
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 August 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.886038
This article is part of the Research Topic Biomedical Application of DNA Modifications View all 5 articles
 Manal Alaamery1,2,3*†
Manal Alaamery1,2,3*† Jahad Alghamdi4†
Jahad Alghamdi4† Salam Massadeh1,2,3†
Salam Massadeh1,2,3† Mona Alsawaji1
Mona Alsawaji1 Nora Aljawini1,2,3
Nora Aljawini1,2,3 Nour Albesher3,5Bader Alghamdi1Mansour Almutairi1Fayez Hejaili6,7
Nour Albesher3,5Bader Alghamdi1Mansour Almutairi1Fayez Hejaili6,7 Majid Alfadhel8
Majid Alfadhel8 Batoul Baz2
Batoul Baz2 Bader Almuzzaini8
Bader Almuzzaini8 Adel F. Almutairi9Mubarak Abdullah10
Adel F. Almutairi9Mubarak Abdullah10 Francisco J. Quintana11Abdullah Sayyari7,10,12*
Francisco J. Quintana11Abdullah Sayyari7,10,12*Despite the enormous economic and societal burden of chronic kidney disease (CKD), its pathogenesis remains elusive, impeding specific diagnosis and targeted therapy. Herein, we sought to elucidate the genetic causes of end-stage renal disease (ESRD) and identify genetic variants associated with CKD and related traits in Saudi kidney disease patients. We applied a genetic testing approach using a targeted next-generation sequencing gene panel including 102 genes causative or associated with CKD. A total of 1,098 Saudi participants were recruited for the study, including 534 patients with ESRD and 564 healthy controls. The pre-validated NGS panel was utilized to screen for genetic variants, and then, statistical analysis was conducted to test for associations. The NGS panel revealed 7,225 variants in 102 sequenced genes. Cases had a significantly higher number of confirmed pathogenic variants as classified by the ClinVar database than controls (i.e., individuals with at least one allele of a confirmed pathogenic variant that is associated with CKD; 279 (0.52) vs. 258 (0.45); p-value = 0.03). A total of 13 genetic variants were found to be significantly associated with ESRD in PLCE1, CLCN5, ATP6V1B1, LAMB2, INVS, FRAS1, C5orf42, SLC12A3, COL4A6, SLC3A1, RET, WNK1, and BICC1, including four novel variants that were not previously reported in any other population. Furthermore, studies are necessary to validate these associations in a larger sample size and among individuals of different ethnic groups.
Chronic kidney disease (CKD) affects over 10% of the adult population worldwide and is now recognized as the most rapidly increasing contributor to the global burden of disease (Thomas et al., 2017). Recent predictions by the Institute for Health Metrics and Evaluation indicate that CKD will become the fifth cause of years of life lost globally by 2040 (Foreman et al., 2018). In Saudi Arabia, end-stage renal disease (ESRD), the terminal manifestation of CKD, affects more than 6% of the population, resulting in substantial morbidity, mortality, and high healthcare costs (Alsuwaida et al., 2010). At present, there are approximately 17,000 patients on dialysis, and this number is increasing at an exponential rate of 8% annually, with a national dialysis incidence of 140 new cases per million population (PMP) (Wang et al., 2016).
Despite the enormous economic and societal burden of CKD, its pathogenesis remains elusive, impeding specific diagnosis and targeted therapy (Romagnani et al., 2017). CKD is a complex disorder comprising numerous pathophysiologically distinct conditions, which share the common feature of leading to persistent anomalies in the kidney structure and/or function (O’Seaghdha and Fox, 2011). Prior research has shown that CKD results from a combination of genetic and environmental factors (Satko and Freedman, 2005). Moreover, familial aggregation of CKD in diverse ethnic groups is well-documented, confirming the role of heritable factors in predisposition to CKD (Cañadas-Garre et al., 2018a). Although CKD can be identified by well-established clinical biomarkers including the estimated glomerular filtration rate (eGFR), albuminuria, or serum creatinine (SCr) levels, the early prediction of individual risk for CKD or the likelihood of later progression to ESRD remains a challenge (Satko and Freedman, 2005; Cañadas-Garre et al., 2018b).
