 João Paulo Kazmierczak de Camargo1
João Paulo Kazmierczak de Camargo1 Giovanna Nazaré de Barros Prezia2
Giovanna Nazaré de Barros Prezia2 Naoye Shiokawa3Mario Teruo Sato3,4
Naoye Shiokawa3Mario Teruo Sato3,4 Roberto Rosati2†
Roberto Rosati2† Angelica Beate Winter Boldt1*†
Angelica Beate Winter Boldt1*†- 1Post-Graduation Program in Genetics, Department of Genetics, Federal University of Paraná, Curitiba, Brazil
- 2Post-Graduation Program in Biotechnology Applied to Child and Adolescent Health, Faculdades Pequeno Príncipe and Pelé Pequeno Príncipe Research Institute, Curitiba, Brazil
- 3Retina and Vitreo Consulting Eye Clinic, Curitiba, Brazil
- 4Department of Ophthalmol/Otorhinolaryngology, Federal University of Paraná, Curitiba, Brazil
Central areolar choroidal dystrophy (CACD) is a rare hereditary disease that mainly affects the macula, resulting in progressive and usually profound visual loss. Being part of congenital retinal dystrophies, it may have an autosomal dominant or recessive inheritance and, until now, has no effective treatment. Given the shortage of genotypic information about the disease, this work systematically reviews the literature for CACD-causing genes. Three independent researchers selected 33 articles after carefully searching and filtering the Scielo, Pubmed, Lilacs, Web of Science, Scopus, and Embase databases. Mutations of six genes (PRPH2, GUCA1A, GUCY2D, CDHR1, ABCA4, and TTLL5) are implicated in the monogenic dominant inheritance of CACD. They are functionally related to photoreceptors (either in the phototransduction process, as in the case of GUCY2D, or the recovery of retinal photodegradation in photoreceptors for GUCA1A, or the formation and maintenance of specific structures within photoreceptors for PRPH2). The identified genetic variants do not explain all observed clinical features, calling for further whole-genome and functional studies for this disease. A network analysis with the CACD-related genes identified in the systematic review resulted in the identification of another 20 genes that may influence CACD onset and symptoms. Furthermore, an enrichment analysis allowed the identification of 13 transcription factors and 4 long noncoding RNAs interacting with the products of the previously mentioned genes. If mutated or dysregulated, they may be directly involved in CACD development and related disorders. More than half of the genes identified by bioinformatic tools do not appear in commercial gene panels, calling for more studies about their role in the maintenance of the retina and phototransduction process, as well as for a timely update of these gene panels.
Systematic Review Registration: website, identifier registration number
1 Introduction
Central areolar choroidal dystrophy (CACD, MIM #215500, #613105 and %613144) is a rare hereditary disease characterized by a bilateral, symmetrical, well-circumscribed loss of choroidal and retinal tissue (Noble, 1977), that mainly affects the macula, resulting in progressive and usually profound visual loss. It is part of the hereditary retinal dystrophies, a highly heterogeneous group of diseases that cause the degeneration of photoreceptors and retinal pigment epithelium (RPE). There is no efficient treatment that prevents the development of CACD and other monogenic macular dystrophies (Michaelides et al., 2003; Prokofyeva et al., 2009).
Four clinical stages of the disease have been described (Hoyng et al., 1996a). In stage 1, subtle focal changes of parafoveal pigmented RPE are evident by ophthalmoscopy. A typical stage 2 finding in the color image is an oval-to-round, mildly atrophic, hypopigmented area. This area shows increased and decreased reflectivity by fundus autofluorescence (FAF) imaging, resulting in a speckled FAF pattern. Stage 3 is characterized by one or more patches of well-demarcated RPE atrophy outside the fovea. In stage 4, the atrophic area involves the fovea, resulting in markedly decreased visual acuity, commonly less than 20/200 (Hoyng et al., 1996a; Boon et al., 2009). The disease begins between the third and fifth decade of life, presenting an area of depigmentation in the parafoveal epithelium area, atrophy of the RPE, and choriocapillaris in the macula center, which gradually progressing with age, leading to a decrease in visual acuity. The visual field presents a large central scotoma. Colour test show a mild protan-deutan defect. The electroretinogram (ERG) and electro-oculogram (EOG) are normal in most of patients, and these findings help in the differential diagnosis with other macular dystrophies (Hoyng et al., 1996a). Even so, CACD is commonly misdiagnosed as other diseases such as age-related macular degeneration (AMD) (Smailhodzic et al., 2011).
CACD may present with autosomal recessive or dominant inheritance; however, autosomal recessive cases are rare (Sorsby, 1939; Iannaccone, 2001). Since this disease has many publications associating genetic variants to the phenotype - but no review with a genetic approach -we systematically reviewed the literature to list genes and variants associated with the CACD phenotype, as well as possible candidates interacting with them, through enrichment analysis. We hope to expand the knowledge of genetic contributions to the pathological mechanisms in this disease, generating new hypotheses for future work.
2 Methods
We followed the recommendations of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA). Three independent investigators carried out the systematic review, using the following Boolean operators in the PubMed, LILACS, SciELO, Web Of Science, Scopus, and Embase databases until 5 May 2021. Although no language restriction was imposed, only literature in English, Spanish or Portuguese was obtained and therefore screened. The search terms were as follows, with syntax adjustments for each database [(“Central areolar choroidal dystrophy” OR “areolar atrophy of the macula” OR CACD OR “central areolar choroidal sclerosis” OR “central areolar choroidal atrophy”) AND (variant OR allele OR gene OR genetic OR “genetic susceptibility” OR “genetic variant” OR “genetic variation” OR genotype OR haplotype OR mutation OR polymorphism OR “single nucleotide polymorphism” OR SNP OR variation)]. The records were screened on the online platform Rayyan (Ouzzani et al., 2016). Articles were included for further analysis if containing genotype data for the CACD patients. We also used the EnrichR tool to screen different databases for enrichment in genes with CACD-associated variants, correcting it for multiple comparisons to uncover new genetic factors that could regulate the expression or function of the genes found in our preliminary systematic review (Maleki et al., 2020).
3 Results
CACD genetic association studies in the literature are scarce, likely due to its rarity. From almost 600 articles reviewed here, only 33 fulfilled all the criteria and truly associated genetic variants to a concise CACD diagnosis. It is crucial to cite here that Albertos-Arranz et al. (2021) (11), re-evaluated patients initially described by Coco-Martins et al. (2020) (12). Cases with phenotypes associated with CACD but not attributed to CACD were excluded to minimize bias.
The 33 publications published between 1995 and 2021 are presented in two tables. Table 1 contains information about the authors, year of publication, techniques used for variant discovery, and the population analyzed. The identification method of each variant on all 33 papers is listed in Table 1, the last column. Table 2 shows detailed information about associated genes and variants related to the CACD phenotype. The filtering process is detailed in Figure 1. We found 17 different mutations in only six genes, reported to cause/likely cause this disease (Figure 3). The peripherin-2 gene (PRPH2) accounts for up to 85% of the mutations cited in the published studies (Figure 2; Tables 2, 3). PRPH2 variants along with Guanylate Cyclase Activator 1A (GUCA1A) and Guanylate Cyclase 2D (GUCY2D) variants, make up 90% of the total identified CACD-related variants. Other genes with CACD-associated variants were ABCA4 (ATP Binding Cassette Subfamily A Member 4), CDHR1 (Cadherin-related family member one precursor), and TTLL5 (Tubulin tyrosine ligase like 5).
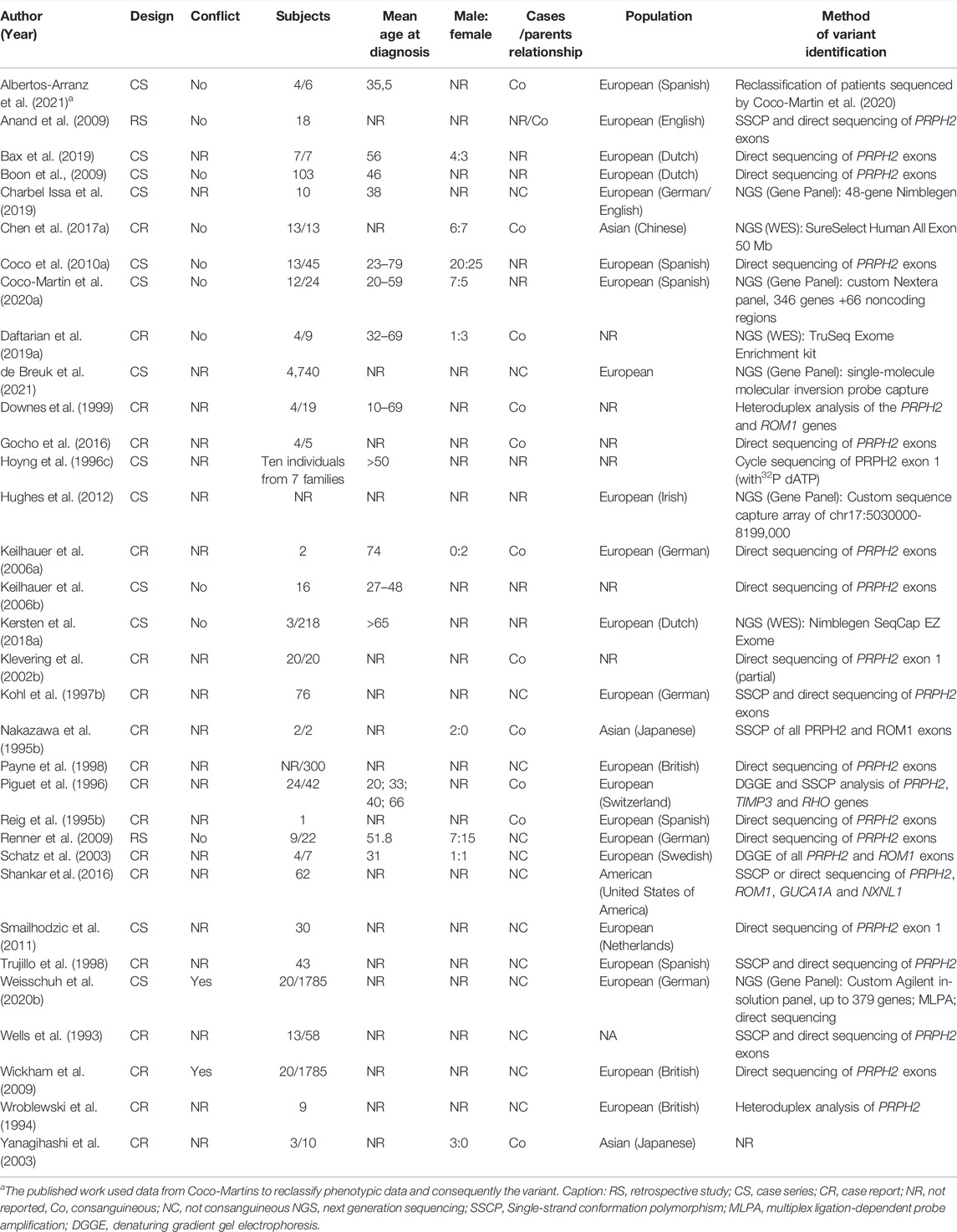
TABLE 1. General characteristics of CACD genetic association studies.

TABLE 2. CACD-associated genes and variants.
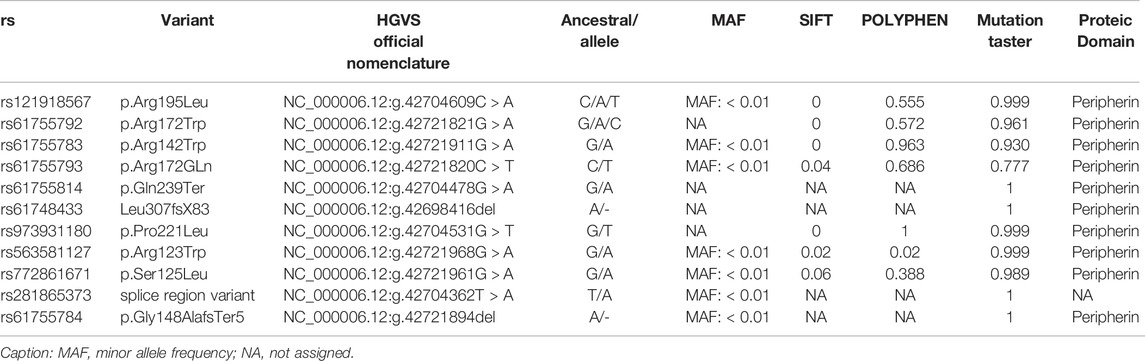
TABLE 3. Prediction of the functional effect of PRPH2 SNPs on different databases and possible proteic domain affected by them.
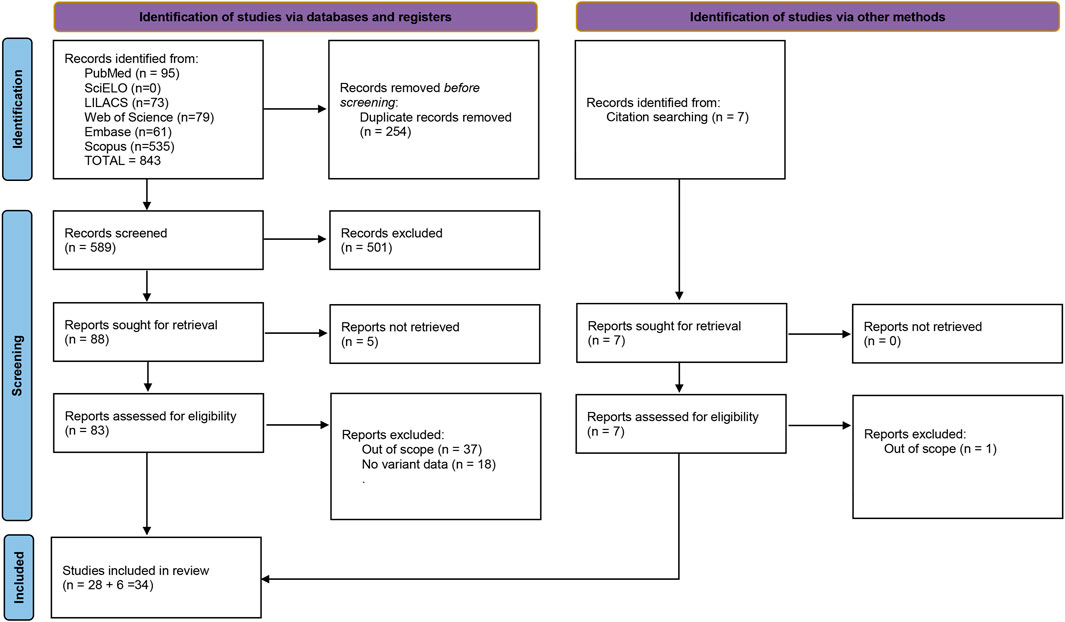
FIGURE 1. Fluxogram summarizing the selection process for this systematic review. Based on (Page et al., 2021).

