 Chenyu Zhao
Chenyu Zhao Xiaoliu Shi
Xiaoliu Shi Yonghong Zhang3
Yonghong Zhang3 Hui Huang
Hui Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 25 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.895247
Background: Dubin–Johnson syndrome (DJS) is a rare autosomal recessive genetic disease which is caused by mutations in the ABCC2 gene; it is characterized by chronic hyperbilirubinemia. Here, we report two pedigrees affected with DJS which were caused by three novel pathogenic ABCC2 mutations.
Case summary: The two patients exhibited intermittent low-grade, predominantly conjugated hyperbilirubinemia and showed no other abnormalities. They were diagnosed clinically with DJS. Three novel pathogenic ABCC2 mutations—c.2980delA, c.1834C>T, and c.4465_4473delinsGGCCCACAG—were identified by whole-exome sequencing. These mutations could be responsible for DJS in the two pedigrees. The genetic test confirmed the diagnosis of DJS.
Conclusion: These results contributed to the genetic diagnosis of the two patients with DJS and expanded the variant database for the ABCC2 gene.
Dubin–Johnson syndrome (DJS, OMIM*237500) is an autosomal recessive genetic disease which is characterized by intermittent, predominantly conjugated hyperbilirubinemia and liver pigmentation. DJS has no other features of hepatobiliary disorder. First described by Dubin and Johnson (1954), It is considered a rare disorder worldwide, although its incidence among Sephardic Jews has been reported as approximately 1/3000 (Shani et al., 1970). Pathogenic mutations in the adenosine triphosphate-binding cassette subfamily C member 2 (ABCC2) gene were first discovered to cause DJS in 1997 (Paulusma et al., 1997). The multidrug resistance-associated protein 2 (MRP2) encoded by the ABCC2 gene is principally located in the hepatocyte canalicular membrane. Loss-of-function variants in the ABCC2 gene disturb the secretion of conjugated bilirubin from hepatocytes. DJS does not affect life expectancy and usually requires no treatment (Memon et al., 2016). To date, 127 mutations in ABCC2 have been described in the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index. php). In the present study, we reported two patients with DJS and identified three novel disease-causing mutations: c.2980delA, c.1834C>T and c.4465_4473delinsGGCCCACAG, in the ABCC2 gene (NM_000392).
In Family 1, a 29-year-old woman (Patient 1) was born to nonconsanguineous parents although her grandparents were cousins (Figure 1A). She was affected with low-grade predominantly conjugated hyperbilirubinemia for more than 6 years. This patient was tested for hyperbilirubinemia in a health physical examination in 2015. There were no other significant abnormalities in her liver function test. She received no other medical treatment then. During her pregnancy, her jaundice was markedly aggravated. She gave birth on 18 August 2018. The obstetrician used reduced glutathione (1.2 g/d, from 16 August 2018 to 20 August 2018) and ursodeoxycholic acid (500 mg/d, from 20 August 2018 to 31 August 2018) to control her jaundice. Her clinical jaundice gradually disappeared after childbirth in 2018. She took oral contraceptives from January 2021 and presented with recurrent mild jaundice again. The jaundice was in remission after she stopped taking oral contraceptives; she was never affected with pruritus. The patient had a previously free medical history. When she came to us as a medical genetic outpatient in July 2021, the physical examination revealed mild splenomegaly. The timeline with relevant data for Patient 1 is shown in Supplementary Figure S1. During the patient’s pregnancy, her level of total bilirubin (TBIL) ranged from 43.1 μmol/L to 82.8 μmol/L (normal range: 3.4–17.1 μmol/L). Her direct bilirubin (DBIL) level ranged from 25.2 μmol/L to 65.1 μmol/L (normal range: 0–6 μmol/L). The ratio of DBIL/TBIL was always greater than 50%. There were no abnormalities in other liver function indexes. She was serologically negative for hepatitis A, B, C, and E viruses. There were no abnormalities in coagulation function, autoimmune liver disease-associated antibodies, immunoglobulin G, or serum ceruloplasmin testing. The abdominal ultrasound examination revealed her portal vein diameter to be about 10 mm and the portal venous maximum velocity was 18 cm/s. The thickness of her spleen was about 49 mm and its inferior margin was about 20 mm below the costal margin. A hepatic hemangioma was also detected by the ultrasound examination (Supplementary Figure S2; Table 1). The whole-exome sequencing (WES) was conducted in July 2021, revealing that Patient 1 carried both the heterozygous c.2980delA and c.1834C>T ABCC2 mutations. Her son also shared the heterozygous c.1834C>T ABCC2 mutation (Figure 1B; Table 2). Considering her symptoms, clinical examinations, and genetic testing, she was diagnosed with DJS. Intrahepatic cholestasis of pregnancy (ICP) was excluded.
