genomics 21, 39. doi:10.1186/s12864-020-6462-y
Takemoto, S., Nakano, M., Fukami, T., and Nakajima, M. (2021). m6A Modification Impacts Hepatic Drug and Lipid Metabolism Properties by Regulating Carboxylesterase 2. Biochem. Pharmacol. 193, 114766. doi:10.1016/j.bcp.2021.114766
Wang, J., Zhang, J., Ma, Y., Zeng, Y., Lu, C., Yang, F., et al. (2021a). WTAP Promotes Myocardial Ischemia/reperfusion Injury by Increasing Endoplasmic Reticulum Stress via Regulating m6A Modification of ATF4 mRNA. Aging 13, 11135–11149. doi:10.18632/aging.202770
Wang, L., Song, C., Wang, N., Li, S., Liu, Q., Sun, Z., et al. (2020). NADP Modulates RNA m6A Methylation and Adipogenesis via Enhancing FTO Activity. Nat. Chem. Biol. 16, 1394–1402. doi:10.1038/s41589-020-0601-2
Wang, P., Wang, Z., Zhang, M., Wu, Q., Shi, F., and Yuan, S. (2021b). KIAA1429 and ALKBH5 Oppositely Influence Aortic Dissection Progression via Regulating the Maturation of Pri-miR-143-3p in an m6A-dependent Manner. Front. Cell Dev. Biol. 9, 668377. doi:10.3389/fcell.2021.668377
Wang, S., Zhang, J., Wu, X., Lin, X., Liu, X.-M., and Zhou, J. (2021c). Differential Roles of YTHDF1 and YTHDF3 in Embryonic Stem Cell-Derived Cardiomyocyte Differentiation. RNA Biol. 18, 1–10. doi:10.1080/15476286.2020.1850628
Wang, X., Feng, J., Xue, Y., Guan, Z., Zhang, D., Liu, Z., et al. (2016). Structural Basis of N6-Adenosine Methylation by the METTL3-METTL14 Complex. Nature 534, 575–578. doi:10.1038/nature18298
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014a). N6-methyladenosine-dependent Regulation of Messenger RNA Stability. Nature 505, 117–120. doi:10.1038/nature12730
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. doi:10.1016/j.cell.2015.05.014
Wang, Y.-J., Yang, B., Lai, Q., Shi, J.-F., Peng, J.-Y., Zhang, Y., et al. (2021d). Reprogramming of m6A Epitranscriptome Is Crucial for Shaping of Transcriptome and Proteome in Response to Hypoxia. RNA Biol. 18, 131–143. doi:10.1080/15476286.2020.1804697
Wang, Y., Li, Y., Toth, J. I., Petroski, M. D., Zhang, Z., and Zhao, J. C. (2014b). N6-methyladenosine Modification Destabilizes Developmental Regulators in Embryonic Stem Cells. Nat. Cell Biol. 16, 191–198. doi:10.1038/ncb2902
Warda, A. S., Kretschmer, J., Hackert, P., Lenz, C., Urlaub, H., Höbartner, C., et al. (2017). Human METTL16 Is a N 6 -methyladenosine (M 6 A) Methyltransferase that Targets Pre-mRNAs and Various Non-coding RNAs. EMBO Rep. 18, 2004–2014. doi:10.15252/embr.201744940
Wei, C.-M., Gershowitz, A., and Moss, B. (1975). Methylated Nucleotides Block 5′ Terminus of HeLa Cell Messenger RNA. Cell 4, 379–386. doi:10.1016/0092-8674(75)90158-0
Wen, J., Lv, R., Ma, H., Shen, H., He, C., Wang, J., et al. (2018). Zc3h13 Regulates Nuclear RNA m6A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell 69, 1028–1038. e6. doi:10.1016/j.molcel.2018.02.015
Wu, S., Zhang, S., Wu, X., and Zhou, X. (2020a). m6A RNA Methylation in Cardiovascular Diseases. Mol. Ther. 28, 2111–2119. doi:10.1016/j.ymthe.2020.08.010
Wu, Y., Zhan, S., Xu, Y., and Gao, X. (2021). RNA Modifications in Cardiovascular Diseases, the Potential Therapeutic Targets. Life Sci. 278, 119565. doi:10.1016/j.lfs.2021.119565