FIGURE 2. PRPH2 gene and CACD-related mutations. Exons are shown as black boxes and introns as lines. Mutations reviewed in the present article are pinned in different colors in the respective exon and relative location.
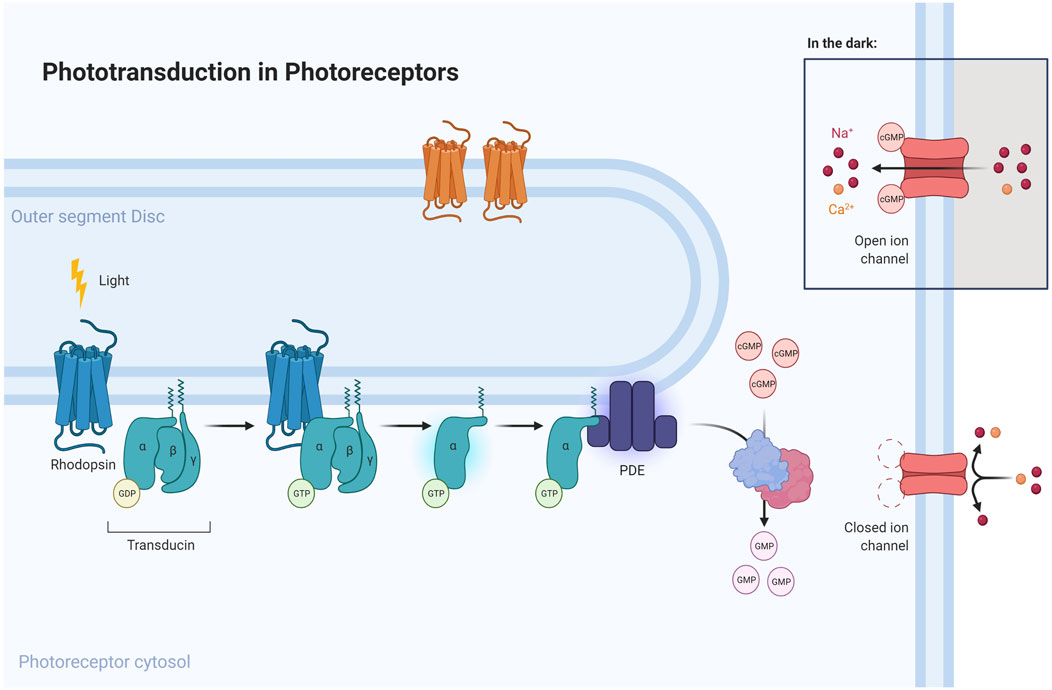
FIGURE 3. Phototransduction pathway emphasizing the role of proteins mutated in CACD. Light is converted into electric signals inside cone and rod cells. An absorbed photon activates rhodopsin, acting through repeated contacts with transducin molecules, catalyzing its activation to release GDP in exchange for cytoplasmic GTP, which expels its β and γ subunits. The G protein transducin activates the phosphodiesterase (PDE), responsible for hydrolyzing cGMP. Afterwards, guanylate cyclase (GC) synthesizes cGMP, the second messenger in the phototransduction cascade. Reduced levels of cGMP close cyclic nucleotide-gated channels, preventing a further influx of Na+ and Ca2+. Mutations in specific genes (represented as light-blue cylinders near respective proteins in the figure above) could disrupt this pathway and lead to RPE degeneration, a sign of CACD.
Enrichment analysis for these six genes indicated that all except TTLL5 are coexpressed with the following transcriptional regulators: NR2E3 (Nuclear Receptor Subfamily 2, Group E, Member 3), VSX 2 (Visual System Homeobox 2), ESRRB (Estrogen Related Receptor, Beta), RAX2 (Retina and Anterior Neural Fold Homeobox 2), NRL (Neural Retina Leucine Zipper), CRX (Cone-Rod Homeobox), ZNF385A (Zinc Finger Protein 385A), and SIX6 (Six Homeobox 6), according to data from the ARCHS4 (Lachmann et al., 2018). Beyond that, PRPH2, CDHR1, ABCA4, and GUCA1A were downregulated in a mouse knockout model of NEUROD1 (Neuronal Differentiation 1) gene (Ochocinska et al., 2012). Perturbations in the transcription factors ONECUT2 (One Cut Homeobox 2), EGR1 (Early Growth Response 1), NRL (Neural Retina Leucine Zipper), and VAX2 (Ventral Anterior Homeobox 2) seems to affect the expression of the genes found in association with CACD in this review, all based in knockout assays in mouse (Alfano et al., 2011; Goetz et al., 2014; Oh et al., 2017; Han et al., 2018; Corso-Díaz et al., 2020). MECP2 (Methyl-CpG-Binding Protein 2) was also included in this group but is the unique factor not associated with retinal diseases or eye development, playing a substantial role in early neurodevelopment (Urdinguio et al., 2008).
The enrichment analysis generated an extensive list of lncRNAs associated with at least one of the genes in this systematic review (Supplementary Table S1). We selected the top three lncRNAs of the list based on the number of genes with which they interact. The antisense WWC2-AS1 was selected for being also associated with PRPH2, the most associated gene found in this review.
4 Discussion
The identification of genetic variants in individuals affected by CACD traditionally relied on chain termination sequencing of the three exons of the PRPH2 gene (Reig et al., 1995a; Hoyng et al., 1996b; Payne et al., 1998), often preceded by exploratory analyses using single-strand conformation polymorphism (Kohl et al., 1997b; Trujillo et al., 1998), denaturing gradient gel electrophoresis (Piguet et al., 1996; Payne et al., 1998) or heteroduplex analysis (Downes, 1999a). Sanger sequencing of the coding region of PRPH2 is a robust technique and continues to prove itself relevant in recent research on CACD (Bax et al., 2019). However, parallel sequencing approaches have gradually emerged as appealing alternatives for nonobvious cases. NGS offers, with gene panels, the opportunity of a streamlined test applicable to a vast range of inherited retinal diseases (IRD), including CACD (Weisschuh et al., 2020a; Coco-Martin et al., 2020b; de Breuk et al., 2020). Weisschuh and collaborators (Weisschuh et al., 2020a) applied several versions of IRD-focused gene panels, covering up to 379 genes to study a total of 2158 IRD cases. They were able to identify three novel causative variants within the 20 CACD cases included in the cohort. However, interestingly, only seven cases (35%) were solved, emphasizing the need for further research in this area. Custom targeted gene panels can be used to precisely explore a genomic region linked with disease; Hughes and collaborators used this approach to scan a 3 Mb genomic region and identify the p. V933A mutation in GUCY2D responsible for CACD in a Northern Irish family (Hughes et al., 2012). The broader approach of whole-exome sequencing has also yielded excellent results in identifying causal variants for CACD (Chen et al., 2017a; Kersten et al., 2018a; Daftarian et al., 2019a).
In the study of Weisschuh et al. (Weisschuh et al., 2020a) mentioned before, only 0.92% of 2,158 individuals were diagnosed with CACD. Furthermore, most published studies point to PRPH2 mutations as responsible for the disease (Hoyng et al., 1996a; Boon et al., 2009; Kohl et al., 1997a; Yanagihashi et al., 2003; Trujillo et al., 1998; Chen et al., 2017a; Weisschuh et al., 2020b; Coco et al., 2010a; Daftarian et al., 2019b; Downes, 1999b; Gamundi et al., 2007; Gocho et al., 2016; Keilhauer, 2006; Kersten et al., 2018b; Klevering et al., 2002a; Nakazawa et al., 1995a; Ouechtati et al., 2009; Reig et al., 1995b; Renner et al., 2009; Schatz et al., 2003; Shankar et al., 2016; Wells et al., 1993; Wickham et al., 2009; Yusuf et al., 2018; Wroblewski et al., 1994). Notwithstanding this, mutations in other genes may be causal, such as those reported in GUCA1A, GUCY2D, ABCA4, CDHR1, and TTLL5. GUCA1A and GUCY2D are coexpressed with PRPH2 (Schadt et al., 2004; Dobbin et al., 2005; Johnson et al., 2003). The mentioned guanylate cyclases also physically interact in the phototransduction pathway of photoreceptor cells (Wu et al., 2010). Furthermore, PRPH2 and ABCA4 share physical protein interaction with CNGB1 (cyclic nucleotide-gated channel beta 1) (Schadt et al., 2004; Johnson et al., 2003; Razick et al., 2008). At least 20 proteins are associated with GUCA1A, GUCY2D, ABCA4, CDHR1, and TTLL5, by direct interaction or coexpression (Supplementary Figures S1, S2). Of those 20, at least 11 are already associated with at least one eye disease (Supplementary Table S2). The usage of GeneMANIA database for prediction of physical interaction was crucial to open new possibilities for research, and to understand pathways associated with the genes retrieved from literature search. Tools like GeneMANIA and STRING are very helpful for protein function prediction, design of protein networks and prioritizing genes or proteins in the pursuit of biological information (Warde-Farley et al., 2010; Han et al., 2020; Szklarczyk et al., 2021; Zhao et al., 2021). In our specific work, at least 11 new possibilities were available for study and research for association with not only CACD but other macular diseases, since some of the genes retrieved from the protein interaction had more than one disease associated in literature. Protein interactome is a growing field with a lot to be discovered, and other bases are providing useful insights, such as the BioGRID database of prediction of protein, genetic and chemical interactions (Oughtred et al., 2021) and CCSB interactome to human, viruses, plants, worm and yeast genomes (Luck et al., 2020).
Several other genes were additionally uncovered by gene set enrichment analysis. Polymorphisms of some of them have been associated with macular conditions. The rs149564368 (g.74267460C > T) of VSX2 was associated with higher susceptibility to ocular sarcoidosis (Garman et al., 2021), and the RAX2 rs76076446 is associated with susceptibility to macular thickness (Gao et al., 2019). In contrast, a heterozygous frameshift mutation in this gene was associated with AMD and cone-rod dystrophy (Yang et al., 2015). NEUROD1 appears to play an essential role as a transcription factor for neuronal development in Xenopus. NRL is proposed as a therapeutic gene therapy target in the retina to prevent secondary cone degeneration and alleviate the symptoms of retinitis pigmentosa, since its disruption in the mature postmitotic rod cells of mice leads them to adopt cone features and resist cell death (Yu et al., 2017). Most of the genes collected from enrichment analysis are not present in commercial sequencing panels. As an example, in NCBI’s Genetic Testing Registry (GTR, www.ncbi.nlm.nih.gov/gtr/, accessed 22 Feb 2022), out of 70778 tests, 13 listed CACD (as MIM # 215500) among the conditions tested; of these, only 8 were NGS panels, with up to 307 genes. Only two of them included all six main genes identified in this review as linked to CACD. Other four panels only missed the TTLL5 gene (The remaining two were in fact offered for other pathologies.) Among the 13 transcription factors identified by our enrichment analysis, all the six valid panels also included CRX, NR2E3, NRL and RAX2; three included VSX2; ESRRB and NEUROD1 were present in one panel each. EGR1, MECP2, ONECUT2, SIX6, VAX2 and ZNF385A were absent. This trend was also present when including all tests in GTR that included at least four out of the six main genes in this review (52 tests in total, Supplementary Table S3). The current signed-off version of Genomics England’s PanelApp Retinal Disorders virtual panel gene list (version v2.195) (Martin et al., 2019) includes all six main CACD genes, plus CRX, NR2E3, NRL, RAX2 and also VSX2, but as low-evidence gene (red list). The latest, unsigned version v2.242 also includes NEUROD1 with borderline evidence (amber list). This absence raises the discussion about possible variants of interest that might be missed with current panels, and reaffirms the need for continuous panel updating, as new works associating different genes with macular diseases are being published.
Despite a clear causal association between some eye diseases and mutations in specific genes (CORD2 for cone-rod distrophy type 2, PRPH2 for CACD, as reinforced in this research; rhodopsin (RHO) for retinitis pigmentosa), mutations in the same gene could lead to different degrees of disease severity and mutations in different genes may lead to similar phenotypes. A primal example of this is CACD, classified into three different types, based on the causal mutation (HOYNG, 1996, HUGHES, 2012). In order to better understand these dynamics, family studies are fundamental to evaluate variable expressivity in the phenotype’s severity based on the identification of mutations disturbing gene regulatory networks. Whole genome investigations would be more informative with this regard in the near future, especially in multigenerational affected families. Due to the rareness of these diseases (cone-rod dystrophies affect one in 40 thousand, retinitis pigmentosa affects one in four thousand (MedlinePlus (2022). Medli, 2022), global consortia as the Commonwealth eye consortium, Global eye genetics consortium, The European eye epidemiology consortium (European Eye Epidemiology, 2022; Global Eye Genetics Conso, 2022; (2022). Publications, 2022) are needed to increase sample size and reach reliable conclusions.