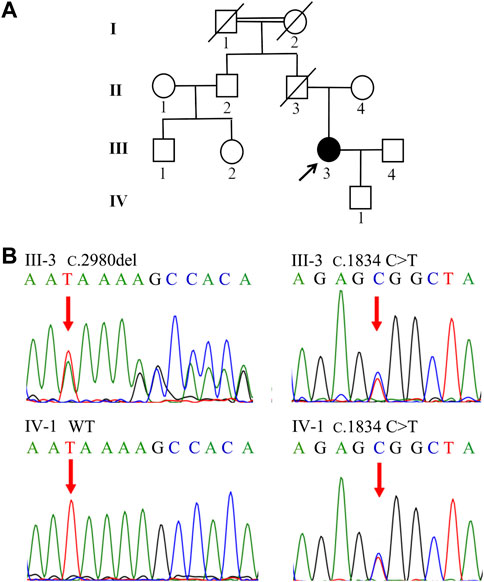
FIGURE 1. Family tree and genetic analysis of Family 1. (A) Proband III-3 was diagnosed with DJS. (B) Sanger sequencing revealed a compound heterozygous (c.2980delA and c.1834 C>T) ABCC2 mutation in III-3. IV-1 shared the heterozygous c.1834 C>T variant.
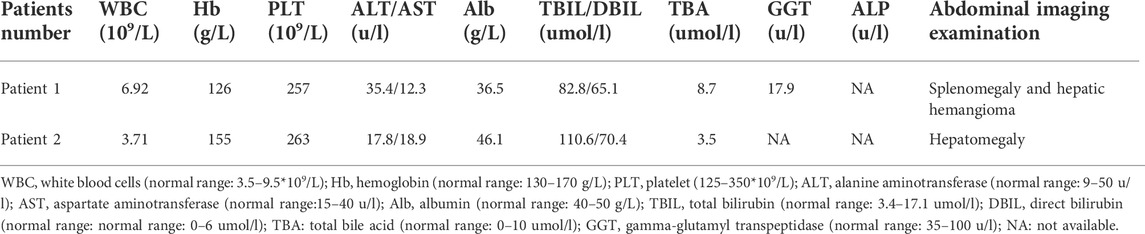
TABLE 1. Clinical laboratory tests and imaging examinations in the patients with Dubin–Johnson syndrome.
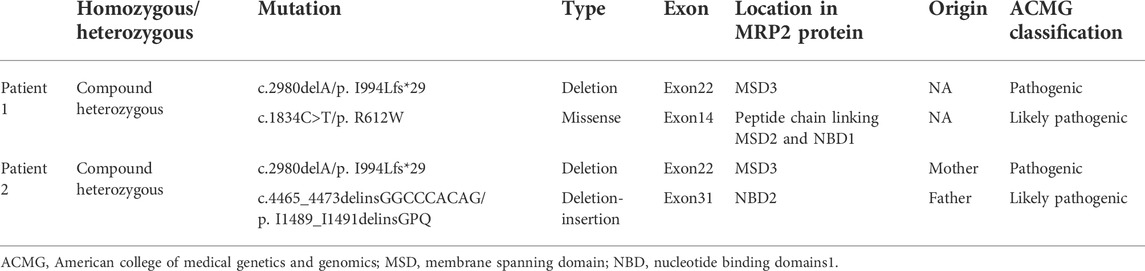
TABLE 2. Variants of ABCC2 in the patients with Dubin–Johnson syndrome.