Genetic studies, initially using candidate gene approaches, and more recently, genome-wide association studies (GWAS) have identified numerous genetic biomarkers conferring susceptibility and disease progression in CKD (Köttgen et al., 2012; Sveinbjornsson et al., 2014; Prokop et al., 2018). However, these genetic biomarkers do not account for all the susceptibility to CKD and explain a minority of the overall heritability. Although there are several causes of kidney failure in Saudi patients on dialysis, as reported in the registry of the Saudi Centre for Organ Transplantation (Bullich et al., 2018), genetic and congenital anomalies of the urinary system are listed as the causes of only 2% and 1.6% of cases, respectively. However, this is an underestimated percentage, and many of the causes listed as “unknown” (7%) or hypertension (38%) may well be due to genetic factors. Globally, a 20% prevalence of family history of kidney disease was reported by incident dialysis patients in the USA (of them, 22.2% in diabetes mellitus; 18.9% in hypertension; 22.7% in glomerulonephritis; and 13.0% in patients with other etiologies) (Fallerini et al., 2014). In another US study, 23% of incident dialysis patients had close relatives with ESRD, while 21% of patients over 55 years old had a family history of ESRD. The prevalence values of family history among patients with diabetes, glomerulonephritis, hypertensive nephrosclerosis, and “other” causes of their CKD were 24.4%, 22.5%, 23.2%, and 17.5%, respectively (Riedhammer et al., 2020). Additionally, a study from Norway reported that individuals with a first-degree relative with ESRD had a 7.2 times higher risk of ESRD than individuals without a first-degree relative with ESRD (Cañadas-Garre et al., 2018a). Importantly, heritability studies of eGFR in twin studies reported an estimate of 50% (Cañadas-Garre et al., 2019). The familial aggregation studies demonstrated that heritability ranged from 36 to 75% for eGFR and from 16% to 49% for albuminuria (Cañadas-Garre et al., 2018a; Adam et al., 2020).
In this study, we used a genetic testing approach based on targeted next-generation sequencing of 104 genes causative or associated with CKD in Saudi patients with ESRD. We sought to elucidate the genetic causes of CKD and identify genetic variants associated with CKD and related traits in the Saudi population, supporting precision medicine for Saudi patients with kidney disease.
Ethical approval for this study and all experimental protocols was obtained from the Institutional Review Board at King Abdullah International Medical Research Center (KAIMRC), Ministry of National Guard—Health Affairs (MNG-HA), and site-specific approvals were obtained from all participating centers. The study and all experimental protocols adhered to the Declaration of Helsinki. All participants or their guardians were consented to by their local team for genetic testing and participation in this study upon recruitment.
This study included a total of 1,098 Saudi subjects, including 534 patients with stage 5 CKD who were referred to the Hemodialysis Unit at King Abdulaziz Medical City (KAMC), MNG-HA between September 2019 and March 2020, or the participating centers. The remaining 564 subjects are healthy CKD-free controls whose medical history was retrieved from the MNGHA health database and carefully revised. The inclusion criteria for the cases were as follows: 1) Saudi adults, who are descendants of Saudi parents and grandparents, 2) having CKD stage 5 (including those on dialysis), and 3) being willing to consent to general genetic research. We excluded patients with terminally ill conditions who were unable to provide informed consent.
Detailed demographic and clinical information was collected for all recruited patients from their electronic health records. All extracted data were collected in a predefined and secure database. Collected variables included age, sex, comorbidities, and family history of renal disease. Clinical information included disease manifestations, phenotypic information, and primary causes of ESRD, which were determined by the treating nephrologist.
