
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 20 May 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.886182
This article is part of the Research TopicRare DyslipidemiasView all 17 articles
 Khalid Al-Waili1
Khalid Al-Waili1 Khalid Al-Rasadi2Muna Al-Bulushi1Mohammed Habais1Abdullah Al-Mujaini3Saif Al-Yaarubi4
Khalid Al-Rasadi2Muna Al-Bulushi1Mohammed Habais1Abdullah Al-Mujaini3Saif Al-Yaarubi4 Antoine Rimbert5Razan Zadjali1
Antoine Rimbert5Razan Zadjali1 Pegah Moradi Khaniabadi1Hamida Al-Barwani6Sana Hasary1Zayana M. Al-Dahmani1Hala Al-Badi1
Pegah Moradi Khaniabadi1Hamida Al-Barwani6Sana Hasary1Zayana M. Al-Dahmani1Hala Al-Badi1 Almundher Al-Maawali7
Almundher Al-Maawali7 Fahad Zadjali1*
Fahad Zadjali1*Familial hypertriglyceridemia (F-HTG) is an autosomal disorder that causes severe elevation of serum triglyceride levels. It is caused by genetic alterations in LPL, APOC2, APOA5, LMF1, and GPIHBP1 genes. The mutation spectrum of F-HTG in Arabic populations is limited. Here, we report the genetic spectrum of six families of F-HTG of Arab ancestry in Oman. Methods: six Omani families affected with triglyceride levels >11.2 mmol/L were included in this study. Ampli-Seq sequencing of the selected gene panels was performed. Whole-exome sequencing and copy number variant analysis were also performed in cases with negative exome results. Three novel pathogenic missense variants in the LPL gene were identified, p.M328T, p.H229L, and p.S286G, along with a novel splice variant c.1322+15T > G. The LPL p.H229L variant existed in double heterozygous mutation with the APOA5 gene p.V153M variant. One family had a homozygous mutation in the LMF1 gene (c.G107A; p.G36D) and a heterozygous mutation in the LPL gene (c.G106A; p.D36N). All affected subjects did not have a serum deficiency of LPL protein. Genetic analysis in one family did not show any pathogenic variants even after whole-exome sequencing. These novel LPL and APOA5 mutations are not reported in other ethnic groups. This suggests that patients with F-HTG in Oman have a founder effect and are genetically unique. This warrants further analysis of patients of F-HTG in the Middle East for preventative and counseling purposes to limit the spread of the disease in a population of high consanguinity.
Hypertriglyceridemia (HTG) is a metabolic disorder characterized by elevated levels of fasting serum triglyceride (TG) (Lewis et al., 2015; Lahoz and Mostaza, 2021). Based on the Endocrine Society Clinical Practice Guideline, HTG is classified as mild (TG 1.7–2.2 mmol/L), moderate (TG 2.3–11.2 mmol/L), severe (TG 11.3–22.5 mmol/L), and very severe (TG ≥ 22.6 mmol/L) (Berglund et al., 2012). Familial hypertriglyceridemia (F-HTG) is a rare genetic disorder that is found in less than 5% of HTG cases. It is caused by mutations in at least five different genes, namely, lipoprotein lipase (LPL), apolipoprotein C-II (APOC2), apolipoprotein A-5 (APOA5), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein (GPIHBP1), and lipase maturation factor 1 (LMF1) genes. Secondary HTGs are more frequent and attributed to obesity-related diabetes mellitus, hypothyroidism, nephrotic syndrome, or drug-induced disorders (Clement et al., 2014; Cruz-Bautista et al., 2021).