In Family 2, an 18-year-old male (Patient 2) presented with intermittent mild jaundice for more than 9 years. This patient showed intermittent jaundice from the age of nine, which would be aggravated by fatigue. He had a previously free medical history and no special personal and family history (Figure 2A). When he came as a medical genetic outpatient to our hospital, he presented with yellow skin and sclera. In the physical examination, his liver could be palpated at the lower margin of the ribs. Patient 2’s TBIL level was 110.6 μmol/L and his DBIL level was 70.4 μmol/L, with no other abnormalities in the liver function test. There were no abnormalities in autoimmune liver disease-associated antibodies or serum hepatitis B and C virus testing. A CT scan revealed hepatomegaly. The lower margin of the liver exceeded the lower costal margin (Table 1, Supplementary Figure S2). The WES indicated that Patient 2 harbored the heterozygous c.2980delA variant inherited from his mother and the heterozygous c.4465_4473delinsGGCCCACAG mutation inherited from his father (Figure 2B; Table 2). He was also diagnosed with DJS and chose to observe his jaundice (without drug interventions). In the telephone follow-up, he still complained of intermittent jaundice.
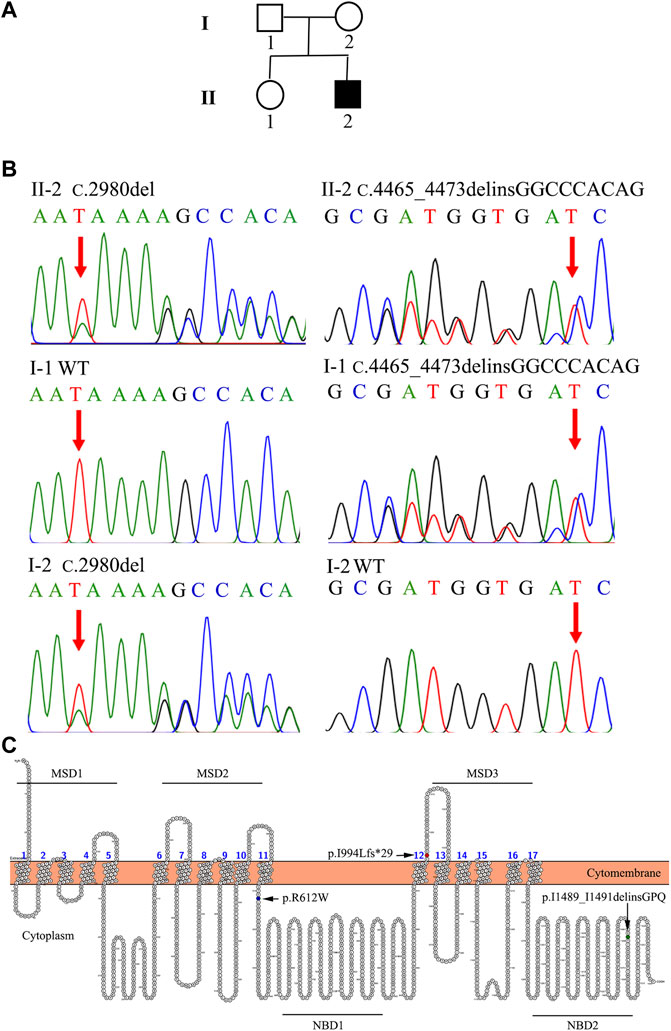
FIGURE 2. The family tree and genetic analysis of Family 2. (A) Proband II-2 suffered from DJS. (B) Direct sequencing showed a compound heterozygous (c.2980delA and c.4465_4473 delinsGGCCCACAG) ABCC2 mutation in II-2. I-1 was affected with the heterozygous c.4465_4473delinsGGCCCACAG mutation. II-2 harbored the heterozygous c.2980delA variant. (C) Locations of the mutations in MRP2. The schematic diagram of MRP2 was generated using Protter software (http://wlab.ethz.ch/protter/start/). MSD: membrane spanning domains, NBD: nucleotide binding domains.