Since the methodological step of this review was finished in May 2021, another article was published, revealing a new mutation in the PRPH2 gene (p.Arg203Pro) found in four family members with symptoms of CACD (Choi et al., 2021). In order to not create a bias in this review, this recent article was not counted in the review and will not appear in the tables but could be included in subsequent reviews. Most of the genes collected from literature searching are not present in commercial sequencing panels. This absence raises the discussion about timely panel updating, as new works associating different genes with macular diseases are being published. The genes found in the systematic review and enrichment analysis results will be described below.
PRPH2
PRPH2 encodes peripherin-2 (ENSG00000112619; MIM: 179605), a transmembrane glycoprotein also named retinal degeneration slow (RDS) (Ensembl (2021). Gene:P, 2021). Highly expressed in the retina, PRPH2 is needed for the morphogenesis, stabilization, and compaction of outer segment discs in both cone and rod cells and the maintenance of the rim’s curvature, a key component of visual transduction (Ensembl (2021). Gene:P, 2021; Stuck et al., 2016). In PRPH2-haploinsufficient mice, retinae lacking PRPH2 present disorganized outer segments, leading to malformed discs and, consequently, photoreceptor loss (Cheng et al., 1997). In Prph2 “null” Rds mice, the lack of peripherin downregulates rhodopsin gene expression, causing a gradual loss of rod cells characteristic of macular diseases as retinitis pigmentosa. Moreover, the injection of a recombinant adenoassociated virus (AAV) carrying a functional copy of PRPH2 regenerated outer segment structures and corrected electrophysiology (Ali et al., 2000).
Peripherin interacts with ROM-1 (rod outer segment membrane protein-1) to generate a functional series of rims in photoreceptors, explaining why PRPH2 mutations cause the malformation of the outer discs of the retina (Bascom et al., 1992). Moreover, opsin, which contains visual pigments responsible for photon capturing, is absent in individuals with impaired peripherin-2. Consequently, the entire outer segment cannot interact with light, and these defects ultimately lead to photoreceptor death. Those facts justify why most case studies associate PRPH2 mutations to macular diseases as CACD. Since the conclusion of the methodological step of this systematic review (May 2021), another publication revealed a new CACD-causing mutation in the PRPH2 gene (p.Arg203Pro). This mutation was identified in four family members with CACD symptoms (Choi et al., 2021). In order not to create a bias, the mutation was not included in the systematic review.
GUCA1A and GUCY2D
The membrane-bound retinal guanylyl cyclase-1 protein (GUCY2D or RetGC-1) is encoded by the guanylate cyclase 2D gene (Ensembl (2021). Gene:C, 2021) (GUCY2D; ENSG00000132518; MIM: 600179), located on chromosome 17p13.1. GUCY2D is responsible for the synthesis of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP), which mediates the recovery of the dark state of photoreceptors following the transduction of a visual stimulus (Zhao et al., 2013; Zobor et al., 2014; Gene:1G00000048, 2020; Peshenko et al., 2015; Abbas et al., 2020; Peshenko et al., 2019; Michaelides et al., 2005; Krizaj and Copenhagen, 2002; Keilhauer et al., 2006a). Guanylate Cyclase Activator 1A (GUCA1A; ENSG00000048545; MIM: 600364) is a gene located on chromosome 6p21.1 that encodes guanylyl cyclase-activating protein 1 (GUCA1A or GCAP1), a retinal protein highly expressed in the inner/outer segments of cones and rods (Downes, 1999b).
The activity of GUCY2D is regulated by calcium feedback through GCAPs as GUCA1A, in response to free intracellular calcium (Ca2+) concentration during photoreceptor excitation and recovery from light (Keilhauer, 2006; Gamundi et al., 2007; Gocho et al., 2016). Briefly, exposure of photoreceptors to a light stimulus promotes the closure of ion channels, blocking the influx of Ca2+ and driving the hyperpolarization necessary for transmitting the visual stimulus to the brain. GCAPs as GUCA1A activate GUCY2D as soon as the concentration of Ca2+ drops, producing cGMP and restoring ion channels to the open conformation. With high Ca2+ concentration, GCAPs inhibit the activity of GUCY2D. This mechanism enables photoreceptors to recover fast and ensures efficient light adaptation. A failure of this process causes a loss in light sensitivity of rods and cones (Reig et al., 1995b; Ouechtati et al., 2009). However, Ca2+ homeostasis tends to be different in the outer segments of rods and cones, with a higher concentration in cones. This differential homeostasis may explain why the expression of GCAPs is also higher in cones than in rods and why mutations of these proteins result more often in cone dystrophies than cone-rod dystrophies. Mutations in GUCY2D, expressed equally in cones and rods, often lead to cone-rod dystrophies (Schatz et al., 2003; Renner et al., 2009). Beyond the well-known relationship between GUCA1A and GUCY2D in the phototransduction pathway, both are also related to olfactory transduction in mice, creating odor preferences (Zimmerman et al., 2020).
Both GUCA1A and GUCY2D variants were causally related to at least five diseases: retinopathy, retinal degeneration, retinal dystrophy, cone-rod dystrophy, and progressive cone dystrophy according to the Open Targets platform (1A (2021).1A | O, 2021; 2D (2021).2D |O, 2021). A heterozygous missense mutation affecting the catalytic domain of GUCY2D (p.Val933Ala) caused CACD in an Irish family (Shankar et al., 2016). Michaelides et al. (2005) identified a Another mutation (p.Tyr99Cys) in the GUCA1A gene, causing autosomal dominant loss of Ca2+ sensitivity. GCAPs with cysteine at position 99 of the protein cannot inactivate RetGCs (GUCY2D) at high Ca2+ concentrations. This obstruction dysregulates intracellular Ca2+ and cGMP levels in photoreceptors, leading to cell degeneration and consequent loss of rod and cone response to light (Renner et al., 2009). In a Chinese family with maculopathies ranging from mild photoreceptor degeneration to CACD, a novel GUCA1A gain-of-function mutation (p.Arg120Leu) kept GUCY2D in its activated state, causing abnormally high Ca2+ concentrations, which led to photoreceptor malfunction and eventually, maculopathy. In a zebrafish model, the mutation was proven to affect the retina and cause atrophy of the ocular vessels (Nollet et al., 2000; Chen et al., 2017b).
CDHR1
Cadherin-related family member-1 gene (CDHR1, MIM: 609502) encodes a protocadherin that belongs to the cadherin superfamily of homophilic cell-adhesion proteins (Wickham et al., 2009), expressed explicitly and abundantly in cone and rod photoreceptor cells. This calcium-dependent adhesion protein plays an essential function in maintaining the morphology of the outer segment of cone and rod cells (Duncan et al., 2012; Stingl et al., 2017; Bessette et al., 2018). Therefore, mutations that disrupt the protein or interfere with protein affinity could lead to several macular diseases, such as retinitis pigmentosa and cone-rod dystrophy, both characterized by dysfunction in cone cells (Wroblewski et al., 1994; Schadt et al., 2004).
Patients with different truncating CDHR1 mutations showed diverse visual symptoms, including glare, reading difficulties, metamorphopsia, and reduced visual acuity, resembling central areolar choroidal dystrophy. Among them, a patient with two truncating mutations showed the most severe retinal morphologic degeneration. Based on these, Charbel Issa et al. (2019) propose a continuum of severity in CDHR1-associated retinal diseases, with homozygous c.783G > A mutations at the mild end and biallelic truncating mutations at the end of this spectrum. This is one of the few works discussing autosomal recessive CACD, also suggesting a new terminology for this disease. Finally, c.783G > A causes exon skipping (exon 8, most precisely) in mRNA processing, which leads to the loss of 48 residues from the protein’s ectodomain and consequently, loss of adhesion in cone and rod cells and a severe loss of retinal function (Charbel Issa et al., 2019).
ABCA4
The ABCA4 (MIM: 601691) gene encodes for a protein belonging to the member 4 of subfamily A of ATP-binding cassette of, and is actively involved in transporting various elements across the cell membranes (Rahman et al., 2020). According to the Human Protein Atlas, this protein is widely produced in photoreceptor cells in the retinal tissue, especially rod photoreceptor cells. This protein acts as an inward-directed retinoid flippase in the visual cycle and imports retinoid substrates from the extracellular or intradiscal (rod) membrane surface to the cytoplasmic membrane surface (Quazi and Molday, 2014). ABCA4 mediates the transport of essential molecules across the photoreceptor cell membrane. The malfunction of this protein leads to the accumulation of lipofuscin, a toxic component to RPE and photoreceptors, whose steady accumulation leads to cell death (Haji Abdollahi and Hirose, 2013). Therefore, mutations in this gene are associated with Stargardt disease, recessive retinitis pigmentosa, cone-rod dystrophy type 3, early-onset severe retinal dystrophy, and age-related macular degeneration type (Cremers et al., 1998; Chen et al., 2012; Birtel et al., 2018; Lenis et al., 2018; Hull et al., 2020). In addition, one intergenic variant near ABCA4 (rs11165052) was associated with “refractive error linked to macular disorders” (Hysi et al., 2020). The genetic rs201471607 variant was related to CACD but considered inconclusive (Weisschuh et al., 2020b). Thus, ABCA4 variants that may cause CACD are still to be identified or require validation.
TTLL5
The Tubulin tyrosine ligase-like 5 (TTLL5, MIM: 612268) gene is found on the long arm of chromosome 14. It encodes minuscule six protein-coding isoforms, including a glucocorticoid receptor that belongs to the tubulin family. This protein interacts with transcriptional intermediary factor 2 (TIF2) and steroid receptor coactivator 1 (SRC1), modulating the induction or repression of glucocorticoids (Westermann and Weber, 2003; He and Simons, 2007). TTLL5 is expressed primarily in the testis and in the inner part of photoreceptors of the retina, in the base of the photoreceptor’s primary cilium (Bedoni et al., 2016a). Few recent associations with cone-rod dystrophies, including one that reports a TTLL5 multi-exon (Reig et al., 1995a; Hoyng et al., 1996b; Kohl et al., 1997b; Payne et al., 1998; Urdinguio et al., 2008; Alfano et al., 2011; Ochocinska et al., 2012; Goetz et al., 2014; Oh et al., 2017; Han et al., 2018; Corso-Díaz et al., 2020) deletion that causes cone-rod dystrophy, were found in a retrospective study with Arab families (Westermann and Weber, 2003; Sergouniotis et al., 2014; Bedoni et al., 2016b; Sun et al., 2016; Dias et al., 2017; Méjécase et al., 2020). Photoreceptors of mice with truncated alleles of TLL5 or of the Retinitis pigmentosa GTPase regulator (RPGR) gene exhibit no tubulin glutamylation (addition of glutamate molecules). TTLL5 pathogenic mutations even lead to the absence of glutamylation on RPGR, compromising its function and leading to a retinal degeneration phenotype. The study also rules out the possibility that other tubulin family members like TTLL6 or TTLL7 could glutamylate RPGR, compensating for the loss of TTLL5. Mutations in these two genes could also affect humans, causing blindness and photoreceptor degeneration, like retinitis pigmentosa or cone/cone-rod dystrophies (Sun et al., 2016). By exome sequencing, four families affected by cone-rod dystrophies were found to harbor likely-causal variants in TTLL5: two frameshifts, two nonsense, and one missense mutations. Those mutations seem to affect the equilibrium of polyglutamylation, causing cone dysfunctions (Sergouniotis et al., 2014). This gene was nevertheless not confirmed as causal for CACD, which raises questions about the exact molecular implications of these mutations, considering that different mutations in the same gene could lead to different phenotypes.
5 Enrichment Analysis Results
5.1 Transcription Factors
Most of the proteins enriched for the previously mentioned CACD-associated genes were transcription factors regulating their expression and identified through loss-of-function whole-genome transcriptomic assays. Each has the potential of critically modifying, if not causing CACD symptoms itself if mutated, and were listed below.
5.1.1 NR2E3
The nuclear receptor subfamily 2, Group E, member three-gene, known as NR2E3 (located at 15q23, MIM: *604485), encodes for a photoreceptor-specific nuclear receptor, also a ligand-dependent transcription factor. It plays a crucial role for photoreceptor development and differentiation, being preferentially expressed in rods, and acting in company with neural retina leucine zipper (NRL), and nuclear receptor subfamily 1, group D, member 1 (NR1D1), and cone-rod homeobox-containing gene (CRX) genes and other transcription factors in rod differentiation (Cheng et al., 2004). Mutations in NR2E3 seem to affect not only the differentiation of rod photoreceptors but also to downregulate mRNA and protein levels of other rod genes like RHO, GNAT1, GRK1A, and PDE6B, as shown in a zebrafish essay (Xie et al., 2019). Those works illustrate this transcription factor’s importance in establishing normal rod proliferation and retinal stability.
5.1.2 VSX2
The Visual system homeobox two genes (VSX2, also known as Chx10, located at 14q24.3, MIM: *142993), encodes a homeobox regulatory protein responsible for the development and maintenance of the neuroretina. It is expressed abundantly in the inner nuclear layer of the retina (Liu et al., 1994). This protein antagonizes the function of PRDM1, being both regulated by the orthodenticle homeobox one transcription factor OTX2 (Goodson et al., 2020), which is on top of the network that controls the development of bipolar and photoreceptor cells, also known as a photoreceptor cell fate. The absence of OTX2 leads to excessive production of amacrine cells connecting bipolar and guanylate cyclase (GC) (Goodson et al., 2020), suggesting a delicate relationship between these transcription factors. There is only one genome-wide study associating VSX2 polymorphisms with eye diseases, specifically ocular sarcoidosis. VSX2 mutations are also associated with autosomal recessive microphthalmia by disrupting the CVC motif (Chx10/Vsx-1 and ceh-10 domain, common in visual system homeobox proteins) of this gene. The disruption of this motif interferes with DNA binding and gene repression, leading to a recessive phenotype with a group of associated ocular abnormalities. This was shown in different species, suggesting that this gene is evolutionarily conserved (Reis et al., 2011).