The most severe forms of HTG are seen in both familial chylomicronemia (type I hyperlipidemia, OMIM#238600) and primary mixed hyperlipidemia (type V hyperlipidemia, OMIM#144650). Type I hyperlipidemia is characterized by defective clearance of chylomicrons and has an isolated elevation of blood chylomicrons, while type V hyperlipidemia shows elevation in both chylomicrons and very-low-density lipoprotein (VLDL) (Hegele, 2009; Brandts and Ray, 2021). The LPL enzyme is expressed on the surface of endothelial cells of capillaries, and it releases fatty acids from triglycerides from the intestine and liver into the bloodstream. A higher density of disease-causing mutation mainly exists in the middle of the LPL gene. The majority of loss-of-function mutations are located in exons 5 and 6 of the LPL gene since these exons encode important functional domains of the enzyme (Murthy et al., 1996; Shakhtshneider et al., 2021; Wu et al., 2021). Up to date, there is no study conducted on common genes associated with F-HTG in the Arabian region, with the exception of a case report from Israel on an Arab decent child with a mutation in the LPL gene (p.Arg270His) and p.Ser286Arg in a Moroccan patient (Bouabdellah et al., 2015; Dron and Hegele, 2017). In this study, we identified six families of F-HTG of Arab ancestry and reported their genetic causes. We further investigated the genetic causes of F-HTG and correlated the genotypes with LPL protein expression.
Blood samples were collected from patients with a clinical diagnosis of F-HTG at the lipid clinic in Sultan Qaboos University Hospital, Oman. Samples were also collected from their relatives. The study involved six families resulting in a cohort of 28 individuals consisting of 12 F-HTG affected patients (11 males and 1 female). Patients with mild or moderate HTG, history of uncontrolled diabetes, hypothyroidism, significant proteinuria or nephrotic syndrome, obesity, alcohol consumption, and paraproteinemia disorders were excluded from the study. Blood samples were collected after overnight fasting from all individuals for molecular and functional studies. Serum samples were collected and stored at −80°C for further analysis. The aliquoted serum samples were used for lipoprotein lipase immunoblotting. The whole blood EDTA samples were stored immediately at −80°C for DNA extraction. Family pedigrees were illustrated using cranefoot_3.2.3 software (Mäkinen et al., 2005).
Genomic DNAs were extracted from whole blood using Qiagen mini kit (QIAamp DNA Mini). Ampli-Seq technology on the Ion Proton platform (Thermo Fisher Scientific, Inc.) was performed to sequence the F-HTG gene panel: LPL, APOC2, APOA5, GPIHBP1, and LMF1. The exon flanking introns and promotor regions were included in the panel. For individuals with negative exome results, DNA samples were sent to a service provider for whole-exome sequencing, Macrogen © South Korea. DNA was barcoded and enriched for the coding exons of targeted genes using hybrid capture technology (Agilent SureSelect Human All-exons-V6). Prepared DNA libraries were then sequenced using Next-Generation Sequencing (NGS) technology (NovaSeq 6000, 150) bp paired-end, at 200X coverage).
Sequencing data were processed by the Torrent Suite and reads were aligned to the hg19 reference genome. Variant call files were then generated using Torrent Variant Caller plugins. Variant annotation was performed using ANNOVAR and variants were linked to ExAC, and Greater Middle East-Arabian Peninsula (GME-AP) databases for allele frequencies. . Effect of amino acid changes was predicted by LRT, CADD, and MutationTaster. Pathogenic variants were identified from allele frequency of <1% or novel, coverage depth >30, the damaging effect from at least two of the three prediction algorithms, and segregation with the disease in the family. For CNV analysis, we used the CNVkit tool for patients with no pathogenic variant identified in the LPL gene (Talevich et al., 2016).
The serum of all affected cases including two serum samples from the control group was examined. Clear serum was collected from the bottom of the tube (milky ring avoided) after maximum centrifugation. A total of 200 μl serum was first cleared from IgG using washed magnetic beads (PureProteome™ Protein A/G Mix Magnetic Beads, Millipore). 1 μl of LPL antibody (LPL Antibody F-1, mouse monoclonal IgG, Santa Cruz Biotechnology) was added to IgG-cleared serum and incubated overnight in the cold room. Then LPL protein was immunoprecipitated using washed IgG beads under rotation for 3 h at room temperature. Then final beads were washed and mixed with reducing buffer. Proteins were separated in 12% Sodium dodecyl sulfate-polyacrylamide gel for electrophoresis. Western blot was performed using primary LPL antibody. We used total protein loading as a normalization technique (Aldridge et al., 2008). Total lane intensity was used to measure total protein loading per sample.
The lipid profiles of the affected family members are described in Figure 1. Table 1 summarizes the identified mutations in the patients.