However, both patients refused the 99mTc-HIDA cholescintigraphy and liver biopsy. The candidate variants in the ABCC2 gene were validated by Sanger sequencing. The primer sequences are listed in Table A1. The methods of genetic testing are described in the supplementary materials. The family members participating in the study signed written informed consent forms. This study complied with the ethical guidelines of the Declaration of Helsinki and was approved by the ethics committee of the Second Xiangya Hospital of Central South University (2014 ethical approval No. S046).

TABLE A1. Primer sequences used for mutation sequencing of ABCC2 gene.
The two patients suffered from chronic low-grade predominantly conjugated hyperbilirubinemia. There were no other features of hepatobiliary disorder. Patient 1’s jaundice increased during pregnancy and while taking oral contraceptives, which was in line with DJS (Erlinger et al., 2014). In the liver biopsy, specimens of DJS had a grossly black appearance and coarse, deep-brown, pigmented granules (Memon et al., 2016). The expression of MRP2 was absent on the hepatocellular membrane in patients with DJS (Morii and Yamamoto, 2016). The MRP2 transporter mediates the canalicular secretion of several organic anions, including many drugs, toxicants, and endogenous compounds (Keppler, 2011). Pigment accumulation is caused by the retention of anionic metabolites of tyrosine, phenylalanine, and tryptophan, which polymerized to form a similar melanin-like pigment in the liver (Kitamura et al., 1992). Postoperative 99mTc-HIDA cholescintigraphy would reveal delayed visualization of the extrahepatic bile ducts (Park et al., 2018). Most patients with DJS have a normal lifespan. Liu et al. even used a graft from a donor with DJS to conduct living donor liver transplantation. The recipient was followed for 1 year and the results were satisfactory (Liu et al., 2012). However, some drugs, such as methotrexate, vincristine, and cisplatin, are excreted from hepatocytes via the MRP2 transporter (Zhang and Fu, 2011). The mutant MRP2 transporter could affect their sensitivity and toxicity (van der Schoor et al., 2015). Therefore, adverse drug reactions need to be carefully noted for DJS patients.
The c.2980delA was a frameshift variant in the ABCC2 gene. Loss of function is a known mechanism of DJS (PVS1). In addition, the c.2980delA was absent from the controls in the 1000G and EXAC databases (PM2). The use of the MutationTaster prediction software (Schwarz et al., 2014) revealed a deleterious effect from c.2980delA (PP3). Patient 1’s phenotype was highly specific for DJS (PP4). Considering the ACMG guidance (Richards et al., 2015), we classified the c.2980delA variant as a pathogenic mutation.
The c.1834C>T mutation was not found in the 1000G or EXAC database (PM2). In addition, c.1834C>T was detected in trans with the pathogenic variant (c.2980delA) in Patient 1, which was confirmed by testing the offspring of her son (PM3). In addition, MutationTaster, PolyPhen-2 (Adzhubei et al., 2010), and SIFT (Kumar et al., 2009) predicted a disease-causing effect for the mutation (PP3). Patient 1 showed highly specific symptoms for DJS (PP4). Based on the ACMG guidelines, c.1834C>T was a likely pathogenic variant. The term “likely pathogenic” indicates a greater than 90% certainty of a variant being disease-causing. The variant had been recorded by gnomAD (with an allele frequency of 4.065e-06) and InterVar (with Uncertain clinical significance) database. But it was clarified for the first time as a likely pathogenic mutation associated with DJS.
The c.2980delA variant was also found in Patient 2. It was a pathogenic variant as analyzed above. The other mutation, c.4465_4473delinsGGCCCACAG, was not found in the 1000G or EXAC database (PM2). For a recessive disorder, c.4465_4473delinsGGCCCACAG was detected in trans with a pathogenic variant (c.2980delA), which was confirmed in his parents (PM3). The c.2980delA and c.4465_4473delinsGGCCCACAG formed a compound heterozygous mutation in Patient 2. The compound heterozygous variant was found to cosegregate in the family members (PP1). In addition, multiple lines of computational evidence supported a deleterious effect of the gene (PP3). Patient 2’s phenotype was highly specific for DJS (PP4). Therefore, c.4465_4473delinsGGCCCACAG was identified as a likely pathogenic variant according to the ACMG guidelines. By scanning the HGMD and previously published works, we concluded that the three mutations are novel pathogenic variants.