5.1.3 ESRRB
The Estrogen-related receptor Beta (located at 14q24.3, MIM: *602167) encodes an estrogen receptor-like transcription factor of road-specific genes. It most likely mediates PRPH2 expression and other transcription factors binding the 5′ flanking region of PRPH2 like OTX, NR2E3, Myocyte-Specific Enhancer Factor 2C (MEF2C), and also Retinoid X receptor family members (Cai et al., 2010). ESRRB also regulates the metabolic demands and long-term survival of photoreceptors, and its mRNA levels fluctuate in a circadian way, according to light intensity. The loss of function of ESRRB seems to cause rod degeneration, as enhanced activity of this gene’s activity rescues photoreceptor defects (Onishi et al., 2010; Kunst et al., 2015).
5.1.4 RAX2
The retina and anterior neural fold homeobox 2 (located at 19p13.3, MIM: *610362) encodes for retina and anterior neural fold homeobox regulatory protein, whose variants may cause macular thickness. RAX2 interacts physically with CRX, recruiting other components of the basal machinery to allow transcription in both inner and outer layers of the retina. RAX2 mutations may alter its affinity with CRX, disturbing normal transcription regulation (Yang et al., 2015). RAX2 and RAX are highly conserved genes in vertebrates, but RAX2 seems more prone to cause severe defects based on its higher nonsynonymous/synonymous mutation ratio in mammals (Orquera and de Souza, 2017; Kon and Furukawa, 2020).
5.1.5 NRL
The neural retina leucine zipper protein encoded by NRL (located at 14q11.2-q12, MIM: *162080) is a transcription factor expressed specifically in rod photoreceptors and the pineal gland, interacting with CRX, NR2E3, and other transcription factors required for retinal development rod photoreceptor differentiation (Kanda et al., 2007). Loss of NRL function deregulates the expression of more than 160 genes, most of which encode proteins associated with signal transduction, transcription regulation, intracellular transport, and other vital processes for cone and rod differentiation (Yoshida et al., 2004). Accumulating evidence also associates NRL mutations with retinitis pigmentosa and with photoreceptor rod differentiation (Bessant et al., 1999; Kanda et al., 2007; Yu et al., 2017). In three independent mouse models of retinal degeneration, CRISPR-Cas9 mediated in vivo knockdown of this gene in mature (postmitotic) rod cells rescued retinal degeneration phenotypes. NRL negative rod cells started presenting cone features with subsequent improved survival rates and thus, without secondary cone degeneration in mice retina. It is possible that the ablation of this gene in germinative embryonal cells or adult rod cells presents different long-term consequences, due to specificities on gene regulatory networks of each cell type. Whereas germline mutations cause retinal degeneration, mutations in mature rod cells disturb a synergistic network of different gene products, with derepression of cone genes (Yu et al., 2017).
5.1.6 CRX
The cone-rod homeobox-containing gene (located at 19q13.33, MIM: *602225) encodes for a protein highly expressed in the retina and plays a role in photoreceptor cell development and maturity (Furukawa et al., 1997). While the overexpression of PRDM1 leads to excessive production of amacrine cells, excessive CRX production increases the number of rod photoreceptors (Furukawa et al., 1997; Goodson et al., 2020). CRX seems to synergize with the OTX2 transcription factor to control the differentiation of early cells in the retina into photoreceptor or bipolar cells (Yamamoto et al., 2020). Rescue assays proved its importance to morphogenesis and showed that CRX mutations turn it incapable of recruiting other homeodomain-interacting proteins (Terrell et al., 2012).
5.1.7 ZNF385A
The zinc finger protein 385A (located at 12q13.13, MIM *609124), also named RZF, is one of the various zinc finger regulatory proteins that work as a transcription factor (Sharma et al., 2004). This family of proteins usually has many functions like DNA recognition, RNA packaging, transcriptional activation, regulation of apoptosis, protein folding and assembly, and lipid binding (Laity et al., 2001). ZNF385A is expressed most predominantly in the retina, restricted to photoreceptors. This specific protein seems to go against the usual functions of a zinc finger protein: The work that isolated and described this protein suggests that RZF may not bind to the nucleic acid as usual zinc finger proteins, based on results from mobility shift assay. Nevertheless, the author does not rule out this possibility completely: since sub-cellular localization showed that a fusion protein RZF-GFP acts inside the nucleoli as a shuttling regulatory protein, it predicted that this protein could regulate the export of protein complexes between nucleolus and cytoplasm, both regions with high presence of this protein. Therefore, it is probable that ZNF385A could be involved in mRNA or protein ligation of photoreceptor-specific genes, having a role in retinal disease mechanisms (Sharma et al., 2004).
5.1.8 SIX6
SIX6 gene encodes the six homeobox protein (located at 14q23.1, MIM *606326), homologous of sine oculis homeobox protein in Drosophila. This protein seems to work with the ventral anterior homeobox (VAX1) (Pandolfi et al., 2020) transcript factor towards developing the suprachiasmatic nucleus, which is the primary circadian clock pacemaker (Clark et al., 2013). Therefore, disrupting one of these proteins could affect neuronal, hormonal, or behavioral homeostasis in the human organism (Pandolfi et al., 2020). SIX6 is also partly responsible for the optic nerve morphology (Ulmer Carnes et al., 2014). Variants in this gene are glaucoma-causative mutations, not only in humans but in other specimens (Ulmer Carnes et al., 2014; Hug et al., 2019). A study with a European population analyzed the retina of patients from EPIC-Norfolk Eye Study, finding a solid association with a common variant (rs33912345) of this transcription factor with a functional effect on glaucoma-associated optic nerve head traits (Khawaja et al., 2018).
5.1.9 NEUROD1
The Neurogenic differentiation 1 gene (located at 2q31.3, MIM *601724) encodes a primary helix-loop-helix transcription factor involved in neural and endocrine structures, a critical factor in regulating the regulation of insulin transcription (Ochocinska et al., 2012; Fu et al., 2013). Repression of NEUROD1 caused severe photoreceptor deficits in a chicken model. Essentially, the outer nuclear layer of the retina fragmented, with regions presenting few or no photoreceptor cells. It seems that the lack of this transcription factor affected the photoreceptors genesis as well (Yan and Wang, 2004). In mice, NEUROD1 has an essential function regulating TRbeta2, a transcription factor responsible for opsin patterning vital for color differentiation in cones. Cones without NEUROD1 express only the short version of opsins (called S-opsin), disrupting the normal function of these cells (Liu et al., 1994).
5.1.10 ONECUT2
The one cut homeobox 2 (ONECUT2 or Oc2, located at 18q21.31, MIM *604894) is a transcription factor known for having a single cut domain and a characteristic homeodomain specific for ONECUT class family members (Jacquemin et al., 1999), essential to cell fate and retinal development in different organisms (Goetz et al., 2014; Sapkota et al., 2014). ONECUT2 is usually expressed with ONECUT 1 (Oc1, MIM *604164) towards developing ganglion cells and horizontal cells (HC) in the retina. It seems that the lack of one or another generates a reduction of HC’s. However, both knockout leads to more profound defects in the development of all early cell types in the retina, also compromising the generation of cones (Sapkota et al., 2014).
5.1.11 EGR1
The Early growth response 1 (located at 5q31.2, MIM *128990) gene encodes for a transcription factor that regulates the growth of cells like epithelial cells, fibroblasts, and lymphocytes in early stages. It seems to work with laminin (encoded by LAMA2), which controls the attachment of cells, primordial for sclera elongation and normal vision (Sukhatme et al., 1988; Lin et al., 2016). EGR1 knockout in a zebrafish model affects the differentiation of amacrine and horizontal cells, delaying the development of other retinal cell types and compromising the integrity of inner and outer plexiform layers (Zhang et al., 2013). EGR1 induces apoptosis associated with p53 and inhibits human cancer cell growth, inducing the expression of transforming growth factor beta-1 (TGFB1) (Das et al., 2001).
5.1.12 MECP2
The Methyl CpG binding protein 2 gene (located at Xq28, MIM *300005) encodes for a chromatin-associated protein that activates or represses transcription of critical genes for postnatal neuronal development (Chahrour et al., 2008; Chen et al., 2003; Martinowich et al., 2003; Zhou et al., 2006; Yasui et al., 2007). Mutations in this gene lead to neurodevelopmental disorders such as Rett syndrome, susceptibility to autism, encephalopathies, and diabetic retinopathy (Swanberg et al., 2009; He et al., 2015; Li et al., 2016). An animal model shows that MECP2 is responsible for translating sensorial experience into synaptic connectivity, mediated by properly binding in methylated cytosines and provoking chromatin remodeling and promoter-mediated transcriptional regulation. This activity regulates specific genes in different expression levels and implies neuronal plasticity (Lee et al., 2014). Knockout or downregulation of MECP2 seems to affect coregulation with EGR2, generating a phenotype of Rett syndrome in women and autistic characteristics in men. This happens because both MECP2 and EGR2 co-regulate each other, where MECP2 binds to an enhancer of EGR2, causing upregulation in the expression of both genes, leading to increased neuronal maturation (Swanberg et al., 2009).
5.1.13 VAX2
The ventral anterior homeobox 2 (located at 2p13.3, MIM *604295) is a transcription factor responsible for the morphogenic process responsible for the correct dorsoventral pattern of the eye (Barbieri et al., 1999). Mutations could alternate the eye dorsal-ventral axis in a mouse model (Barbieri et al., 2002), or the distribution of retinoic acid metabolism, contributing to the expression of cone opsins in humans (Alfano et al., 2011). In agreement, one VAX2 variant (rs3771395) was also associated with astigmatism (Lopes et al., 2013).
5.2 lncRNAs
LINC02865 encodes two transcripts, LINC02865-201 and LINC02865-202, with 381 bp and 535 bp, respectively. According to the FANTOM5 Project (Lizio et al., 2019), this long non-coding RNA (lncRNA) is highly expressed in the eye at the fetal stage (expression level of 69 TPM). This RNA appears to be frequently coexpressed with rhodopsin (RHO) and Aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) genes. Perturbations of its function may cause several phenotypes of retinal disorders in rats, like abnormal retinal cone cell morphology, absent photoreceptor outer segment, absence of retinal rod cells, among many other conditions affecting the eye (listed in the lncHUB database) (Lachmann et al., 2019). The LINC02865 is predicted as a member of the phototransduction pathway as well, according to lncHUB Database.
LINC00575 encodes three transcripts with 1,055 bp, 910 bp, and 848 bp. Data retrieved from the lncHUB database (Lachmann et al., 2019) indicates that this lncRNA is strongly associated with the phototransduction pathway, as well as with phenotypes like abnormal morphology of retinal cone cells, abnormal morphology of the outer segment of retinal rod cells and absence of photoreceptors in the outer segment. LINC00575 is expressed in the cerebral cortex and choroid plexus and predicted to affect diverse gene ontology-mediated biological processes, like regulation of rhodopsin, rhodopsin-mediated signaling, and phototransduction.
LINC02733 encodes two transcripts with 2,938 bp and 788 bp. Expressed in the diencephalon, midbrain, and the choroid plexus, this lncRNA is also associated with the phototransduction pathway and various phenotypes related to visual impairment, like abnormal morphology of cone cells in the retina, absence of photoreceptors in the outer segment, abnormal morphology in the ocular fundus. Like the previous RNAs, it is also predicted to affect the signaling mediated by the rhodopsin pathway.
WWC2-AS1 is an antisense RNA of 404 bp for the WWC2 gene, associated with the phototransduction pathway (lncHUB) and expressed in the eye and choroid plexus. WWC2-AS1 downregulates miRNA mir16 levels by acting as a competing endogenous RNA, regulating the expression of fibroblast growth factor 2 (FGF2), a protein implicated in corneal vascularization and regeneration of tissues in the retina (Lee and Kay, 2006; de Oliveira Dias et al., 2011; Zhou et al., 2019; Chen et al., 2020). This miRNA is responsible for uveal melanoma and primary open-angle glaucoma, along with other miRNAs. Disturbance in the balance between WWC2-AS1, miRNA16, and proper regulation of FGF2 could lead to several implications starting from the proper development of tissues in the eye to lesions in the eye caused by melanoma or glaucoma (Stark et al., 2019; Tamkovich et al., 2019).
6 Conclusion
CACD is a rare hereditary disease with dominant inheritance. Candidate genes for this disease encode proteins functionally related to photoreceptors, either in the phototransduction process, as in the case of GUCY2D, recovery of retinal photodegradation photoreceptors in phototransduction pathway in the case of GUCA1A, or the formation and maintenance of specific structures within the photoreceptors, as in the case of PRPH2, TTLL5 and CDHR1. Enrichment analysis was crucial to compile a set of other genetic factors that could also be related to CACD, despite not appearing in the systematic review process. They are not commonly related to CACD or visual diseases and most of them do not appear in commercial gene panels; therefore, more studies about these genes need to be executed, and those panels reviewed in due time.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
AB administered the project and supervised this work. RR contributed to the conception of the work and curated and analyzed the data. JK, GP and RR performed the investigation. NS and MT provided ophthalmologic information about the CACD disease. JK, GP, AB and RR drafted and edited the manuscript, after critical review for intellectual content, by all co-authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants of CAPES/PROAP Finance Code 001. AB receives a research productivity scholarship from CNPq (protocol number 314288/2018-0).
Conflict of Interest
Authors NS and MTS are employed by Retina and Vitreo Consulting Eye Clinic, Curitiba, Brazil.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Alessandro Max, Sara Cristina Lobo Alves and Liana Alves de Oliveira for helpful discussions about the genetic etiology of CACD.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.886461/full#supplementary-material
Supplementary Figure S1 | Network for protein interactions with CACD candidate gene products. Generated using GeneMania (64).
Supplementary Figure S2 | Network for coexpression of CACD candidate genes. Generated using GeneMania (64).