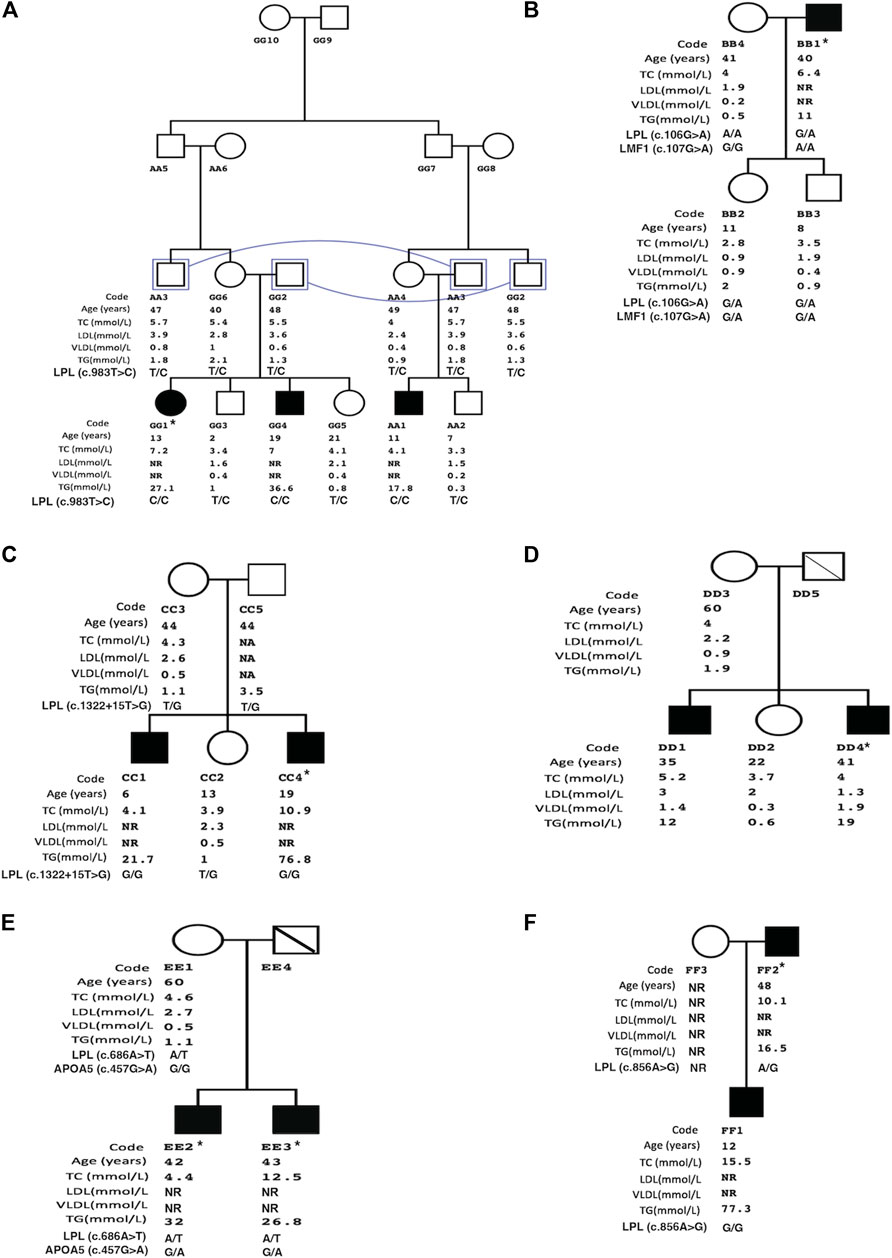
FIGURE 1. Family pedigree structures of patients with familial hypertriglyceridemia. Illustrates family trees of study groups (A= Family1 to F= Family 6). Study group data includes age, total cholesterol (TC), low-density lipoprotein (LDL), very-low-density lipoprotein (VLDL), and triglycerides (TG). Squares and circles represent male and female family members, respectively, and filled symbols represent affected subjects with familial hypertriglyceridemia. * Index case, NR: not recorded, connecting line: same individual, diagonal line: deceased subject.