Beside the three ABCC2 mutations identified during the steps in the filtering strategies in the WES-yielded data, we also detected a heterozygous mutation, p. G71R, in the UGT1A1 gene in Patient 1 (Table A2). UGT1A1 is the disease-causing gene of Gilbert syndrome (GS), Criggler–Najjar syndrome I (CNS-I), and Criggler–Najjar syndrome II (CNS-II). However, the three diseases are autosomal recessive diseases and cause predominantly unconjugated hyperbilirubinemia. This is not consistent with the patient’s symptoms. In addition, the minor allele frequency of the variant is greater than 0.01 in the 1000G, ExAC, and gnomAD databases. Therefore, we excluded the variant. DJS and Rotor syndrome have similar symptoms, but no pathogenic variant was detected in the OATP1B1 or OATP1B3 genes, which are causative genes for Rotor syndrome. Therefore, Rotor syndrome was excluded. Furthermore, there were no candidate mutations in the ATP8B1, ABCB4, or ABCB11 genes, which could cause benign recurrent intrahepatic cholestasis (BRIC) or ICP.

TABLE A2. Other bilirubin-metabolism related mutation in the patients
MRP2 consists of three membrane spanning domains (MSDs) which include 17 transmembrane helices and two intracellular highly conserved nucleotide binding domains (NBDs). The NBD has an ATP binding site and is important for transporting some organic anions out of the hepatocyte. The transport process by MRP2 relies on the energy generated by the hydrolysis of ATP. There have been studies on genotype-phenotype correlations in DJS (Lee et al., 2006; Togawa et al., 2018; Kim et al., 2020). Mutations located in the NBD domain have been associated with early-onset DJS (Lee et al., 2006). Our two patients carried the same heterozygous c.2980delA/p.I994Lfs*29 mutation in the MSD3 domain. However, Patient 2 was affected by a heterozygous deletion-insertion mutation—c.4465_4473delinsGGCCCACAG/p.I1489_I1491delinsGPQ—in the NBD2 domain. Patient 1 carried a heterozygous missense mutation—c.1834C>T/p.R612W—in the peptide chain linking MSD2 and NBD1 (Wen et al., 2017; Xiang et al., 2017) (Figure 2C). Patient 2 carried the variant in the NBD2 domain. He suffered from early-onset and more serious hyperbilirubinemia than Patient 1, consistent with the above study. In addition, the combination of missense and truncating variants may be essential for the phenotype of neonatal DJS (Togawa et al., 2018). Patient 1 was affected by missense and truncating variants but she did not show neonatal DJS.
Because of the favorable prognosis of DJS, the two patients’ stresses were significantly relieved. Patient 1 could also avoid using oral contraceptives, which might aggravate her jaundice. DJS could not cause splenomegaly. The etiology of splenomegaly requires further analysis and follow-up. Patient 2 believed that the genetic testing could facilitate his search for work, as an unclear diagnosis of his liver disease would restrict the range of his job choices. Both patients were satisfied with the outcome of the genetic testing. However, there were several limitations in our research. The 99mTc-HIDA cholescintigraphy and biopsy could not be conducted because of the patients’ rejections. In addition, functional experiments of the novel pathogenic ABCC2 mutations need further investigation.
In conclusion, this study identified three novel pathogenic ABCC2 mutations in two patients with DJS. These results contribute to the genetic diagnosis and counseling of families with DJS and expand the spectrum of ABCC2 mutations.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the ethics committee of the Second Xiangya Hospital of Central South University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
CZ: Formal analysis, methodology, software, writing—original draft XS: Writing—review and editing YZ: Writing—review and editing. HH: Conceptualization, Supervision, validation, visualization, writing—review and editing.
This work was supported by the Natural Science Foundation of Hunan (2020JJ5810 HH).