Blood samples were collected in EDTA tubes from all recruited subjects. Genomic DNA was extracted from whole blood using the Gentra Puregene Blood Kit C instrument (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The isolated DNA was then quantified using a NanoDropTM spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and/or Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States) using standard procedures. DNA with the A260/280 ratio between 1.8 and 2.0, and A260/230 ratio ≥ was used for NGS library preparation.
We used a predesigned panel that contains 102 genes that have known or suspected associations with CKD. The gene list was curated by a multi-disciplinary team of nephrologists, clinical geneticists, and laboratory scientists (Cyrus et al., 2018). The used panel has shown 57% overall clinical sensitivity, i.e., detection of a likely causal variant that is subsequently confirmed by Sanger sequencing, and the sensitivity values in selected subgroups of patients with glomerular/tubular disorders, cystic kidney diseases, and kidney malformations were 41%, 63%, and 69%, respectively (Cyrus et al., 2018). The complete list of the included genes and description of gene panel design information is provided by the Saudi Mendeliome Group (Cyrus et al., 2018).
NGS libraries were constructed using the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s standard protocol. Sequencing was performed on the Ion Chef System instrument (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s workflow. A complete library preparation, NGS methodology, and data processing and bioinformatics analysis are described in the SHGP (Cyrus et al., 2018). Additionally, the Ensembl Variant Effect Predictor (VEP, v104) tool was used to annotate all the identified variants using Ensembl release 100 and assembly GRCh37/hg 19 of the human reference genome (Lee et al., 2007). Variants were also compared against the Genome Aggregation Database (GenomAD). Functional assessment of the identified variants was conducted using SIFT (v5.2.2), PolyPhen-2 (v2.2.2), and CADD (v1.6) (Cyrus et al., 2018; O'Seaghdha et al., 2014; Santín et al., 2011).
Baseline characteristics and known risk factors for CKD were summarized by case and control status using frequency (%) and mean (standard deviation). The difference between case and control was compared by the Wilcoxon rank-sum test for continuous variables and by the chi-squared test for categorical variables. The direct association between identified variants and the risk of CKD status was tested by using Fisher’s exact test as implemented in PLINK (Cyrus et al., 2018). A p-value that is corrected for multiple comparisons using the Bonferroni method was considered significant for the genetic association test. For all statistical analyses, the significant value is considered at a two-sided 5% level unless stated otherwise.
A total of 1,098 participants fulfilled the study eligibility criteria and were included in all subsequent analyses. Of the recruited subjects, 49% were cases (n = 534) and the remaining 51% were controls (n = 564). The baseline characteristics of the two groups are shown in Tables 1, 2. There were no significant differences between the two groups in terms of gender or body weight level, but the control group was younger than the case group (44 vs. 59 years; p-value <0.05). Existing other comorbidities were more common among cases than in controls, especially for cardiovascular diseases and diabetes (Table 2). The proportion of the common CKD subtypes among cases is provided in Table 3.