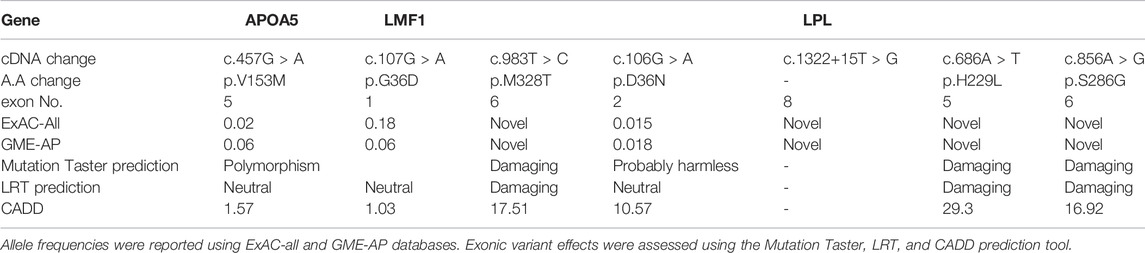
TABLE 1. List of pathogenic variants in the families.
Family 1 (Figure 1A) is made of double cousin marriage with three affected individuals AA1 (age 11-yrs), GG1 (12-yrs), and GG4 (19yrs) with a severe elevation of triglycerides. Clinically, three patients suffered from recurrent pancreatitis and did not have lipemia retinalis, hepatosteatosis, and a history of cardiovascular disease, obesity, alcoholism, renal disease, or diabetes mellitus. Sequencing of the 5 candidate genes showed a novel LPL gene variant (c.983T > C, p.M328T) with all affected siblings being homozygous. The rest of the family members were found heterozygous carriers. The LPL p.M328T mutation is not reported in the LPL-Leiden Open Variation Database [14] and is located in exon 6 of the gene with the predicted damaging effects, Table 1.
In family 2, only one affected member (father, BB1) had clinical criteria for F-HTG with severe triglyceride levels. The father had obesity with no signs of lipemia retinalis. There was no history of acute pancreatitis, diabetes mellitus, renal disease, cardiovascular disease, or steatohepatitis in the family. The analysis of both targeted and whole-exome sequencing data did not identify pathogenic variants. The condition in the father could be due to double alteration in LPL (c.G106A; p.D36N) and LMF1 (c.G107A; p.G36D). The LPL variant p.D36N is likely a benign variant (rs1801177) shown by the prediction algorithms, Table 1. The family’s two siblings were heterozygous in two genes variants, however, they showed normal triglyceride levels. Yet, the father was homozygous for the LMF1 (NM_001352020) p.G36D variant which may explain his severe phenotype. Furthermore, analysis of whole-exome sequencing data did not identify any CNV nor pathogenic variants in glycerol-3-phosphate dehydrogenase 1 (GPD1) to exclude possible Transient Infantile Hypertriglyceridemia.
In Family 3, two of the five members were affected with severe HTG. The affected sons, CC1(6 yrs) and CC4 (19 yrs) had fasting TG levels of 21.7 mmol/L and 76.8 mmol/L, respectively. Recurrent pancreatitis was present in the older son only. Both affected siblings did not have signs of lipemia retinalis and had normal blood glucose levels. Genetic analysis of family 3 showed a splice variant in the exon8-intron8 junction of the LPL gene. Both affected sons were homozygous, while the unaffected sibling and parents were heterozygous carriers.
In family 4, fasting TG levels of the affected males DD1 (35 yrs) and DD4 (41 yrs) were severely high at 12 mmol/L and 19 mmol/L, respectively. TG level of the mother (DD3) was normal. There was no history of acute pancreatitis and signs of lipemia retinalis in the family. Patient DD1 had obesity and steatohepatitis associated with hepatitis B infection. Patient DD4 had a history of transient ischemic heart disease and stage 2 chronic kidney disease. Using filtering strategy, no CNV nor pathogenic variants were identified in the Ampli-Seq panel or whole-exome sequencing.
Family 5 consisted of two affected patients with severe TG levels and a mother with normal TG levels (Figure 1). Recurrent acute pancreatitis was present in the affected patients but not in the mother. Genetic analysis of the two affected patients revealed a novel pathogenic LPL gene variant (c.686A > T, p.H229L) according to the predictions algorithm. However, the healthy mother was also heterozygous like the affected siblings. Therefore, we analyzed the family using a double heterozygous model. We identified a polymorphic variant in the APOA5 gene in affected patients but not in the mother. Therefore, this family had a compound heterozygous mutation in LPL and APOA5 genes.