References
Abbas, S., Marino, V., Bielefeld, L., Koch, K. W., and Dell'Orco, D. (2020). Constitutive Activation of Guanylate Cyclase by the G86R GCAP1 Variant Is Due to "Locking" Cation-π Interactions that Impair the Activator-To-Inhibitor Structural Transition. Int. J. Mol. Sci. 21 (3), 752. doi:10.3390/ijms21030752
Albertos-Arranz, H., Sánchez-Sáez, X., Martínez-Gil, N., Ruiz-Pastor, M. J., Pinilla, I., Coco-Martín, R. M., et al. (2021). Phenotypic Differences in the PRPH2 Mutation in Members of the Same Family Assessed with OCT and OCTA. Invest. Ophthalmol. Vis. Sci. 62 (8), 2443.
Alfano, G., Conte, I., Caramico, T., Avellino, R., Arnò, B., Pizzo, M. T., et al. (2011). Vax2 Regulates Retinoic Acid Distribution and Cone Opsin Expression in the Vertebrate Eye. Development 138 (2), 261–271. doi:10.1242/dev.051037
Ali, R. R., Sarra, G.-M., Stephens, C., Alwis, M. d., Bainbridge, J. W. B., Munro, P. M., et al. (2000). Restoration of Photoreceptor Ultrastructure and Function in Retinal Degeneration Slow Mice by Gene Therapy. Nat. Genet. 25 (3), 306–310. doi:10.1038/77068
Anand, S., Sheridan, E., Cassidy, F., Inglehearn, C., Williams, G., Springell, K., et al. (2009). Macular Dystrophy Associated with the Arg172Trp Substitution in Peripherin/rds. Retina 29 (5), 682–688. doi:10.1097/iae.0b013e318198dbed
Barbieri, A. M., Broccoli, V., Bovolenta, P., Alfano, G., Marchitiello, A., Mocchetti, C., et al. (2002). Vax2inactivation in Mouse Determines Alteration of the Eye Dorsal-Ventral axis, Misrouting of the Optic Fibres and Eye Coloboma. Development 129 (3), 805–813. doi:10.1242/dev.129.3.805
Barbieri, A. M., Lupo, G., Bulfone, A., Andreazzoli, M., Mariani, M., Fougerousse, F., et al. (1999). A Homeobox Gene, Vax2 , Controls the Patterning of the Eye Dorsoventral axis. Proc. Natl. Acad. Sci. U.S.A. 96 (19), 10729–10734. doi:10.1073/pnas.96.19.10729
Bascom, R. A., Manara, S., Collins, L., Molday, R. S., Kalnins, V. I., and Mclnnes, R. R. (1992). Cloning of the cDNA for a Novel Photoreceptor Membrane Protein (Rom-1) Identifies a Disk Rim Protein Family Implicated in Human Retinopathies. Neuron 8 (6), 1171–1184. doi:10.1016/0896-6273(92)90137-3
Bax, N. M., Valkenburg, D., Lambertus, S., Klevering, B. J., Boon, C. J. F., Holz, F. G., et al. (2019). Foveal Sparing in Central Retinal Dystrophies. Invest. Ophthalmol. Vis. Sci. 60 (10), 3456. doi:10.1167/iovs.18-26533
Bedoni, N., Haer-Wigman, L., Vaclavik, V., Tran, V. H., Farinelli, P., Balzano, S., et al. (2016). Mutations in the Polyglutamylase Gene TTLL5 , Expressed in Photoreceptor Cells and Spermatozoa, Are Associated with Cone-Rod Degeneration and Reduced Male Fertility. Hum. Mol. Genet. 25 (20), 4546–4555. doi:10.1093/hmg/ddw282
Bedoni, N., Haer-Wigman, L., Vaclavik, V., Tran, V. H., Farinelli, P., Balzano, S., et al. (2016). Mutations in the Polyglutamylase Gene TTLL5, Expressed in Photoreceptor Cells and Spermatozoa, Are Associated with Cone-Rod Degeneration and Reduced Male Fertility. Hum. Mol. Genet. 25 (20), 4546–4555. doi:10.1093/hmg/ddw282
Bessant, D. A. R., Payne, A. M., Mitton, K. P., Wang, Q.-L., Swain, P. K., Plant, C., et al. (1999). A Mutation in NRL Is Associated with Autosomal Dominant Retinitis Pigmentosa. Nat. Genet. 21 (4), 355–356. doi:10.1038/7678
Bessette, A. P., DeBenedictis, M. J., and Traboulsi, E. I. (2018). Clinical Characteristics of Recessive Retinal Degeneration Due to Mutations in the CDHR1 Gene and a Review of the Literature. Ophthalmic Genet. 39 (1), 51–55. doi:10.1080/13816810.2017.1363244
Birtel, J., Eisenberger, T., Gliem, M., Müller, P. L., Herrmann, P., Betz, C., et al. (2018). Clinical and Genetic Characteristics of 251 Consecutive Patients with Macular and Cone/cone-Rod Dystrophy. Sci. Rep. 8, 4824. doi:10.1038/s41598-018-22096-0
Boon, C. J. F., Klevering, B. J., Cremers, F. P. M., Zonneveld-Vrieling, M. N., Theelen, T., Den Hollander, A. I., et al. (2009). Central Areolar Choroidal Dystrophy. Ophthalmology 116 (4), 771–782. doi:10.1016/j.ophtha.2008.12.019
Cai, X., Conley, S. M., Cheng, T., Al-Ubaidi, M. R., and Naash, M. I. (2010). A 350 Bp Region of the Proximal Promoter of Rds Drives Cell-type Specific Gene Expression. Exp. Eye Res. 91 (2), 186–194. doi:10.1016/j.exer.2010.04.017
CEHC (2022). Publications [Internet]. London, UK: CEHC. Available from: https://cehc.lshtm.ac.uk/publications/.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., et al. (2008). MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 320 (5880), 1224–1229. doi:10.1126/science.1153252
Charbel Issa, P., Gliem, M., Yusuf, I. H., Birtel, J., Müller, P. L., Mangold, E., et al. (2019). A Specific Macula-Predominant Retinal Phenotype Is Associated with the CDHR1 Variant c.783G>A, a Silent Mutation Leading to In-Frame Exon Skipping. Invest. Ophthalmol. Vis. Sci. 60 (10), 3388–3397. doi:10.1167/iovs.18-26415
Chen, M., Bao, L., Zhao, M., Cao, J., and Zheng, H. (2020). Progress in Research on the Role of FGF in the Formation and Treatment of Corneal Neovascularization. Front. Pharmacol. 11, 111. doi:10.3389/fphar.2020.00111
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF Transcription Involves Calcium-dependent Phosphorylation of MeCP2. Science 302 (5646), 885–889. doi:10.1126/science.1086446
Chen, X., Sheng, X., Zhuang, W., Sun, X., Liu, G., Shi, X., et al. (2017). GUCA1A Mutation Causes Maculopathy in a Five-Generation Family with a Wide Spectrum of Severity. Genet. Med. 19 (8), 945–954. doi:10.1038/gim.2016.217
Chen, X., Sheng, X., Zhuang, W., Sun, X., Liu, G., Shi, X., et al. (2017). GUCA1A Mutation Causes Maculopathy in a Five-Generation Family with a Wide Spectrum of Severity. Genet. Med. 19 (8), 945–954. doi:10.1038/gim.2016.217
Chen, Y., Okano, K., Maeda, T., Chauhan, V., Golczak, M., Maeda, A., et al. (2012). Mechanism of All-Trans-Retinal Toxicity with Implications for Stargardt Disease and Age-Related Macular Degeneration. J. Biol. Chem. 287 (7), 5059–5069. doi:10.1074/jbc.m111.315432
Cheng, H., Khanna, H., Oh, E. C. T., Hicks, D., Mitton, K. P., and Swaroop, A. (2004). Photoreceptor-specific Nuclear Receptor NR2E3 Functions as a Transcriptional Activator in Rod Photoreceptors. Hum. Mol. Genet. 13 (15), 1563–1575. doi:10.1093/hmg/ddh173
Cheng, T., Peachey, N. S., Li, S., Goto, Y., Cao, Y., and Naash, M. I. (1997). The Effect of Peripherin/rds Haploinsufficiency on Rod and Cone Photoreceptors. J. Neurosci. 17 (21), 8118–8128. doi:10.1523/jneurosci.17-21-08118.1997
Choi, H., Cloutier, A., and Lally, D. (2021). PRPH2-Associated Macular Dystrophy in 4 Family Members with a Novel Mutation. Ophthalmic Genet. 0 (0), 1–5. doi:10.1080/13816810.2021.2015790
Clark, D. D., Gorman, M. R., Hatori, M., Meadows, J. D., Panda, S., and Mellon, P. L. (2013). Aberrant Development of the Suprachiasmatic Nucleus and Circadian Rhythms in Mice Lacking the Homeodomain Protein Six6. J. Biol. Rhythms 28 (1), 15–25. doi:10.1177/0748730412468084
Coco, R. M., Tellena, J. J., Sanabria, M. R., Rodríguez-Rúa, E., and García, M. T. (2010). PRPH2 (Peripherin/RDS) Mutations Associated with Different Macular Dystrophies in a Spanish Population: A New Mutation. Eur. J. Ophthalmol. 20 (4), 724–732. doi:10.1177/112067211002000413
Coco, R. M., Tellena, J. J., Sanabria, M. R., Rodríguez-Rúa, E., and García, M. T. (2010). PRPH2 (Peripherin/RDS) Mutations Associated with Different Macular Dystrophies in a Spanish Population: a New Mutation. Eur. J. Ophthalmol. 20 (4), 724–732. doi:10.1177/112067211002000413
Coco-Martin, R. M., Sanchez-Tocino, H. T., Desco, C., Usategui-Martín, R., and Tellería, J. J. (2020). PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes (Basel) 11 (7), E773. doi:10.3390/genes11070773
Coco-Martin, R. M., Sanchez-Tocino, H. T., Desco, C., Usategui-Martín, R., and Tellería, J. J. (2020). PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes (Basel) 11 (7), 773. doi:10.3390/genes11070773
Corso-Díaz, X., Gentry, J., Rebernick, R., Jaeger, C., Brooks, M. J., van Asten, F., et al. (2020). Genome-wide Profiling Identifies DNA Methylation Signatures of Aging in Rod Photoreceptors Associated with Alterations in Energy Metabolism. Cel Rep. 31 (3), 107525. doi:10.1016/j.celrep.2020.107525
Cremers, F., van de Pol, D. J. R., van Driel, M., den Hollander, A. I., van Haren Fjj, , Knoers, N. V. A. M., et al. (1998). Autosomal Recessive Retinitis Pigmentosa and Cone-Rod Dystrophy Caused by Splice Site Mutations in the Stargardt's Disease Gene ABCR. Hum. Mol. Genet. 7 (3), 355–362. doi:10.1093/hmg/7.3.355
Daftarian, N., Mirrahimi, M., Sabbaghi, H., Moghadasi, A., Zal, N., Dehghan Banadaki, H., et al. (2019). PRPH2 Mutation as the Ical Manifestations in a Family Affected with Inherited Retinal Dystrophy. Ophthalmic Genet. 40 (5), 436–442. doi:10.1080/13816810.2019.1678178
Daftarian, N., Mirrahimi, M., Sabbaghi, H., Moghadasi, A., Zal, N., Dehghan Banadaki, H., et al. (2019). PRPH2 Mutation as the Cause of Various Clinical Manifestations in a Family Affected with Inherited Retinal Dystrophy. Ophthalmic Genet. 40 (5), 436–442. doi:10.1080/13816810.2019.1678178
Das, A., Chendil, D., Dey, S., Mohiuddin, M., Mohiuddin, M., Milbrandt, J., et al. (2001). Ionizing Radiation Down-Regulates P53 Protein in Primary Egr-1−/− Mouse Embryonic Fibroblast Cells Causing Enhanced Resistance to Apoptosis. J. Biol. Chem. 276 (5), 3279–3286. doi:10.1074/jbc.m008454200
de Breuk, A., Acar, I. E., Kersten, E., Schijvenaars, M. M. V. A. P., Colijn, J. M., Haer-Wigman, L., et al. (2020). Development of a Genotype Assay for Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 128, 1604–1617. doi:10.1016/j.ophtha.2020.07.037
de Breuk, A., Acar, I. E., Kersten, E., Schijvenaars, M. M. V. A. P., Colijn, J. M., Haer-Wigman, L., et al. (2021). Development of a Genotype Assay for Age-Related Macular Degeneration. Ophthalmology 128 (11), 1604–1617. doi:10.1016/j.ophtha.2020.07.037
de Oliveira Dias, J. R., Rodrigues, E. B., Maia, M., Magalhães, O., Penha, F. M., and Farah, M. E. (2011). Cytokines in Neovascular Age-Related Macular Degeneration: Fundamentals of Targeted Combination Therapy. Br. J. Ophthalmol. 95 (12), 1631–1637. doi:10.1136/bjo.2010.186361
Dias, M. S., Hamel, C. P., Meunier, I., Varin, J., Blanchard, S., Boyard, F., et al. (2017). Novel Splice-Site Mutation in TTLL5 Causes Cone Dystrophy in a Consanguineous Family. Mol. Vis. 23, 131–139.
Dobbin, K. K., Beer, D. G., Meyerson, M., Yeatman, T. J., Gerald, W. L., Jacobson, J. W., et al. (2005). Interlaboratory Comparability Study of Cancer Gene Expression Analysis Using Oligonucleotide Microarrays. Clin. Cancer Res. 11 (2 Pt 1), 565–572.