We would like to thank the members of the patients’ families for their participation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.895247/full#supplementary-material
Supplementary Figure S1 | (A) Ttimelines with relevant data from Patient 1. She was tested for hyperbilirubinemia in 2015 without obvious symptoms. She fell pregnant in November 2017 and, from then on, her jaundice gradually aggravated. The jaundice was in remission after her parturition. However, the hyperbilirubinemia deteriorated again after she took oral contraceptives. She finally received genetic testing in July 2021. (B) The bilirubin concentrations were during Patient 1’s pregnancy and drug interventions. She gave birth on 18 August 2018. The drug interventions were conducted from 16 August 2018 to 31 August 2018. TBIL: total bilirubin, DBIL: direct bilirubin.
Supplementary Figure S2 | The image data of the patients. (A-C) The results of Patient 1's ultrasound examinations. (A) The thickness of her spleen was about 49 mm. (B) The inferior margin of the spleen was about 20 mm below the costal margin. (C) The hemangioma in the liver (about 7.4 mm*7.6 mm). (D) The CT scan showed that Patient 2 was affected by hepatomegaly. The left lobe of the liver extended to the lower left.
The genomic DNA was isolated from peripheral blood using a Blood gDNA Miniprep Kit (Hangzhou Beiwo Meditech Co., Ltd., Hangzhou, China). The WES was conducted on Patients 1 and 2, which was performed by AmCare Genomics Lab (Guangzhou, China). High-throughput sequencing was performed on the MGISEQ-2000 platform (MGI Tech Co., Ltd., Shenzhen, China). Basic bioinformatics analysis, including read, mapping and variant detection, was also completed by AmCare Genomics Lab Limited.
The methods of WES data filtering were as follows: (1) We excluded variants in intergenic, intronic, and untranslated regions (UTRs) and synonymous variants. (2) Variants in the databases with a minor allele frequency>0.01 were excluded. The databases included the 1000 Genomes project (1000G; http://browser.1000genomes.org/), Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/), Exome Sequencing Project 6500 (esp6500; http://evs.gs.washington.edu/EVS/), and Genome Aggregation (gnomAD; http://gnomad.broadinstitute.org/). (3) The candidate pathogenetic variants in bilirubin-metabolism related genes were retained. (4) The pathogenicity of the variants was analyzed according to the American College of Medical Genetics and Genomics (ACMG) guidance for the interpretation of sequence variants.
The potential variants from WES were validated by Sanger sequencing. The primer sequences were listed in Table A1. PCR products were sequenced on a Superyears Classic Genetic Analyzer (Nanjing Superyears Gene Technology Co., Ltd., Nanjing, China).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Dubin, I. N., and Johnson, F. B. (1954). Chronic idiopathic jaundice with unidentified pigment in liver cells; a new clinicopathologic entity with a report of 12 cases. Med. Baltim. 33 (3), 155–197. doi:10.1097/00005792-195409000-00001
Erlinger, S., Arias, I. M., and Dhumeaux, D. (2014). Inherited disorders of bilirubin transport and conjugation: New insights into molecular mechanisms and consequences. Gastroenterology 146 (7), 1625–1638. doi:10.1053/j.gastro.2014.03.047
Keppler, D. (2011). Multidrug resistance proteins (MRPs, ABCCs): Importance for pathophysiology and drug therapy. Handb. Exp. Pharmacol. 201, 299–323. doi:10.1007/978-3-642-14541-4_8
Kim, K. Y., Kim, T. H., Seong, M. W., Park, S. S., Moon, J. S., and Ko, J. S. (2020). Mutation spectrum and biochemical features in infants with neonatal Dubin-Johnson syndrome. BMC Pediatr. 20 (1), 369. doi:10.1186/s12887-020-02260-0