In family 6, the TG levels of an affected son (FF1) was 77.3 mmol/L which was higher than the affected father (FF2) with a TG level of 16.5 mmol/L. Both patients had recurrent attacks of acute pancreatitis and no sign of lipemia retinalis. The affected father had a history of diabetes mellitus with nephropathy. There was no history of steatohepatitis and cardiovascular disease in the family. A novel LPL gene variation (c.856A > G, p.S286G) was identified in the existing panel screening. The affected son with severely elevated TG was homozygous at the mutation while the father with a lower TG level was heterozygous (Figure 1). The mother did not attend the clinic for investigation.
We then analyzed the LPL protein expression in serum to investigate if patients had an absolute deficiency of enzyme. To normalize the LPL expression, we used total protein loading in each Western blot lane, Figure 2. LPL was immunoprecipitated and expression was compared to two control cases. We found that LPL protein was expressed in all cases of F-HTG, which indicates that mutations impact the protein’s functioning rather than its synthesis or stability.
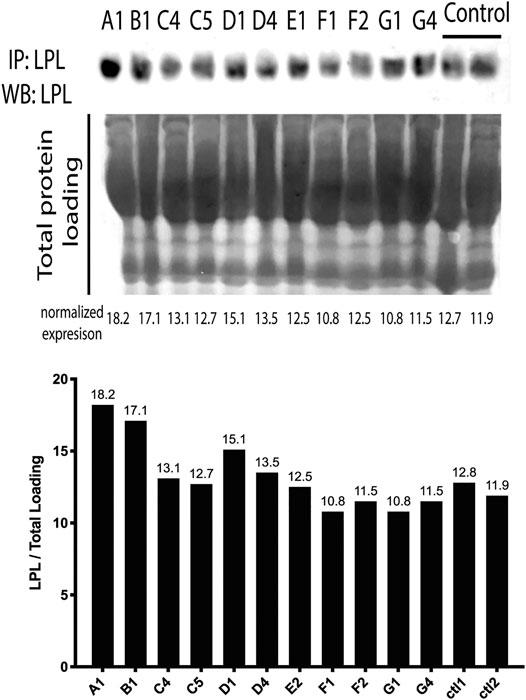
FIGURE 2. LPL protein levels in the serum of affected individuals. The LPL densities were normalized to total protein loading. Both Ctl1 and Ctl2 are normal control samples. Individuals ‘labels are as follows: A1:AA1, B1:BB1, C4:CC4, C5:CC5, D1:DD1, D4:DD4, E2: EE2, F1: FF1, F2: FF2, G1: GG1, G4: GG4.
Primary disorders of triglyceride metabolism result from genetic defects in triglyceride synthesis and metabolism. Here, we report the genetic spectrum of six families with severe familial hypertriglyceridemia of Arab ancestry in Oman. Three families presented with novel pathogenic variants in the LPL gene: two missense variants p.M328T and p.S286G and one splice variant c.1322+15T > G. One family had double heterozygous variants in LPL (p.H229L) and APOA5 gene (p.V153M). Another family had double heterozygosity between LPL (p.G36N) and homozygous LMF1 mutation (p.G36D). All affected subjects did not have serum deficiency of LPL protein level and most likely the variants affect the lipolytic activity of LPL. Previous studies on other disease-causing mutations also the variable effect of protein levels of LPL (Kozaki et al., 1993; Hooper et al., 2008). Genetic analysis in one family did not show any pathogenic variants even with whole-exome sequencing.
The novel pathogenic missense variant in the LPL gene (rs1181582051, c.983T > C, p.M328T) causes a substitution in the carboxy-terminal structural region of LPL, which spans 313–448 amino acids, on the highly conserved beta-strand (uniport: LIPL_HUMAN). The LPL-mediated lipoprotein absorption through the cell surface receptor catalyzes the first contact between LPL and the substrate in this domain (Murthy et al., 1996). Furthermore, beta-strands are crucial secondary structural elements that affect protein folding, structure, and topology, as well as the protein’s functional confirmation and activity (Henderson et al., 1993; Jin et al., 2021). When compared to controls, this variant was detected in family 1 members who had normal LPL expression. This indicates that the mutation had no effect on the stability of the protein.