Downes, S. M. (1999). Clinical Features of Codon 172 RDSMacular Dystrophy: Similar Phenotype in 12 Families. Arch. Ophthalmol. 117 (10), 1373. doi:10.1001/archopht.117.10.1373
Downes, S. M. (1999). Clinical Features of Codon 172 RDSMacular Dystrophy. Arch. Ophthalmol. 117 (10), 1373. doi:10.1001/archopht.117.10.1373
Downes, S. M., Fitzke, F. W., Holder, G. E., Payne, A. M., Bessant, D. A., Bhattacharya, S. S., et al. (1999). Clinical Features of Codon 172 RDSMacular Dystrophy. Arch. Ophthalmol. 117 (10), 1373–1383. doi:10.1001/archopht.117.10.1373
Duncan, J. L., Roorda, A., Navani, M., Vishweswaraiah, S., Syed, R., Soudry, S., et al. (2012). Identification of a Novel Mutation in the CDHR1 Gene in a Family with Recessive Retinal Degeneration. Arch. Ophthalmol. 130 (10), 1301–1308. doi:10.1001/archophthalmol.2012.1906
Ensembl (2021). Gene: GUCY2D (ENSG00000132518) - Summary - Homo_sapiens - Ensembl Genome Browser 103 [Internet]. Hinxton, Cambridgeshire, UK: Ensembl. Available from: https://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000132518;r=17:8002615-8020342.
Ensembl (2021). Gene: PRPH2 (ENSG00000112619) - Summary - Homo_sapiens - Ensembl Genome Browser 103 [Internet]. Ernst-Abbe-Straße, Bonn: Ensembl. Available from: https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000112619;r=6:42696598-42722597;t=ENST00000230381.
European Eye Epidemiology (2022). E3: European Eye Epidemiology Consortium [Internet]. Hinxton, Cambridgeshire, UK: European Eye Epidemiology. Available from: http://www.eye-epi.eu/.
Fu, Z., R. Gilbert, E., and Liu, D. (2013). Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 9 (1), 25–53. doi:10.2174/157339913804143225
Furukawa, T., Morrow, E. M., and Cepko, C. L. (1997). Crx, a Novel Otx-like Homeobox Gene, Shows Photoreceptor-specific Expression and Regulates Photoreceptor Differentiation. Cell 91 (4), 531–541. doi:10.1016/s0092-8674(00)80439-0
Gamundi, M. J., Hernan, I., Muntanyola, M., Trujillo, M. J., García-Sandoval, B., Ayuso, C., et al. (2007). High Prevalence of Mutations in Peripherin/RDS in Autosomal Dominant Macular Dystrophies in a Spanish Population. Mol. Vis. 13, 1031–1037.
Gao, X. R., Huang, H., and Kim, H. (2019). Genome-wide Association Analyses Identify 139 Loci Associated with Macular Thickness in the UK Biobank Cohort. Hum. Mol. Genet. 28 (7), 1162–1172. doi:10.1093/hmg/ddy422
Garman, L., Pezant, N., Pastori, A., Savoy, K. A., Li, C., Levin, A. M., et al. (2021). Genome-Wide Association Study of Ocular Sarcoidosis Confirms HLA Associations and Implicates Barrier Function and Autoimmunity in African Americans. Ocul. Immunol. Inflamm. 29 (2), 244–249. doi:10.1080/09273948.2019.1705985
Gene: GUCA1A ENSG00000048545 (2020). ENSEMBL. Gene: GUCA1A (ENSG00000048545) Human (GRCh38.P13) [Internet]. Tokyo, Japan: Gene: GUCA1A ENSG00000048545. Available from: http://www.ensembl.org/Homo_sapiens/Gene/StructuralVariation_Gene?db=core.
Global Eye Genetics Consortium (2022). Global Eye Genetics Consortium [Internet]. Tokyo, Japan: Global Eye Genetics Consortium. Available from: https://www.gegc.org/gegc.org/.
Gocho, K., Akeo, K., Itoh, N., Kameya, S., Hayashi, T., Katagiri, S., et al. (2016). High-Resolution Adaptive Optics Retinal Image Analysis at Early Stage Central Areolar Choroidal Dystrophy with PRPH2 Mutation. Ophthalmic Surg. Lasers Imaging Retina 47 (12), 1115–1126. doi:10.3928/23258160-20161130-05
Goetz, J. J., Martin, G. M., Chowdhury, R., and Trimarchi, J. M. (2014). Onecut1 and Onecut2 Play Critical Roles in the Development of the Mouse Retina. PLoS One 9 (10), e110194. doi:10.1371/journal.pone.0110194
Goodson, N. B., Kaufman, M. A., Park, K. U., and Brzezinski, J. A. (2020). Simultaneous Deletion of Prdm1 and Vsx2 Enhancers in the Retina Alters Photoreceptor and Bipolar Cell Fate Specification, yet Differs from Deleting Both Genes. Development 147 (13), dev190272. doi:10.1242/dev.190272
GUCA1A (2021). GUCA1A | Open Targets Platform [Internet]. Available from: https://platform.opentargets.org/target/ENSG00000048545?view=sec:mouse_phenotypes.
GUCY2D (2021). GUCY2D | Open Targets Platform [Internet]. Available from: https://platform.opentargets.org/target/ENSG00000132518.
Haji Abdollahi, S., and Hirose, T. (2013). Stargardt-Fundus Flavimaculatus: Recent Advancements and Treatment. Semin. Ophthalmol. 28 (5–6), 372–376. doi:10.3109/08820538.2013.825286
Han, H., Cho, J.-W., Lee, S., Yun, A., Kim, H., Bae, D., et al. (2018). TRRUST V2: an Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res. 46 (D1), D380–D386. doi:10.1093/nar/gkx1013
Han, J., Wan, M., Ma, Z., Hu, C., and Yi, H. (2020). Prediction of Targets of Curculigoside A in Osteoporosis and Rheumatoid Arthritis Using Network Pharmacology and Experimental Verification. Dddt 14, 5235–5250. doi:10.2147/dddt.s282112
He, S., Barron, E., Ishikawa, K., Nazari Khanamiri, H., Spee, C., Zhou, P., et al. (2015). Inhibition of DNA Methylation and Methyl-CpG-Binding Protein 2 Suppresses RPE Transdifferentiation: Relevance to Proliferative Vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 56 (9), 5579–5589. doi:10.1167/iovs.14-16258
He, Y., and Simons, S. S. (2007). STAMP, a Novel Predicted Factor Assisting TIF2 Actions in Glucocorticoid Receptor-Mediated Induction and Repression. Mol. Cel Biol 27 (4), 1467–1485. doi:10.1128/mcb.01360-06
Hoyng, C. B., Heutink, P., Testers, L., Pinckers, A., Deutman, A. F., and Oostra, B. A. (1996). Autosomal Dominant Central Areolar Choroidal Dystrophy Caused by a Mutation in Codon 142 in the Peripherin/RDS Gene. Am. J. Ophthalmol. 121 (6), 623–629. doi:10.1016/s0002-9394(14)70627-0
Hoyng, C. B., Heutink, P., Testers, L., Pinckers, A., Deutman, A. F., and Oostra, B. A. (1996). Autosomal Dominant Central Areolar Choroidal Dystrophy Caused by a Mutation in Codon 142 in the Peripherin/RDS Gene. Am. J. Ophthalmol. 121 (6), 623–629. doi:10.1016/s0002-9394(14)70627-0
Hoyng, C. B., Heutink, P., Testers, L., Pinckers, A., Deutman, A. F., and Oostra, B. A. (1996). Autosomal Dominant central Areolar Choroidal Dystrophy Caused by a Mutation in Codon 142 in the Peripherin/RDS Gene. Am. J. Ophthalmol. 121 (6), 623–629. doi:10.1016/s0002-9394(14)70627-0
Hug, P., Anderegg, L., Dürig, N., Lepori, V., Jagannathan, V., Spiess, B., et al. (2019). A SIX6 Nonsense Variant in Golden Retrievers with Congenital Eye Malformations. Genes (Basel) 10 (6), E454. doi:10.3390/genes10060454
Hughes, A. E., Meng, W., Lotery, A. J., and Bradley, D. T. (2012). A NovelGUCY2DMutation, V933A, Causes Central Areolar Choroidal Dystrophy. Invest. Ophthalmol. Vis. Sci. 53 (8), 4748. doi:10.1167/iovs.12-10061
Hull, S., Kiray, G., Chiang, J. P. W., and Vincent, A. L. (2020). Molecular and Phenotypic Investigation of a New Zealand Cohort of Childhood‐onset Retinal Dystrophy. Am. J. Med. Genet. 184 (3), 708–717. doi:10.1002/ajmg.c.31836
Hysi, P. G., Choquet, H., Choquet, H., Khawaja, A. P., Wojciechowski, R., Tedja, M. S., et al. (2020). Meta-analysis of 542,934 Subjects of European Ancestry Identifies New Genes and Mechanisms Predisposing to Refractive Error and Myopia. Nat. Genet. 52 (4), 401–407. doi:10.1038/s41588-020-0599-0
Iannaccone, A. (2001). Genotype-phenotype Correlations and Differential Diagnosis in Autosomal Dominant Macular Disease. Doc Ophthalmol. 102 (3), 197–236. doi:10.1023/a:1017566600871
Jacquemin, P., Lannoy, V. J., Rousseau, G. G., and Lemaigre, F. P. (1999). OC-2, a Novel Mammalian Member of the ONECUT Class of Homeodomain Transcription Factors Whose Function in Liver Partially Overlaps with that of Hepatocyte Nuclear Factor-6. J. Biol. Chem. 274 (5), 2665–2671. doi:10.1074/jbc.274.5.2665
Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P. M., Armour, C. D., et al. (2003). Genome-wide Survey of Human Alternative Pre-mRNA Splicing with Exon junction Microarrays. Science 302 (5653), 2141–2144. doi:10.1126/science.1090100
Kanda, A., Friedman, J. S., Nishiguchi, K. M., and Swaroop, A. (2007). Retinopathy Mutations in the bZIP Protein NRL Alter Phosphorylation and Transcriptional Activity. Hum. Mutat. 28 (6), 589–598. doi:10.1002/humu.20488
Keilhauer, C. N. (2006). Clinical Findings in a Multigeneration Family with Autosomal Dominant Central Areolar Choroidal Dystrophy Associated with an Arg195Leu Mutation in the Peripherin/RDS Gene. Arch. Ophthalmol. 124 (7), 1020. doi:10.1001/archopht.124.7.1020
Keilhauer, C. N., Meigen, T., and Weber, B. H. F. (2006). Clinical Findings in a Multigeneration Family with Autosomal Dominant central Areolar Choroidal Dystrophy Associated with an Arg195Leu Mutation in the Peripherin/RDS Gene. Arch. Ophthalmol. 124 (7), 1020–1027. doi:10.1001/archopht.124.7.1020
Keilhauer, C. N., Meigen, T., Stöhr, H., and Weber, B. H. F. (2006). Late-onset central Areolar Choroidal Dystrophy Caused by a Heterozygous Frame-Shift Mutation Affecting Codon 307 of the Peripherin/RDS Gene. Ophthalmic Genet. 27 (4), 139–144. doi:10.1080/13816810600976822
Kersten, E., Geerlings, M. J., Pauper, M., Corominas, J., Bakker, B., Altay, L., et al. (2018). Genetic Screening for Macular Dystrophies in Patients Clinically Diagnosed with Dry Age‐related Macular Degeneration. Clin. Genet. 94 (6), 569–574. doi:10.1111/cge.13447
Kersten, E., Geerlings, M. J., Pauper, M., Corominas, J., Bakker, B., Altay, L., et al. (2018). Genetic Screening for Macular Dystrophies in Patients Clinically Diagnosed with Dry Age‐related Macular Degeneration. Clin. Genet. 94 (6), 569–574. doi:10.1111/cge.13447
Khawaja, A. P., Chan, M. P. Y., Yip, J. L. Y., Broadway, D. C., Garway-Heath, D. F., Viswanathan, A. C., et al. (2018). A Common Glaucoma-Risk Variant of SIX6 Alters Retinal Nerve Fiber Layer and Optic Disc Measures in a European Population: The EPIC-Norfolk Eye Study. J. Glaucoma 27 (9), 743–749. doi:10.1097/ijg.0000000000001026
Klevering, B. J., van Driel, M., van Hogerwou, A. J. M., van de Pol, D. J. R., Deutman, A. F., Pinckers, A. J. L. G., et al. (2002). Central Areolar Choroidal Dystrophy Associated with Dominantly Inherited Drusen. Br. J. Ophthalmol. 86 (1), 91–96. doi:10.1136/bjo.86.1.91
Klevering, B. J., van Driel, M., van Hogerwou, A. J. M., van De Pol, D. J. R., Deutman, A. F., Pinckers, A. J. L. G., et al. (2002). Central Areolar Choroidal Dystrophy Associated with Dominantly Inherited Drusen. Br. J. Ophthalmol. 86 (1), 91–96. doi:10.1136/bjo.86.1.91
Kohl, S., Christ-Adler, M., Apfelstedt-Sylla, E., Kellner, U., Eckstein, A., Zrenner, E., et al. (1997). RDS/peripherin Gene Mutations Are Frequent Causes of central Retinal Dystrophies. J. Med. Genet. 34 (8), 620–626. doi:10.1136/jmg.34.8.620
Kohl, S., Christ-Adler, M., Apfelstedt-Sylla, E., Kellner, U., Eckstein, A., Zrenner, E., et al. (1997). RDS/peripherin Gene Mutations Are Frequent Causes of central Retinal Dystrophies. J. Med. Genet. 34 (8), 620–626. doi:10.1136/jmg.34.8.620
Kon, T., and Furukawa, T. (2020). Origin and Evolution of the Rax Homeobox Gene by Comprehensive Evolutionary Analysis. FEBS Open Bio 10 (4), 657–673. doi:10.1002/2211-5463.12832
Krizaj, D., and Copenhagen, D. R. (2002). Calcium Regulation in Photoreceptors. Front. Biosci. 7, d2023–44. doi:10.2741/a896
Kunst, S., Wolloscheck, T., Grether, M., Trunsch, P., Wolfrum, U., and Spessert, R. (2015). Photoreceptor Cells Display a Daily Rhythm in the Orphan Receptor Esrrβ. Mol. Vis. 21, 173–184.