Wu, Z., Shi, Y., Lu, M., Song, M., Yu, Z., Wang, J., et al. (2020b). METTL3 Counteracts Premature Aging via m6A-dependent Stabilization of MIS12 mRNA. Nucleic Acids Res. 48, 11083–11096. doi:10.1093/nar/gkaa816
Xia, C., Wang, J., Wu, Z., Miao, Y., Chen, C., Li, R., et al. (2021). METTL3-mediated M6A Methylation Modification Is Involved in Colistin-Induced Nephrotoxicity through Apoptosis Mediated by Keap1/Nrf2 Signaling Pathway. Toxicology 462, 152961. doi:10.1016/j.tox.2021.152961
Xu, S., Xu, X., Zhang, Z., Yan, L., Zhang, L., and Du, L. (2021). The Role of RNA m6A Methylation in the Regulation of Postnatal Hypoxia-Induced Pulmonary Hypertension. Respir. Res. 22, 121. doi:10.1186/s12931-021-01728-6
Yang, Y., Hsu, P. J., Chen, Y.-S., and Yang, Y.-G. (2018). Dynamic Transcriptomic m6A Decoration: Writers, Erasers, Readers and Functions in RNA Metabolism. Cell Res. 28, 616–624. doi:10.1038/s41422-018-0040-8
Ye, F., Wang, X., Tu, S., Zeng, L., Deng, X., Luo, W., et al. (2021). The Effects of NCBP3 on METTL3-Mediated m6A RNA Methylation to Enhance Translation Process in Hypoxic Cardiomyocytes. J. Cell. Mol. Med. 25, 8920–8928. doi:10.1111/jcmm.16852
Ylä-Herttuala, S., and Baker, A. H. (2017). Cardiovascular Gene Therapy: Past, Present, and Future. Mol. Ther. 25, 1095–1106. doi:10.1016/j.ymthe.2017.03.027
Yoon, K.-J., Ringeling, F. R., Vissers, C., Jacob, F., Pokrass, M., Jimenez-Cyrus, D., et al. (2017). Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 171, 877–889. e17. doi:10.1016/j.cell.2017.09.003
Yu, R., Li, Q., Feng, Z., Cai, L., and Xu, Q. (2019). m6A Reader YTHDF2 Regulates LPS-Induced Inflammatory Response. Int. J. Mol. Sci. 20, 1323. doi:10.3390/ijms20061323
Yue, Y., Liu, J., Cui, X., Cao, J., Luo, G., Zhang, Z., et al. (2018). VIRMA Mediates Preferential m6A mRNA Methylation in 3′UTR and Near Stop Codon and Associates with Alternative Polyadenylation. Cell Discov. 4, 10. doi:10.1038/s41421-018-0019-0
Zaccara, S., Ries, R. J., and Jaffrey, S. R. (2019). Reading, Writing and Erasing mRNA Methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624. doi:10.1038/s41580-019-0168-5
Zaky, A., Deem, S., Bendjelid, K., and Treggiari, M. M. (2014). Characterization of Cardiac Dysfunction in Sepsis. Shock Augusta Ga 41, 12–24. doi:10.1097/SHK.0000000000000065
Zeng, Y., Huang, T., Zuo, W., Wang, D., Xie, Y., Wang, X., et al. (2021). Integrated Analysis of m6A mRNA Methylation in Rats with Monocrotaline-Induced Pulmonary Arterial Hypertension. Aging 13, 18238–18256. doi:10.18632/aging.203230
Zhang, B.-F., Wu, Z.-H., Deng, J., Jin, H.-J., Chen, W.-B., Zhang, S., et al. (2022). M6A Methylation-Mediated Elevation of SM22α Inhibits the Proliferation and Migration of Vascular Smooth Muscle Cells and Ameliorates Intimal Hyperplasia in Type 2 Diabetes Mellitus. Biol. Chem. 403, 317–329. doi:10.1515/hsz-2021-0296
Zhang, B., Jiang, H., Wu, J., Cai, Y., Dong, Z., Zhao, Y., et al. (2021a). m6A Demethylase FTO Attenuates Cardiac Dysfunction by Regulating Glucose Uptake and Glycolysis in Mice with Pressure Overload-Induced Heart Failure. Sig Transduct. Target Ther. 6, 377. doi:10.1038/s41392-021-00699-w
Zhang, B., Xu, Y., Cui, X., Jiang, H., Luo, W., Weng, X., et al. (2021b). Alteration of m6A RNA Methylation in Heart Failure with Preserved Ejection Fraction. Front. Cardiovasc. Med. 8, 647806. doi:10.3389/fcvm.2021.647806