The novel missense mutation p.S286G in exon 6 of the LPL gene occurs in a less-conserved region of the gene. The 286 residues are located very close to the S-S bridge between the two cysteine residues (278–283) which plays an important role in stabilizing the heparin-binding (R279-K280 and R282) (Murthy et al., 1996). A previous case with F-HTG was reported in a Moroccan patient but with homozygous c.858T > A/p.S286R mutation (Bouabdellah et al., 2015). The heterozygous p.S286G carriers in the study family had triglycerides half levels of those of homozygote cases. suggesting that the mutation has a dose effect.
A missense mutation (c.686A > T, p.H229L) in the LPL gene has been detected in a family of 4 members even in the mother with normal TG levels. To the best of our knowledge, this mutation is not reported in any population. Amino acid residues from 228 to 234 of LPL make up the ß-strand in the N -terminal domain, UniProt. Studies by Dugi et al. (1992) and Henderson et al. (1993) highlighted the region from Cys218 to Cys 239 as the lid/loop domain which is stabilized by one of the four disulfide bonds of LPL protein. The lid/loop is involved in the hydrolysis of some triglycerides and phospholipids, the initial identification of the lipase substrate, and giving excess to the catalytic triad (Goldberg and Chait, 2020). At the lid proximal and distal regions, the charge and periodicity play a critical role in maintaining the catalytic activity and the apical residues of the loop contribute minimally to LPL function (Dron and Hegele, 2017). Therefore, to have a dominant effect on serum TG levels, the patient phenotype suggested the presence of a compound heterozygous model. Earlier reports show that more than two-thirds of F-HTG is caused by an autosomal recessive variant in the LPL gene [22, 23]. Less abundant cases are due to autosomal recessive or double heterozygous LPL variants with variants in LMF1, APOA5, APOC2, or GPIHBP1 gene (Basel-Vanagaite et al., 2012; Goldberg and Chait, 2020). The double heterozygosity affects the function and maturity of LPL leading to elevated serum TG. LPL activity requires apoC2 and apoA5, while the maturity and transport of LPL are controlled by LMF1. GPIHBP1 is required for surface attachment of LPL at the endothelial cell surface. The compound heterozygosity in family B explains that the presence of homozygous p.G36D variant in LMF1 along with heterozygous changes in LPL and GPIHBP1 causes the severe elevation in the affected father BB1. The p.G36D of LMF1 was earlier reported in a Thai family with F-HTG with one patient being homozygous and 4 were heterozygous (Plengpanich et al., 2020).
None of the cases presented clinically as transient infantile hypertriglyceridemia. These patients usually present with moderate hypertriglyceridemia at birth and gradually decrease with growth [24]. The condition is caused by an autosomal recessive mutation in the glycerol-3-phosphate dehydrogenase 1 gene (GPD1).
Hypertriglyceridemia can present as a monogenic (familial) or polygenic disease. Monogenic hypertriglyceridemia conditions are very rare and commonly cause severe elevation of serum TG. Here, we described six families of F-HTG in Oman. Four pathogenic variants in the LPL gene were identified: p.M328T, c.1322+15T > G. p.S286G, and p.H229L. The latter variant existed as double heterozygous with the APOA5 variant p.V153M variant. One family with severe hypertriglyceridemia resulted in negative exome data. Further whole-genome sequencing and epigenetic studies are needed to explore the genetic basis in these families. The reported novel mutations in the study suggest that patients with F-HTG in Oman may have a founder effect and are genetically unique. This warrants further analysis of patients with F-HTG in the Middle East for the preventative and counseling purpose to limit the spread of the disease in a population of high consanguinity.
The datasets for novel mutation presented in this study can be found in online repositories: Global Variome shared LOVD (https://databases.lovd.nl/shared/individuals/LPL) with accession individual I.D numbers 00408363, 00408402, 00408403, 00408404.
The studies involving human participants were reviewed and approved by the Medical Research Ethics Committee (SQU-EC/006/19 #1842), Sultan Qaboos University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
The article was written by FZ and MA. The revision was carried out by KA-R, RZ, and PK. Data acquisition was performed by KA-W, ABDA-M, SA-Y, AR, HAMA-B, SH, ZMA-D, HALA-B, MH, and MH. Data analysis was performed by FZ, KA-W, MH, AR, and ALMA-M. Funding was provided by FZ and KA-W.