Lachmann, A., Schilder, B. M., Wojciechowicz, M. L., Torre, D., Kuleshov, M. V., Keenan, A. B., et al. (2019). Geneshot: Search Engine for Ranking Genes from Arbitrary Text Queries. Nucleic Acids Res. 47 (W1), W571–W577. doi:10.1093/nar/gkz393
Lachmann, A., Torre, D., Keenan, A. B., Jagodnik, K. M., Lee, H. J., and Wang, L. (2018). Massive Mining of Publicly Available RNA-Seq Data from Human and Mouse. Nat. Commun. 9 (1), 1366. doi:10.1038/s41467-018-03751-6
Laity, J. H., Lee, B. M., and Wright, P. E. (2001). Zinc finger Proteins: New Insights into Structural and Functional Diversity. Curr. Opin. Struct. Biol. 11 (1), 39–46. doi:10.1016/s0959-440x(00)00167-6
Lee, J. G., and Kay, E. P. (2006). FGF-2-induced Wound Healing in Corneal Endothelial Cells Requires Cdc42 Activation and Rho Inactivation through the Phosphatidylinositol 3-kinase Pathway. Invest. Ophthalmol. Vis. Sci. 47 (4), 1376–1386. doi:10.1167/iovs.05-1223
Lee, W., Yun, J.-M., Woods, R., Dunaway, K., Yasui, D. H., Lasalle, J. M., et al. (2014). MeCP2 Regulates Activity-dependent Transcriptional Responses in Olfactory Sensory Neurons. Hum. Mol. Genet. 23 (23), 6366–6374. doi:10.1093/hmg/ddu358
Lenis, T. L., Hu, J., Ng, S. Y., Jiang, Z., Sarfare, S., Lloyd, M. B., et al. (2018). Expression of ABCA4 in the Retinal Pigment Epithelium and its Implications for Stargardt Macular Degeneration. Proc. Natl. Acad. Sci. U S A. 115 (47), E11120–E11127. doi:10.1073/pnas.1802519115
Li, X., Liu, X., Guo, H., Zhao, Z., Li, Y. S., and Chen, G. (2016). The Significance of the Increased Expression of Phosphorylated MeCP2 in the Membranes from Patients with Proliferative Diabetic Retinopathy. Sci. Rep. 6, 32850. doi:10.1038/srep32850
Lin, F.-y., Huang, Z., Lu, N., Chen, W., Fang, H., and Han, W. (2016). Controversial Opinion: Evaluation of EGR1 and LAMA2 Loci for High Myopia in Chinese Populations. J. Zhejiang Univ. Sci. B 17 (3), 225–235. doi:10.1631/jzus.b1500233
Liu, I. S. C., Chen, J.-d., Ploder, L., Vidgen, D., van der Kooy, D., Kalnins, V. I., et al. (1994). Developmental Expression of a Novel Murine Homeobox Gene (Chx10): Evidence for Roles in Determination of the Neuroretina and Inner Nuclear Layer. Neuron 13 (2), 377–393. doi:10.1016/0896-6273(94)90354-9
Lizio, M., Abugessaisa, I., Noguchi, S., Kondo, A., Hasegawa, A., Hon, C. C., et al. (2019). Update of the FANTOM Web Resource: Expansion to Provide Additional Transcriptome Atlases. Nucleic Acids Res. 47 (D1), D752–D758. doi:10.1093/nar/gky1099
Lopes, M. C., Hysi, P. G., Verhoeven, V. J. M., Macgregor, S., Hewitt, A. W., Montgomery, G. W., et al. (2013). Identification of a Candidate Gene for Astigmatism. Invest. Ophthalmol. Vis. Sci. 54 (2), 1260–1267. doi:10.1167/iovs.12-10463
Luck, K., Kim, D. K., Lambourne, L., Spirohn, K., Begg, B. E., Bian, W., et al. (2020). A Reference Map of the Human Binary Protein Interactome. Nature 580 (7803), 402–408. doi:10.1038/s41586-020-2188-x
Maleki, F., Ovens, K., Hogan, D. J., and Kusalik, A. J. (2020). Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 11, 654. doi:10.3389/fgene.2020.00654
Martin, A. R., Williams, E., Foulger, R. E., Leigh, S., Daugherty, L. C., Niblock, O., et al. (2019). PanelApp Crowdsources Expert Knowledge to Establish Consensus Diagnostic Gene Panels. Nat. Genet. 51 (11), 1560–1565. doi:10.1038/s41588-019-0528-2
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA Methylation-Related Chromatin Remodeling in Activity-dependent BDNF Gene Regulation. Science 302 (5646), 890–893. doi:10.1126/science.1090842
MedlinePlus (2022). MedlinePlus - Health Information from the National Library of Medicine[Internet]. Bethesda, MD: MedlinePlus. Available from: https://medlineplus.gov/.
Méjécase, C., Kozak, I., and Moosajee, M. (2020). The Genetic Landscape of Inherited Eye Disorders in 74 Consecutive Families from the United Arab Emirates. Am. J. Med. Genet. 184 (3), 762–772. doi:10.1002/ajmg.c.31824
Michaelides, M., Hunt, D., and Moore, A. (2003). The Genetics of Inherited Macular Dystrophies. J. Med. Genet. 40 (9), 641–650. doi:10.1136/jmg.40.9.641
Michaelides, M., Wilkie, S. E., Jenkins, S., Holder, G. E., Hunt, D. M., Moore, A. T., et al. (2005). Mutation in the Gene GUCA1A, Encoding Guanylate Cyclase-Activating Protein 1, Causes Cone, Cone-Rod, and Macular Dystrophy. Ophthalmology 112 (8), 1442–1447. doi:10.1016/j.ophtha.2005.02.024
Nakazawa, M., Wada, Y., and Tamai, M. (1995). Macular Dystrophy Associated with Monogenic Arg172trp Mutation of the Peripherin/RDS. Gene A Jpn. Fam. Retina 15 (6), 518–523.
Nakazawa, M., Wada, Y., and Tamai, M. (1995). Macular Dystrophy Associated with Monogenic Arg172Trp Mutation of the Peripherin/RDS Gene in a Japanese Family. Retina 15 (6), 518–523. doi:10.1097/00006982-199515060-00011
Noble, K. G. (1977). Central Areolar Choroidal Dystrophy. Am. J. Ophthalmol. 84 (3), 310–318. doi:10.1016/0002-9394(77)90670-5
Nollet, F., Kools, P., and van Roy, F. (2000). Phylogenetic Analysis of the Cadherin Superfamily Allows Identification of Six Major Subfamilies besides Several Solitary Members 1 1Edited by M. Yaniv. J. Mol. Biol. 299 (3), 551–572. doi:10.1006/jmbi.2000.3777
Ochocinska, M. J., Muñoz, E. M., Veleri, S., Weller, J. L., Coon, S. L., Pozdeyev, N., et al. (2012). NeuroD1is Required for Survival of Photoreceptors but Not Pinealocytes: Results from Targeted Gene Deletion Studies. J. Neurochem. 123 (1), 44–59. doi:10.1111/j.1471-4159.2012.07870.x
Oh, J., Wang, Y., Chen, S., Li, P., Du, N., Yu, Z. X., et al. (2017). Genetic Background-dependent Role of Egr1 for Eyelid Development. Proc. Natl. Acad. Sci. U S A. 114 (34), E7131–E7139. doi:10.1073/pnas.1705848114
Onishi, A., Peng, G.-H., Poth, E. M., Lee, D. A., Chen, J., Alexis, U., et al. (2010). The Orphan Nuclear Hormone Receptor ERR β Controls Rod Photoreceptor Survival. Proc. Natl. Acad. Sci. U.S.A. 107 (25), 11579–11584. doi:10.1073/pnas.1000102107
Orquera, D. P., and de Souza, F. S. J. (2017). Evolution of the Rax Family of Developmental Transcription Factors in Vertebrates. Mech. Development 144 (Pt B), 163–170. doi:10.1016/j.mod.2016.11.002
Ouechtati, F., Belhadj Tahar, O., Mhenni, A., Chakroun, S., Chouchene, I., Oueslati, S., et al. (2009). Central Areolar Choroidal Dystrophy Associated with Inherited Drusen in a Multigeneration Tunisian Family: Exclusion of the PRPH2 Gene and the 17p13 Locus. J. Hum. Genet. 54 (10), 589–594. doi:10.1038/jhg.2009.82
Oughtred, R., Rust, J., Chang, C., Breitkreutz, B. J., Stark, C., Willems, A., et al. (2021). TheBioGRIDdatabase: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 30 (1), 187–200. doi:10.1002/pro.3978
Ouzzani, M., Hammady, H., Fedorowicz, Z., and Elmagarmid, A. (2016). Rayyan-a Web and mobile App for Systematic Reviews. Syst. Rev. 5 (1), 210. doi:10.1186/s13643-016-0384-4
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The PRISMA 2020 Statement: an Updated Guideline for Reporting Systematic Reviews. BMJ 372, n71. doi:10.1136/bmj.n71
Pandolfi, E. C., Breuer, J. A., Nguyen Huu, V. A., Talluri, T., Nguyen, D., Lee, J. S., et al. (2020). The Homeodomain Transcription Factors Vax1 and Six6 Are Required for SCN Development and Function. Mol. Neurobiol. 57 (2), 1217–1232. doi:10.1007/s12035-019-01781-9
Payne, A. M., Downes, S. M., Bessant, D. A. R., Bird, A. C., and Bhattacharya, S. S. (1998). Founder Effect, Seen in the British Population, of the 172 Peripherin/RDS Mutation-And Further Refinement of Genetic Positioning of the Peripherin/RDS Gene. Am. J. Hum. Genet. 62 (1), 192–195. doi:10.1086/301679
Peshenko, I. V., Cideciyan, A. V., Sumaroka, A., Olshevskaya, E. V., Scholten, A., Abbas, S., et al. (2019). A G86R Mutation in the Calcium-Sensor Protein GCAP1 Alters Regulation of Retinal Guanylyl Cyclase and Causes Dominant Cone-Rod Degeneration. J. Biol. Chem. 294 (10), 3476–3488. doi:10.1074/jbc.ra118.006180
Peshenko, I. V., Olshevskaya, E. V., and Dizhoor, A. M. (2015). Evaluating the Role of Retinal Membrane Guanylyl Cyclase 1 (RetGC1) Domains in Binding Guanylyl Cyclase-Activating Proteins (GCAPs). J. Biol. Chem. 290 (11), 6913–6924. doi:10.1074/jbc.m114.629642
Piguet, B., Héon, E., Munier, F. L., Grounauer, P. A., Niemeyer, G., Butler, N., et al. (1996). Full Characterization of the Maculopathy Associated with an Arg-172-Trp Mutation in the RDS/peripherin Gene. Ophthalmic Genet. 17 (4), 175–186. doi:10.3109/13816819609057891
Prokofyeva, E., Wilke, R., Lotz, G., Troeger, E., Strasser, T., and Zrenner, E. (2009). An Epidemiological Approach for the Estimation of Disease Onset in Central Europe in central and Peripheral Monogenic Retinal Dystrophies. Graefes Arch. Clin. Exp. Ophthalmol. 247 (7), 885–894. doi:10.1007/s00417-009-1059-9
Quazi, F., and Molday, R. S. (2014). ATP-binding Cassette Transporter ABCA4 and Chemical Isomerization Protect Photoreceptor Cells from the Toxic Accumulation of Excess 11- Cis -retinal. Proc. Natl. Acad. Sci. U.S.A. 111 (13), 5024–5029. doi:10.1073/pnas.1400780111
Rahman, N., Georgiou, M., Khan, K. N., and Michaelides, M. (2020). Macular Dystrophies: Clinical and Imaging Features, Molecular Genetics and Therapeutic Options. Br. J. Ophthalmol. 104 (4), 451–460. doi:10.1136/bjophthalmol-2019-315086
Razick, S., Magklaras, G., and Donaldson, I. M. (2008). iRefIndex: A Consolidated Protein Interaction Database with Provenance. BMC Bioinformatic 9, 405. doi:10.1186/1471-2105-9-405
Reig, C., Serra, A., Gean, E., Vidal, M., Arumi, J., De la Calzada, M. D., et al. (1995). A point Mutation in the RDS-Peripherin Gene in a Spanish Family with central Areolar Choroidal Dystrophy. Ophthalmic Genet. 16 (2), 39–44. doi:10.3109/13816819509056911
Reig, C., Serra, A., Gean, E., Vidal, M., Arumi, J., De la Calzada, M. D., et al. (1995). A point Mutation in the RDS-Peripherin Gene in a Spanish Family with central Areolar Choroidal Dystrophy. Ophthalmic Genet. 16 (2), 39–44. doi:10.3109/13816819509056911
Reis, L. M., Khan, A., Kariminejad, A., Ebadi, F., Tyler, R. C., and Semina, E. V. (2011). VSX2 Mutations in Autosomal Recessive Microphthalmia. Mol. Vis. 17, 2527–2532.