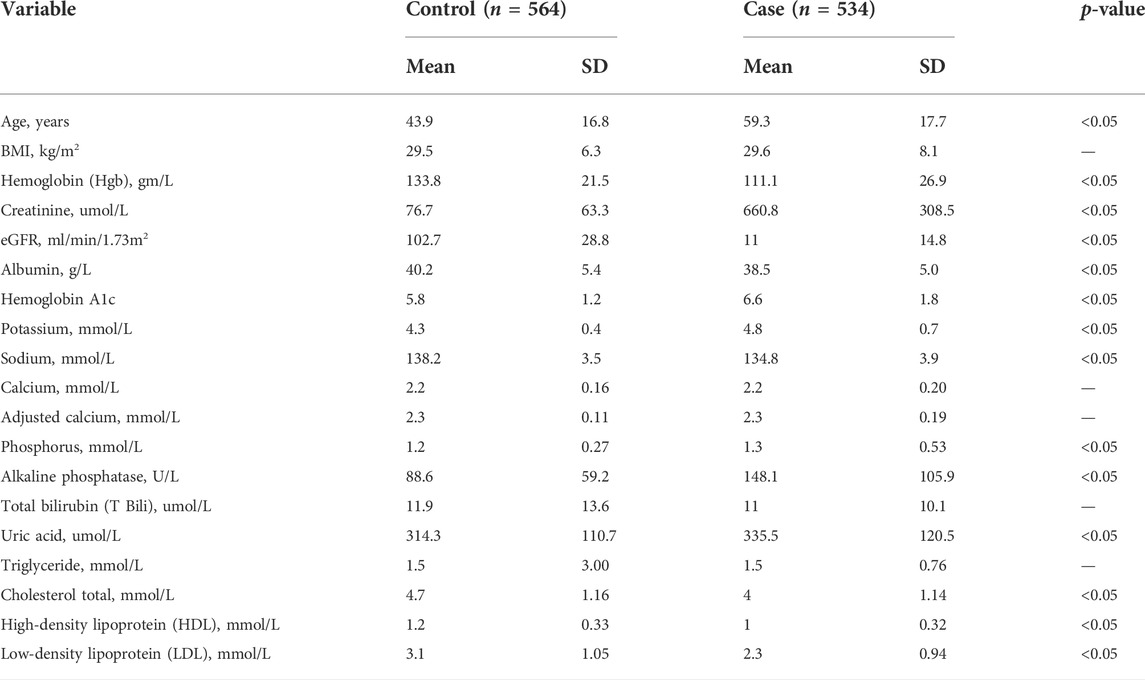
TABLE 1. Baseline characteristics of the participants.
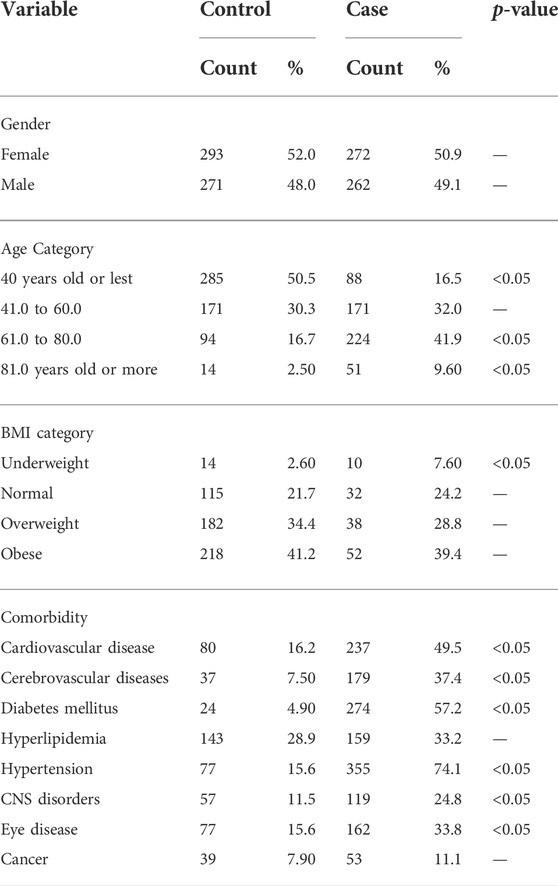
TABLE 2. Categorical baseline characteristics of the participants.