This study was supported by the internal grants of Sultan Qaboos University IG/MED/BIOC/18/02. MA-B has received a young investigator fund from the Wael AlMahmeed & IAS Research Training Grants and Fellowships for the MENA Region"
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor FS declared a past co-authorship with the authors KAL, KAR, HAB, FZ.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aldridge, G. M., Podrebarac, D. M., Greenough, W. T., and Weiler, I. J. (2008). The Use of Total Protein Stains as Loading Controls: An Alternative to High-Abundance Single-Protein Controls in Semi-quantitative Immunoblotting. J. Neurosci. Methods 172 (2), 250–254. doi:10.1016/j.jneumeth.2008.05.003
Basel-Vanagaite, L., Zevit, N., Zahav, A. H., Guo, L., Parathath, S., Pasmanik-Chor, M., et al. (2012). Transient Infantile Hypertriglyceridemia, Fatty Liver, and Hepatic Fibrosis Caused by Mutated GPD1, Encoding Glycerol-3-Phosphate Dehydrogenase 1. Am. J. Hum. Genet. 90 (1), 49–60. doi:10.1016/j.ajhg.2011.11.028
Berglund, L., Brunzell, J. D., Goldberg, A. C., Goldberg, I. J., Sacks, F., Murad, M. H., et al. (2012). Evaluation and Treatment of Hypertriglyceridemia: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 97 (9), 2969–2989. doi:10.1210/jc.2011-3213
Bouabdellah, M., Iraqi, H., Benlian, P., Berqia, I., Benchekroun, L., Chraïbi, A., et al. (2015). Familial Hypertriglyceridemia: Biochemical, Clinical and Molecular Study in a Moroccan Family. Ann. Biol. Clin. (Paris) 73 (4), 474–484. doi:10.1684/abc.2015.1058
Brandts, J., and Ray, K. K. (2021). Familial Hypercholesterolemia: JACC Focus Seminar 4/4. J. Am. Coll. Cardiol. 78 (18), 1831–1843. doi:10.1016/j.jacc.2021.09.004
Clement, L. C., Macé, C., Avila-Casado, C., Joles, J. A., Kersten, S., and Chugh, S. S. (2014). Circulating Angiopoietin-like 4 Links Proteinuria with Hypertriglyceridemia in Nephrotic Syndrome. Nat. Med. 20 (1), 37–46. doi:10.1038/nm.3396
Cruz-Bautista, I., Huerta-Chagoya, A., Moreno-Macías, H., Rodríguez-Guillén, R., Ordóñez-Sánchez, M. L., Segura-Kato, Y., et al. (2021). Familial Hypertriglyceridemia: An Entity with Distinguishable Features from Other Causes of Hypertriglyceridemia. Lipids Health Dis. 20 (1), 14. doi:10.1186/s12944-021-01436-6
Dron, J. S., and Hegele, R. A. (2017). Genetics of Triglycerides and the Risk of Atherosclerosis. Curr. Atheroscler. Rep. 19 (7), 31. doi:10.1007/s11883-017-0667-9
Dugi, K. A., Dichek, H. L., Talley, G. D., Brewer, H. B., and Santamarina-Fojo, S. (1992). Human Lipoprotein Lipase: The Loop Covering the Catalytic Site Is Essential for Interaction with Lipid Substrates. J. Biol. Chem. 267 (35), 25086–25091. doi:10.1016/s0021-9258(19)74009-5
Goldberg, R. B., and Chait, A. (2020). A Comprehensive Update on the Chylomicronemia Syndrome. Front. Endocrinol. 11, 593931. doi:10.3389/fendo.2020.593931
Hegele, R. A. (2009). Plasma Lipoproteins: Genetic Influences and Clinical Implications. Nat. Rev. Genet. 10 (2), 109–121. doi:10.1038/nrg2481
Henderson, H., Ma, Y., Liu, M., Clark-Lewis, I., Maeder, D., Kastelein, J., et al. (1993). Structure-function Relationships of Lipoprotein Lipase: Mutation Analysis and Mutagenesis of the Loop Region. J. Lipid Res. 34 (9), 1593–1602. doi:10.1016/s0022-2275(20)36952-2