Kitamura, T., Alroy, J., Gatmaitan, Z., Inoue, M., Mikami, T., Jansen, P., et al. (1992). Defective biliary excretion of epinephrine metabolites in mutant (TR-) rats: Relation to the pathogenesis of black liver in the Dubin-Johnson syndrome and corriedale sheep with an analogous excretory defect. Hepatology 15 (6), 1154–1159. doi:10.1002/hep.1840150629
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4 (7), 1073–1081. doi:10.1038/nprot.2009.86
Lee, J. H., Chen, H. L., Chen, H. L., Ni, Y. H., Hsu, H. Y., and Chang, M. H. (2006). Neonatal Dubin-Johnson syndrome: Long-term follow-up and MRP2 mutations study. Pediatr. Res. 59 (1), 584–589. doi:10.1203/01.pdr.0000203093.10908.bb
Liu, C., Niu, D. M., Hsia, C. Y., Loong, C. C., Lin, N. C., Tsai, H. L., et al. (2012). Living donor liver transplantation using a graft from a donor with Dubin-Johnson syndrome. Pediatr. Transpl. 16 (1), E25–E29. doi:10.1111/j.1399-3046.2010.01385.x
Memon, N., Weinberger, B. I., Hegyi, T., and Aleksunes, L. M. (2016). Inherited disorders of bilirubin clearance. Pediatr. Res. 79 (3), 378–386. doi:10.1038/pr.2015.247
Morii, K., and Yamamoto, T. (2016). Images in clinical medicine. Dubin-johnson syndrome. N. Engl. J. Med. 375 (1), e1. doi:10.1056/NEJMicm1509529
Park, S. W., Jun, C. H., Choi, S. K., Kim, H. J., and Kim, G. E. (2018). Hepatobiliary and pancreatic: A black liver of dubin-johnson syndrome. J. Gastroenterol. Hepatol. 33 (3), 562. doi:10.1111/jgh.14036
Paulusma, C. C., Kool, M., Bosma, P. J., Scheffer, G. L., ter Borg, F., Scheper, R. J., et al. (1997). A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology 25 (6), 1539–1542. doi:10.1002/hep.510250635
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11 (4), 361–362. doi:10.1038/nmeth.2890
Shani, M., Seligsohn, U., Gilon, E., Sheba, C., and Adam, A. (1970). Dubin-Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q. J. Med. 39 (156), 549–567.
Togawa, T., Mizuochi, T., Sugiura, T., Kusano, H., Tanikawa, K., Sasaki, T., et al. (2018). Clinical, pathologic, and genetic features of neonatal dubin-johnson syndrome: A multicenter study in Japan. J. Pediatr. 196, 161–167. e161. doi:10.1016/j.jpeds.2017.12.058
van der Schoor, L. W., Verkade, H. J., Kuipers, F., and Jonker, J. W. (2015). New insights in the biology of ABC transporters ABCC2 and ABCC3: Impact on drug disposition. Expert Opin. Drug Metab. Toxicol. 11 (2), 273–293. doi:10.1517/17425255.2015.981152
Wen, X., Joy, M. S., and Aleksunes, L. M. (2017). In vitro transport activity and trafficking of MRP2/ABCC2 polymorphic variants. Pharm. Res. 34 (8), 1637–1647. doi:10.1007/s11095-017-2160-0
Xiang, R., Li, J. J., Fan, L. L., Jin, J. Y., Xia, K., and Wang, F. (2017). Identification of a compound heterozygous mutation of ABCC2 in a patient with hyperbilirubinemia. Mol. Med. Rep. 16 (3), 2830–2834. doi:10.3892/mmr.2017.6926
Keywords: Dubin–Johnson syndrome, ABCC2, multidrug resistance-associated protein 2, mutation, hyperbilirubinemia
Citation: Zhao C, Shi X, Zhang Y and Huang H (2022) Case Report: Three novel pathogenic ABCC2 mutations identified in two patients with Dubin–Johnson syndrome. Front. Genet. 13:895247. doi: 10.3389/fgene.2022.895247
Received: 13 March 2022; Accepted: 04 July 2022;
Published: 25 August 2022.
Edited by:
Mahmood Rasool, King Abdulaziz University, Saudi ArabiaReviewed by:
Donghu Zhou, Affiliated Beijing Friendship Hospital, Capital Medical University, ChinaCopyright © 2022 Zhao, Shi, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Huang, aHVpaHVhbmcwOTE2QGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.