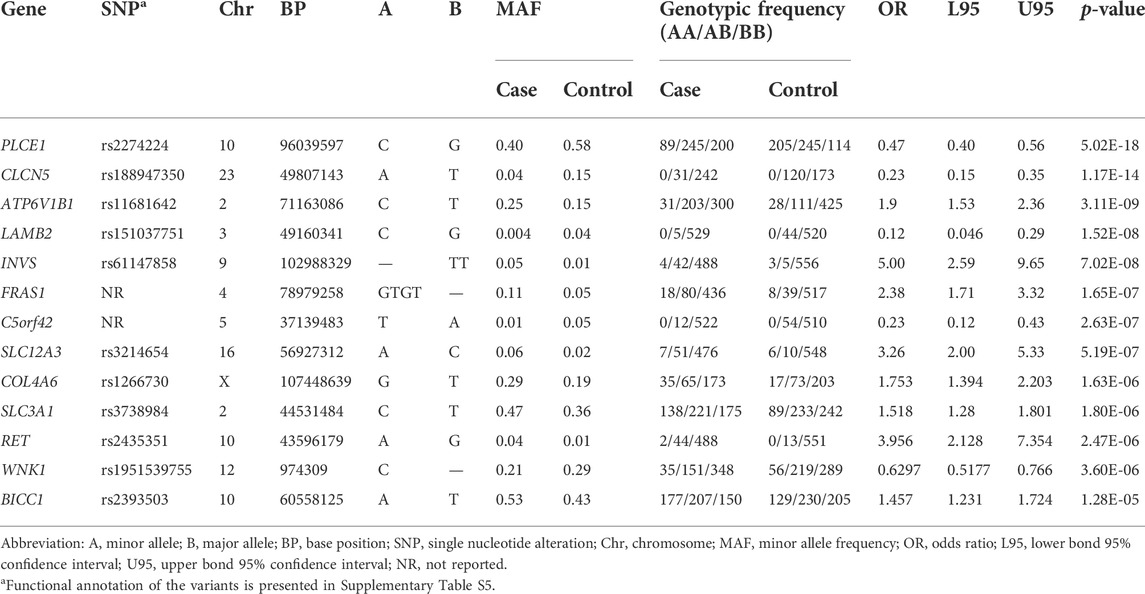
TABLE 3. SNPs associated with ESRD.
The NGS panel revealed 7,225 variants in the 104 sequenced genes. More than half of the identified variants are extremely rare variants (MAF <0.001; 52.9%), and only 830 (11.5%) variants were common with an allelic frequency above 0.05 (Supplementary Figure S1). We identified 1,348 (18.7%) new variants that were not previously reported in other databases. These new variants were more common in FRAS1, C5orf42, PKD1, CEP290, and PKHD1 (Supplementary Figure S2, Supplementary Table S1). Most of the variants identified were intronic variants (0.46; Supplementary Table S3), followed by missense variants (0.25). Genes with the highest proportion of possibly or probably damaging variants as predicted by the PolyPhen score (Supplementary Table S4); GRHPR (21%) and FXYD2 (20%); UMOD (19%); LAMB2 (18.2%) and FREM2 (17.4%). Cases had a significantly higher number of the confirmed pathogenic variant as classified by the ClinVar database than control (i.e., individuals with at least one allele of a confirmed pathogenic variant that is associated with CKD; 279 (0.52) vs. 258 (0.45); p-value = 0.03).
Variants with extremely rare allele frequency (MAF <0.001) were excluded from this analysis (n = 3,827 variants), as well as variants that extremely deviated from HWE among the control group only (n = 102 variants); thus, a total of 3,294 variants were included in the association test. Supplementary Table S4 shows the statistically significant SNPs for CKD stage 5 and dialysis. The top three significant SNPs were rs2274224 in PLCE1 gene (OR = 0.47; 95%CI: 0.39–0.56; p-value = 5.02E-18), rs188947350 in CLCN5 gene (OR = 0.23; 95%CI: 0.15–0.35; p-value = 1.17E-14), and rs11681642 in ATP6V1B1 gene (OR = 1.9; 95%CI: 1.53–2.36; p-value = 3.11E-09).
Herein, we screened a relatively large cohort of ESRD patients to identify genetic factors contributing to extremely severe forms of CKD. We applied an advanced genetic testing approach using a targeted NGS gene panel including 102 genes causative or associated with CKD to characterize a cohort of 534 Saudi patients. We identified 13 statistically significant variants within genes implicated in kidney function. All 13 variants were not previously reported to be associated with kidney diseases. Among these 13 variants, four were novel variants that were not previously reported in any other population. Unlike most GWASs that explored the role of genetic factors in kidney function among the general population, this study uniquely reported the association between genetic variants and severe forms of kidney disease among a homogenous disease-specific population.