Hooper, A. J., Crawford, G. M., Brisbane, J. M., Robertson, K., Watts, G. F., van Bockxmeer, F. M., et al. (2008). Familial Lipoprotein Lipase Deficiency Caused by Known (G188E) and Novel (W394X) LPL Gene Mutations. Ann. Clin. Biochem. 45 (Pt 1), 102–105. doi:10.1258/acb.2007.007080
Jin, N., Matter, W. F., Michael, L. F., Qian, Y., Gheyi, T., Cano, L., et al. (2021). The Angiopoietin-like Protein 3 and 8 Complex Interacts with Lipoprotein Lipase and Induces LPL Cleavage. ACS Chem. Biol. 16 (3), 457–462. doi:10.1021/acschembio.0c00954
Kozaki, K., Gotoda, T., Kawamura, M., Shimano, H., Yazaki, Y., Ouchi, Y., et al. (1993). Mutational Analysis of Human Lipoprotein Lipase by Carboxy-Terminal Truncation. J. Lipid Res. 34 (10), 1765–1772. doi:10.1016/s0022-2275(20)35739-4
Lahoz, C., and Mostaza, J. M. (2021). Hipertrigliceridemia Familiar/hipertrigliceridemia Poligénica. Clin. Investig. Arterioscler. 33 (Suppl. 2), 37–42. doi:10.1016/j.arteri.2020.12.014
Lewis, G. F., Xiao, C., and Hegele, R. A. (2015). Hypertriglyceridemia in the Genomic Era: A New Paradigm. Endocr. Rev. 36 (1), 131–147. doi:10.1210/er.2014-1062
Mäkinen, V.-P., Parkkonen, M., Wessman, M., Groop, P.-H., Kanninen, T., and Kaski, K. (2005). High-throughput Pedigree Drawing. Eur. J. Hum. Genet. 13 (8), 987–989. doi:10.1038/sj.ejhg.5201430
Murthy, V., Julien, P., and Gagné, C. (1996). Molecular Pathobiology of the Human Lipoprotein Lipase Gene. Pharmacol. Ther. 70 (2), 101–135. doi:10.1016/0163-7258(96)00005-8
Plengpanich, W., Muanpetch, S., Charoen, S., Kiateprungvej, A., and Khovidhunkit, W. (2020). Genetic and Functional Studies of the LMF1 Gene in Thai Patients with Severe Hypertriglyceridemia. Mol. Genet. Metab. Rep. 23, 100576. doi:10.1016/j.ymgmr.2020.100576
Shakhtshneider, E., Ivanoshchuk, D., Timoshchenko, O., Orlov, P., Semaev, S., Valeev, E., et al. (2021). Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia). J. Pers Med. 11 (11). doi:10.3390/jpm11111232
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: Genome-wide Copy Number Detection and Visualization from Targeted DNA Sequencing. Plos Comput. Biol. 12 (4), e1004873. doi:10.1371/journal.pcbi.1004873
Keywords: familial hypertriglyceridemia, lipoprotein lipase, gene variant, gene mutation, LPL
Citation: Al-Waili K, Al-Rasadi K, Al-Bulushi M, Habais M, Al-Mujaini A, Al-Yaarubi S, Rimbert A, Zadjali R, Khaniabadi PM, Al-Barwani H, Hasary S, Al-Dahmani ZM, Al-Badi H, Al-Maawali A and Zadjali F (2022) The Genetic Spectrum of Familial Hypertriglyceridemia in Oman. Front. Genet. 13:886182. doi: 10.3389/fgene.2022.886182
Received: 28 February 2022; Accepted: 11 April 2022;
Published: 20 May 2022.
Edited by:
Fouzia Sadiq, Shifa Tameer-e-Millat University, PakistanReviewed by:
Matej Mlinaric, University Medical Centre Ljubljana, SloveniaCopyright © 2022 Al-Waili, Al-Rasadi, Al-Bulushi, Habais, Al-Mujaini, Al-Yaarubi, Rimbert, Zadjali, Khaniabadi, Al-Barwani, Hasary, Al-Dahmani, Al-Badi, Al-Maawali and Zadjali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fahad Zadjali, ZmFoYWR6QHNxdS5lZHUub20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.