Zhang, B. Y., Han, L., Tang, Y. F., Zhang, G. X., Fan, X. L., Zhang, J. J., et al. (2020a). METTL14 Regulates M6A Methylation-Modified Primary miR-19a to Promote Cardiovascular Endothelial Cell Proliferation and Invasion. Eur. Rev. Med. Pharmacol. Sci. 24, 7015–7023. doi:10.26355/eurrev_202006_21694
Zhang, J., Ao, Y., Zhang, Z., Mo, Y., Peng, L., Jiang, Y., et al. (2020b). Lamin A Safeguards the M 6 A Methylase METTL14 Nuclear Speckle Reservoir to Prevent Cellular Senescence. Aging Cell 19, e13215. doi:10.1111/acel.13215
Zhang, W., Qian, Y., and Jia, G. (2021c). The Detection and Functions of RNA Modification m6A Based on m6A Writers and Erasers. J. Biol. Chem. 297, 100973. doi:10.1016/j.jbc.2021.100973
Zhang, X., Li, X., Jia, H., An, G., and Ni, J. (2021d). The m6A Methyltransferase METTL3 Modifies PGC-1α mRNA Promoting Mitochondrial Dysfunction and oxLDL-Induced Inflammation in Monocytes. J. Biol. Chem. 297, 101058. doi:10.1016/j.jbc.2021.101058
Zhao, B. S., Roundtree, I. A., and He, C. (2017). Post-transcriptional Gene Regulation by mRNA Modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42. doi:10.1038/nrm.2016.132
Zhao, J., Han, D.-X., Wang, C.-B., and Wang, X.-L. (2020a). Zbtb7b Suppresses Aseptic Inflammation by Regulating m6A Modification of IL6 mRNA. Biochem. Biophysical Res. Commun. 530, 336–341. doi:10.1016/j.bbrc.2020.07.011
Zhao, K., Yang, C.-x., Li, P., Sun, W., and Kong, X.-q. (2020b). Epigenetic Role of N6-Methyladenosine (m6A) RNA Methylation in the Cardiovascular System. J. Zhejiang Univ. Sci. B 21, 509–523. doi:10.1631/jzus.B1900680
Zhao, X., Yang, L., and Qin, L. (2021a). Methyltransferase-like 3 (METTL3) Attenuates Cardiomyocyte Apoptosis with Myocardial Ischemia-Reperfusion (I/R) Injury through miR-25-3p and miR-873-5p. Cell Biol. Int. 46, 992. doi:10.1002/cbin.11706
Zhao, Y., Hu, J., Sun, X., Yang, K., Yang, L., Kong, L., et al. (2021b). Loss of m6A Demethylase ALKBH5 Promotes Post-ischemic Angiogenesis via Post-transcriptional Stabilization of WNT5A. Clin. Transl. Med. 11, e402. doi:10.1002/ctm2.402
Zhao, Y., Shi, Y., Shen, H., and Xie, W. (2020c). m6A-binding Proteins: the Emerging Crucial Performers in Epigenetics. J. Hematol. Oncol. 13, 35. doi:10.1186/s13045-020-00872-8
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C.-M., Li, C. J., et al. (2013). ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 49, 18–29. doi:10.1016/j.molcel.2012.10.015
Zhou, K. I., Shi, H., Lyu, R., Wylder, A. C., Matuszek, Ż., Pan, J. N., et al. (2019). Regulation of Co-transcriptional Pre-mRNA Splicing by m6A through the Low-Complexity Protein hnRNPG. Mol. Cell 76, 70–81. e9. doi:10.1016/j.molcel.2019.07.005
Zhou, T., Han, D., Liu, J., Shi, J., Zhu, P., Wang, Y., et al. (2021a). Factors Influencing Osteogenic Differentiation of Human Aortic Valve Interstitial Cells. J. Thorac. Cardiovasc. Surg. 161, e163–e185. doi:10.1016/j.jtcvs.2019.10.039
Zhou, X.-L., Huang, F.-J., Li, Y., Huang, H., and Wu, Q.-C. (2021b). SEDT2/METTL14-mediated m6A Methylation Awakening Contributes to Hypoxia-Induced Pulmonary Arterial Hypertension in Mice. Aging 13, 7538–7548. doi:10.18632/aging.202616