Several GWASs were performed for variable kidney function traits. The Chronic Kidney Disease Genetic Consortium (CKDGen) performed a GWAS on more than 67,000 individuals of the European ancestry and reported 20 loci associated with renal function and CKD (Prokop et al., 2018). The study showed that a genetic risk score (GRS) including 16 SNPs explained a 1.4% variation in the renal function, as explained by serum creatinine (eGFRcrea). Later GWASs which included larger sample sizes were able to bring the number of variants that are associated with eGFRcrea up to 63, explaining up to 3.99% of the phenotypic variance (Fallerini et al., 2014; Bullich et al., 2018). As expected, variants identified from these GWAS were of modest effect size and common with allele frequency above 5%. Since these studies were performed on the general population with the majority of study participants having eGFR greater than 60 ml/min/1.732, transferring such results into concrete risk estimation, for the development of ESRD remains uncertain. Hence, genotyping these common variants provides minimal clinically relevant information to individual patients. Importantly, Parsa et al. (2013) examined systematically the association of eGFR with common variants in 258 genes responsible for Mendelian forms of kidney abnormalities using the CKDGen Consortium GWAS data [30]. This study, in addition to others, has been largely unsuccessful in identifying common variants in genes that cause rare monogenic forms of CKD, except for a few genes such as UMOD, LPR2, and SLC7A4 [30–32]. Although such an approach has proven its success in other complex traits such as diabetes and lipid levels, it suggests that this failure might be a result of relying on the GWAS panel and subjects from the general population [30]. This is specifically true as CKDGen encompasses a general population that may not easily identify new variants beyond what has already been identified by their GWAS approach. These challenges imply that further research is needed by performing targeted sequencing of these genes or whole-exome sequencing in a disease-specific cohort.
As such, our study has targeted patients with ESRD and identified 13 novel genetic variants associated with ESRD in the Saudi population. The top significant variant (NM_016341.4:c.4724G>C) is a missense variant within the PLCE1 gene that results in the substitution of arginine for proline at position 1,575 (p.R1575P) and is classified by ClinVar as uncertain significance for association with nephrotic syndrome type 3. When the same variant was not reported before for association with nephrotic syndrome type 3, several other variants within PLCE1 have been reported in familial and sporadic variants within PLCE1 (O'Seaghdha et al., 2014). The gene product of PLCE1 is expressed in the developing kidney in glomerular podocytes, and sequence alteration may lead to abnormal protein products that cause an arrest in normal glomerular development (O'Seaghdha et al., 2014). The second top variant (NM_001127898.4:c.163 + 72T>A) is a rare intronic variant within the CLCN5 gene, 0.00101 allele frequency in gnomAD, which has not been reported before for association with any phenotype. Several variants within CLCN5 are associated with different types of renal tubular disorders, such as Dent’s disease (Santín et al., 2011). The third variant (NM_001692.4:c.2T>C) causes a missense mutation, resulting in the substitution of methionine for threonine at position 1 (p.M1T) within the ATP6V1B1 gene, which encodes a component of vacuolar ATPase (V-ATPase), a multisubunit enzyme that mediates acidification of intracellular organelles. ATP6V1B1 variants are associated with an autosomal recessive form of distal renal tubular acidosis 2 (Santín et al., 2011). In addition, we have identified variants in LAMB2, INVS, FRAS1, C5orf42, SLC12A3, COL4A6, SLC3A1, RET, WNK1, and BICC1; which all play major pathophysiological roles in the pathogenesis of kidney functions (Kestilä et al., 1998).