Renner, A. B., Fiebig, B. S., Weber, B. H. F., Wissinger, B., Andreasson, S., Gal, A., et al. (2009). Phenotypic Variability and Long-Term Follow-Up of Patients with Known and Novel PRPH2/RDS Gene Mutations. Am. J. Ophthalmol. 147 (3), 518–530. doi:10.1016/j.ajo.2008.09.007
Sapkota, D., Chintala, H., Wu, F., Fliesler, S. J., Hu, Z., and Mu, X. (2014). Onecut1 and Onecut2 Redundantly Regulate Early Retinal Cell Fates during Development. Proc. Natl. Acad. Sci. U S A. 111 (39), E4086–E4095. doi:10.1073/pnas.1405354111
Schadt, E. E., Edwards, S. W., GuhaThakurta, D., Holder, D., Ying, L., Svetnik, V., et al. (2004). A Comprehensive Transcript index of the Human Genome Generated Using Microarrays and Computational Approaches. Genome Biol. 5 (10), R73. doi:10.1186/gb-2004-5-10-r73
Schatz, P., Abrahamson, M., Eksandh, L., Ponjavic, V., and Andréasson, S. (2003). Macular Appearance by Means of OCT and Electrophysiology in Members of Two Families with Different Mutations in RDS (The Peripherin/RDS Gene). Acta Ophthalmologica Scand. 81 (5), 500–507. doi:10.1034/j.1600-0420.2003.00134.x
Sergouniotis, P. I., Chakarova, C., Murphy, C., Becker, M., Lenassi, E., Arno, G., et al. (2014). Biallelic Variants in TTLL5, Encoding a Tubulin Glutamylase, Cause Retinal Dystrophy. Am. J. Hum. Genet. 94 (5), 760–769. doi:10.1016/j.ajhg.2014.04.003
Shankar, S. P., Hughbanks-Wheaton, D. K., Birch, D. G., Sullivan, L. S., Conneely, K. N., Bowne, S. J., et al. (2016). Autosomal Dominant Retinal Dystrophies Caused by a Founder Splice Site Mutation, c.828+3A>T, in PRPH2 and Protein Haplotypes in Trans as Modifiers. Invest. Ophthalmol. Vis. Sci. 57 (2), 349–359. doi:10.1167/iovs.15-16965
Sharma, S., Dimasi, D., Higginson, K., and Della, N. G. (2004). RZF, a Zinc-finger Protein in the Photoreceptors of Human Retina. Gene 342 (2), 219–229. doi:10.1016/j.gene.2004.08.015
Smailhodzic, D., Fleckenstein, M., Theelen, T., Boon, C. J. F., van Huet, R. A. C., van de Ven, J. P. H., et al. (2011). Central Areolar Choroidal Dystrophy (CACD) and Age-Related Macular Degeneration (AMD): Differentiating Characteristics in Multimodal Imaging. Invest. Ophthalmol. Vis. Sci. 52 (12), 8908–8918. doi:10.1167/iovs.11-7926
Sorsby, A. (1939). Choroidal Angio-Sclerosis with Special Reference to its Hereditary Character. Br. J. Ophthalmol. 23 (7), 433–444. doi:10.1136/bjo.23.7.433
Stark, M. S., Gray, E. S., Isaacs, T., Chen, F. K., Millward, M., McEvoy, A., et al. (2019). A Panel of Circulating MicroRNAs Detects Uveal Melanoma with High Precision. Trans. Vis. Sci. Tech. 8 (6), 12. doi:10.1167/tvst.8.6.12
Stingl, K., Mayer, A. K., Llavona, P., Mulahasanovic, L., Rudolph, G., Jacobson, S. G., et al. (2017). CDHR1 Mutations in Retinal Dystrophies. Sci. Rep. 7, 6992. doi:10.1038/s41598-017-07117-8
Stuck, M. W., Conley, S. M., and Naash, M. I. (2016). PRPH2/RDS and ROM-1: Historical Context, Current Views and Future Considerations. Prog. Retin. Eye Res. 52, 47–63. doi:10.1016/j.preteyeres.2015.12.002
Sukhatme, V. P., Cao, X., Chang, L. C., Tsai-Morris, C.-H., Stamenkovich, D., Ferreira, P. C. P., et al. (1988). A Zinc finger-encoding Gene Coregulated with C-Fos during Growth and Differentiation, and after Cellular Depolarization. Cell 53 (1), 37–43. doi:10.1016/0092-8674(88)90485-0
Sun, X., Park, J. H., Gumerson, J., Wu, Z., Swaroop, A., Qian, H., et al. (2016). Loss of RPGR Glutamylation Underlies the Pathogenic Mechanism of Retinal Dystrophy Caused by TTLL5 Mutations. Proc. Natl. Acad. Sci. U S A. 113 (21), E2925–E2934. doi:10.1073/pnas.1523201113
Swanberg, S. E., Nagarajan, R. P., Peddada, S., Yasui, D. H., and LaSalle, J. M. (2009). Reciprocal Co-regulation of EGR2 and MECP2 Is Disrupted in Rett Syndrome and Autism. Hum. Mol. Genet. 18 (3), 525–534. doi:10.1093/hmg/ddn380
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/measurement Sets. Nucleic Acids Res. 49 (D1), D605–D612. doi:10.1093/nar/gkaa1074
Tamkovich, S., Grigor'eva, A., Eremina, A., Tupikin, A., Kabilov, M., Chernykh, V., et al. (2019). What Information Can Be Obtained from the Tears of a Patient with Primary Open Angle Glaucoma? Clinica Chim. Acta 495, 529–537. doi:10.1016/j.cca.2019.05.028
Terrell, D., Xie, B., Workman, M., Mahato, S., Zelhof, A., Gebelein, B., et al. (2012). OTX2 and CRX rescue Overlapping and Photoreceptor-specific Functions in theDrosophilaeye. Dev. Dyn. 241 (1), 215–228. doi:10.1002/dvdy.22782
Trujillo, M., Bueno, J., Osorio, A., Sanz, R., Garcia-Sandoval, B., Ramos, C., et al. (1998). Three novel RDS-peripherin mutations (689delT, 857del17, G208D) in Spanish familes affected with autosomal dominant retinal degenerations. Hum. Mutat. 12 (1), 70. doi:10.1002/(sici)1098-1004(1998)12:1<70::aid-humu11>3.0.co;2-m
Ulmer Carnes, M., Liu, Y. P., Allingham, R. R., Whigham, B. T., Havens, S., Garrett, M. E., et al. (2014). Discovery and Functional Annotation of SIX6 Variants in Primary Open-Angle Glaucoma. Plos Genet. 10 (5), e1004372. doi:10.1371/journal.pgen.1004372
Urdinguio, R. G., Lopez-Serra, L., Lopez-Nieva, P., Alaminos, M., Diaz-Uriarte, R., Fernandez, A. F., et al. (2008). Mecp2-null Mice Provide New Neuronal Targets for Rett Syndrome. PLoS One 3 (11), e3669. doi:10.1371/journal.pone.0003669
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P., et al. (2010). The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 38, W214–W220. doi:10.1093/nar/gkq537
Weisschuh, N., Obermaier, C. D., Battke, F., Bernd, A., Kuehlewein, L., Nasser, F., et al. (2020). Genetic Architecture of Inherited Retinal Degeneration in Germany: A Large Cohort Study from a Single Diagnostic center over a 9‐year Period. Hum. Mutat. 41 (9), 1514–1527. doi:10.1002/humu.24064
Weisschuh, N., Obermaier, C. D., Battke, F., Bernd, A., Kuehlewein, L., Nasser, F., et al. (2020). Genetic Architecture of Inherited Retinal Degeneration in Germany: A Large Cohort Study from a Single Diagnostic center over a 9‐year Period. Hum. Mutat. 41 (9), 1514–1527. doi:10.1002/humu.24064
Wells, J., Wroblewski, J., Keen, J., Inglehearn, C., Jubb, C., Eckstein, A., et al. (1993). Mutations in the Human Retinal Degeneration Slow (RDS) Gene Can Cause Either Retinitis Pigmentosa or Macular Dystrophy. Nat. Genet. 3 (3), 213–218. doi:10.1007/978-1-4615-2974-3_6
Westermann, S., and Weber, K. (2003). Post-translational Modifications Regulate Microtubule Function. Nat. Rev. Mol. Cel Biol 4 (12), 938–948. doi:10.1038/nrm1260
Wickham, L., Chen, F. K., Lewis, G. P., Uppal, G. S., Neveu, M. M., Wright, G. A., et al. (2009). Clinicopathological Case Series of Four Patients with Inherited Macular Disease. Invest. Ophthalmol. Vis. Sci. 50 (8), 3553. doi:10.1167/iovs.08-2715
Wroblewski, J. J., Wells, J. A., Eckstein, A., Fitzke, F., Jubb, C., Keen, T. J., et al. (1994). Macular Dystrophy Associated with Mutations at Codon 172 in the Human Retinal Degeneration Slow Gene. Ophthalmology 101 (1), 12–22. doi:10.1016/s0161-6420(94)31377-7
Wu, G., Feng, X., and Stein, L. (2010). A Human Functional Protein Interaction Network and its Application to Cancer Data Analysis. Genome Biol. 11 (5), R53. doi:10.1186/gb-2010-11-5-r53
Xie, S., Han, S., Qu, Z., Liu, F., Li, J., Yu, S., et al. (2019). Knockout of Nr2e3 prevents rod photoreceptor differentiation and leads to selective L-/M-cone photoreceptor degeneration in zebrafish. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1865 (6), 1273–1283. doi:10.1016/j.bbadis.2019.01.022
Yamamoto, H., Kon, T., Omori, Y., and Furukawa, T. (2020). Functional and Evolutionary Diversification of Otx2 and Crx in Vertebrate Retinal Photoreceptor and Bipolar Cell Development. Cel Rep. 30 (3), 658–671. doi:10.1016/j.celrep.2019.12.072
Yan, R.-T., and Wang, S.-Z. (2004). Requirement ofNeuroDfor Photoreceptor Formation in the Chick Retina. Invest. Ophthalmol. Vis. Sci. 45 (1), 48–58. doi:10.1167/iovs.03-0774
Yanagihashi, S., Nakazawa, M., Kurotaki, J., Sato, M., Miyagawa, Y., and Ohguro, H. (2003). Autosomal Dominant central Areolar Choroidal Dystrophy and a Novel Arg195Leu Mutation in the Peripherin/RDS Gene. Arch. Ophthalmol. 121 (10), 1458–1461. doi:10.1001/archopht.121.10.1458
Yang, P., Chiang, P.-W., Weleber, R. G., and Pennesi, M. E. (2015). Autosomal Dominant Retinal Dystrophy with Electronegative Waveform Associated with a NovelRAX2Mutation. JAMA Ophthalmol. 133 (6), 653–661. doi:10.1001/jamaophthalmol.2015.0357
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., et al. (2007). Integrated Epigenomic Analyses of Neuronal MeCP2 Reveal a Role for Long-Range Interaction with Active Genes. Proc. Natl. Acad. Sci. U.S.A. 104 (49), 19416–19421. doi:10.1073/pnas.0707442104
Yoshida, S., Mears, A. J., Friedman, J. S., Carter, T., He, S., Oh, E., et al. (2004). Expression Profiling of the Developing and Mature Nrl −/− Mouse Retina: Identification of Retinal Disease Candidates and Transcriptional Regulatory Targets of Nrl. Hum. Mol. Genet. 13 (14), 1487–1503. doi:10.1093/hmg/ddh160
Yu, W., Mookherjee, S., Chaitankar, V., Hiriyanna, S., Kim, J.-W., Brooks, M., et al. (2017). Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 8, 14716. doi:10.1038/ncomms14716
Yusuf, I. H., Gliem, M., Birtel, J., Muller, P. L., Mangold, E., Bolz, H., et al. (2018). The Retinal Phenotype Associated with the Frequent, Synonymous c.783G>A Mutation in CDHR1. Invest. Ophthalmol. Vis. Sci. 59 (9), 3146.
Zhang, L., Cho, J., Ptak, D., and Leung, Y. F. (2013). The Role of Egr1 in Early Zebrafish Retinogenesis. PLoS One 8 (2), e56108. doi:10.1371/journal.pone.0056108
Zhao, X., Ren, Y., Zhang, X., Chen, C., Dong, B., and Li, Y. (2013). A Novel GUCY2D Mutation in a Chinese Family with Dominant Cone Dystrophy. Mol. Vis. 19, 1039–1046.
Zhao, X., Zhang, L., Wang, J., Zhang, M., Song, Z., Ni, B., et al. (2021). Identification of Key Biomarkers and Immune Infiltration in Systemic Lupus Erythematosus by Integrated Bioinformatics Analysis. J. Transl Med. 19 (1), 35. doi:10.1186/s12967-020-02698-x
Zhou, J.-M., Liang, R., Zhu, S.-Y., Wang, H., Zou, M., Zou, W.-J., et al. (2019). LncRNA WWC2-AS1 Functions AS a Novel Competing Endogenous RNA in the Regulation of FGF2 Expression by Sponging miR-16 in Radiation-Induced Intestinal Fibrosis. BMC Cancer 19 (1), 647. doi:10.1186/s12885-019-5754-6
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W.-n., Ho, H.-y. H., Schmidt, L., et al. (2006). Brain-specific Phosphorylation of MeCP2 Regulates Activity-dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron 52 (2), 255–269. doi:10.1016/j.neuron.2006.09.037
Zimmerman, A. D., Nagy, C. R., and Munger, S. D. (2020). Sensory Neurons Expressing the Atypical Olfactory Receptor Guanylyl Cyclase D Are Required for the Acquisition of Odor Preferences by Mice in Diverse Social Contexts. Physiol. Behav. 227, 113150. doi:10.1016/j.physbeh.2020.113150
Keywords: macular disease, CACD, central areolar choroidal distrophy, PRPH2, GUCA1A, GUCY2D
Citation: Kazmierczak de Camargo JP, Prezia GNdB, Shiokawa N, Sato MT, Rosati R and Beate Winter Boldt A (2022) New Insights on the Regulatory Gene Network Disturbed in Central Areolar Choroidal Dystrophy—Beyond Classical Gene Candidates. Front. Genet. 13:886461. doi: 10.3389/fgene.2022.886461
Received: 28 February 2022; Accepted: 04 April 2022;
Published: 17 May 2022.
Edited by:
Helen Louise May-Simera, Johannes Gutenberg University Mainz, GermanyReviewed by:
Qingnan Liang, Baylor College of Medicine, United StatesJiaxing Wang, Emory University, United States
Copyright © 2022 Kazmierczak de Camargo, Prezia, Shiokawa, Sato, Rosati and Beate Winter Boldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelica Beate Winter Boldt, YW5nZWxpY2Fib2xkdEBnbWFpbC5jb20=
†These authors have contributed equally to this work