Zhu, B., Gong, Y., Shen, L., Li, J., Han, J., Song, B., et al. (2020). Total Panax Notoginseng Saponin Inhibits Vascular Smooth Muscle Cell Proliferation and Migration and Intimal Hyperplasia by Regulating WTAP/p16 Signals via m6A Modulation. Biomed. Pharmacother. 124, 109935. doi:10.1016/j.biopha.2020.109935
Glossary
CVD cardiovascular disease
m6A n6-methyladenosine
mRNA messenger RNA
tRNA transfer RNA
circRNA circular RNA circular RNA
lncRNA long non-coding RNA long noncoding RNA
rRNA ribosomal RNA
UTRs untranslated regions
METTL 3 methyltransferase-like 3
METTL14 methyltransferase-like 14
WTAP Wilms tumor 1-associated protein
SAM S-adenosylmethionine
VIRMA Vir-like m6A methyltransferase-associated protein
RBM15 RNA binding motif protein 15
HAKAI Cbl photo oncogene like 1
ZC3H13 zinc finger CCCH-type containing 13
FTO fat mass and obesity-associated protein
ALKBH5 ALKB homologue 5 protein
CHD coronary heart disease
SRSF3 YTHDC, TH domain-containing protein; serine- and arginine-rich splicing factor 3
NXF1 nuclear RNA-export factor 1
IGF2BPs insulin-like growth factor 2 mRNA-binding proteins
HNRNPC heterogeneous nuclear ribonucleoprotein C
HNRNPA2B1 heterogeneous nuclear ribonucleoprotein A2/B1
MEPCE methylphosphate capping enzyme
TREX transcription–export
eIF eukaryotic initiation factor
CCR4 carbon catabolite-repression 4
NOT negative on TATA-less
HRSP12 heat-responsive protein 12
RNase ribonuclease
MRP mitochondrial RNA-processing
DGCR8 DiGeorge syndrome critical region 8
DROSHA Drosha ribonuclease III
miRNA microRNA
pri-miRNA primary-miRNA
lncRNA long non-coding RNA long noncoding RNA
circRNA circular RNA circular RNA
VSMC vascular smooth muscle cell
ESCs embryonic stem cells
I/R ischemia-reperfusion
TFEB transcription factor EB
ULK1 unc-51-like kinase 1
IL6 interleukin-6
ox-LDL oxidized-low density lipoprotein
GEO Gene Expression Omnibus
TNF tumor necrosis factor
ICAM-1 intercellular adhesion molecule-1
VCAM-1 vascular cell adhesion molecule-1
PGC-1α peroxisome proliferators-activated receptor γ coactivator l alpha
NLRP1 nucleotide-binding domain leucine-rich repeat pyrin domain containing 1
KLF4 kruppel-like factor 4
STAT1 signal transducer and activator of transcription 1
KLF5 Krueppel-like factor 5
ATF4 activation of transcription factor 4
GO Gene Ontology
KEGG Kyoto Encyclopedia of Genes and Genomes
IL17 interleukin-17
TAC transverse aortic constriction
m6A-SNP m6A-single nucleotide polymorphism
PGD2 prostaglandin D2
L-PGDS lipocalin-type prostaglandin D synthase
PPAR γ peroxisome proliferator-activated receptor γ
CD36 cluster of differentiation 36
SR -B1 scavenger receptor B-type 1
PDX1 pancreatic and duodenal homeobox 1.
Keywords: epigenetics, cardiovascular pathophysiology, cardiovascular diseases, m6A demethylase, m6A methyltransferase, m6A
Citation: Li L, Xu N, Liu J, Chen Z, Liu X and Wang J (2022) m6A Methylation in Cardiovascular Diseases: From Mechanisms to Therapeutic Potential. Front. Genet. 13:908976. doi: 10.3389/fgene.2022.908976
Received: 09 April 2022; Accepted: 07 June 2022;
Published: 28 June 2022.
Edited by:
Jing Huang, National Cancer Institute, United StatesCopyright © 2022 Li, Xu, Liu, Chen, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junnan Wang, amRleXdqbkAxNjMuY29t