From both clinical and economic viewpoints, understanding the genetic etiology behind CKD is essential before opting for renal transplantation. Such information may provide novel insights into disease recurrence risk post-transplantation. This study was performed in a unique and novel (Philippe et al., 2008) population that has not been previously explored—the uniqueness of the Saudi population lay in the high consanguinity rate—which provides a perfect population to study the genetic basis of diseases. This has been seen previously in a study that detected the highest rate of causative genes in the Saudi population (45.2%) compared to only 13.7% in Al-Hamed et al. (2016), which showed a strong correlation between the rate of consanguinity and the detection rate of disease-causing genes (R2 = 0.9414). Nonetheless, findings from this study might be limited by some important factors; first, the results are reported from a very homogenous population originating from the Saudi population, and it must be replicated in other diverse ethnic groups to validate the reported association. Second, the control group was younger and healthier than the case group, which could confound the reported results. However, given the fact that these genes are implicated in kidney function, they have pathophysiological roles that could mitigate some of these concerns. Third, we have only tested the disease status as a binomial trait, and future studies should be performed to test other kidney-related functions.
In summary, utilizing a comprehensive kidney-disease gene panel using a case-control study in a Saudi Arabian cohort, we identified 13 genetic variants significantly associated with ESRD in this cohort. Since this study was conducted on a disease-specific population and not on a general population, like most of the previously conducted GWAS, it provides more concrete evidence of the roles of these variants in the pathogenesis of ESRD. Furthermore, studies to replicate the findings of this study, as well as functional analysis of the identified variants, may provide measures to reduce the burden of CKD in KSA. In particular, that elucidating the genetic etiology of CKD may have important ramifications when considering disease recurrence risk post-transplantation. Further studies both to replicate these findings in larger sample sizes and among individuals of different ethnic groups and to functionally validate these candidate genetic variants are imperative. This may provide a comprehensive priority list of molecular targets for translational research and eventually help reduce the burden of CKD in KSA (Franceschini et al., 2006).
Information for existing publicly accessible datasets are contained within the article.
The studies involving human participants were reviewed and approved by the study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board at King Abdullah International Medical Research Centre, Riyadh; (RC 19/104 approved on the 7th of July 2019). The patients/participants provided their written informed consent to participate in this study.
Conceptualization, AA, MA, SM, and JA; methodology, AA, MA, JA, SM, MAS, BA, and MSA; validation, MA, SM, and JA; formal analysis, MA, JA, and SM; investigation, MA, SM, BA, and MSA; resources, MA, BB, and AA; data curation, MA, SM, JA, MAS, BA, and MSA; writing—original draft preparation, MA, JA, SM, NB, and NA; writing—review and editing, SM, MA, JA, NA, NB, FH, MAF, BB, BAM, AFA, MAB, FJQ, and AA; visualization, MA, JA, AA, and SM; supervision, MA and SM; project administration, SM and MA; funding acquisition, MA. All authors have read and agreed to the published version of the manuscript.
The authors would like to thank all study participants and their families for contributing to this study. In addition, we thank all the developmental medicine department laboratory members for the DNA extraction of the samples. Also, we acknowledge the efforts of Ms. Amaal Alharbi to recruit patients from the western region. Furthermore, MA and SM gratefully acknowledge King Abdulaziz City for Science and Technology and the Saudi Human Genome Project for the technical and financial support (https://shgp.kacst.edu.sa). Also, the authors acknowledge the KACST-Centre of excellence in biomedicine (CEBM) for the support. Additionally, the authors like to acknowledge the support of King Abdulaziz City for Science and Technology through the Therapy Development Research Project (TDRP). Lastly, all authors would like to dedicate this work for the soul of Professor Abdullah Sayyari, who passed away on the 8th of February 2022.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.886038/full#supplementary-material
Adam, K. M., Mohammed, A. M., and Elamin, A. A. (2020). Non-diabetic end-stage renal disease in Saudis associated with polymorphism of MYH9 gene but not UMOD gene. Med. Baltim. 99 (3), e18722. doi:10.1097/MD.0000000000018722
Al-Hamed, M. H., Kurdi, W., Alsahan, N., Alabdullah, Z., Abudraz, R., Tulbah, M., et al. (2016). Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. J. Med. Genet. 53 (5), 338–347. doi:10.1136/jmedgenet-2015-103469
Alsuwaida, A. O., Farag, Y. M., Al Sayyari, A. A., Mousa, D., Alhejaili, F., Al-Harbi, A., et al. (2010). Epidemiology of chronic kidney disease in the Kingdom of Saudi Arabia (SEEK-Saudi investigators) - A pilot study. Saudi J. Kidney Dis. Transpl. 21 (6), 1066–1072.
Bullich, G., Domingo-Gallego, A., Vargas, I., Ruiz, P., Lorente-Grandoso, L., Furlano, M., et al. (2018). A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 94 (2), 363–371. doi:10.1016/j.kint.2018.02.027
Cañadas-Garre, M., Anderson, K., Cappa, R., Skelly, R., Smyth, L. J., McKnight, A. J., et al. (2019). Genetic susceptibility to chronic kidney disease – some more pieces for the heritability puzzle. Front. Genet. 10, 453. doi:10.3389/fgene.2019.00453
Cañadas-Garre, M., Anderson, K., McGoldrick, J., Maxwell, A. P., and McKnight, A. J. (2018a). Genomic approaches in the search for molecular biomarkers in chronic kidney disease. J. Transl. Med. 16, 292. doi:10.1186/s12967-018-1664-7
Cañadas-Garre, M., Anderson, K., McGoldrick, J., Maxwell, A. P., and McKnight, A. J. (2018b). Proteomic and metabolomic approaches in the search for biomarkers in chronic kidney disease. J. Proteomics 193, 93–122. doi:10.1016/j.jprot.2018.09.020
Cyrus, C., Al-Mueilo, S., Vatte, C., Chathoth, S., Li, Y. R., Qutub, H., et al. (2018). Assessing known chronic kidney disease associated genetic variants in Saudi Arabian populations. BMC Nephrol. 19, 88. doi:10.1186/s12882-018-0890-9
Fallerini, C., Dosa, L., Tita, R., Del Prete, D., Feriozzi, S., Gai, G., et al. (2014). Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 86 (3), 252. doi:10.1111/cge.12258
Foreman, K. J., Marquez, N., Dolgert, A., Fukutaki, K., Fullman, N., McGaughey, M., et al. (2018). Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet 392, 2052–2090. doi:10.1016/S0140-6736(18)31694-5
Franceschini, N., North, K. E., Kopp, J. B., McKenzie, L., and Winkler, C. (2006). NPHS2 gene, nephrotic syndrome and focal segmental glomerulosclerosis: A HuGE review. Genet. Med. 8 (2), 63–75. doi:10.1097/01.gim.0000200947.09626.1c
Kestilä, M., Lenkkeri, U., Männikkö, M., Herva, R., McCready, P., Putaala, H., et al. (1998). Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol. Cell. 1 (4), 575–582. doi:10.1016/s1097-2765(00)80057-x
Köttgen, A., Yang, Q., Shimmin, L. C., Tin, A., Schaeffer, C., Coresh, J., et al. (2012). Association of estimated glomerular filtration rate and urinary uromodulin concentrations with rare variants identified by UMOD gene region sequencing. PLoS One 7, e38311. doi:10.1371/journal.pone.0038311
Lee, C., Scherr, H. M., Wallingford, J. B., Delalle, I., Caviness, V. S., Silver, J., et al. (2007). Shroom family proteins regulate gamma-tubulin distribution and microtubule architecture during epithelial cell shape change. Development 134 (7), 1431–1441. doi:10.1242/dev.02828
O'Seaghdha, C. M., Tin, A., Yang, Q., Katz, R., Liu, Y., Harris, T., et al. (2014). Association of a cystatin C gene variant with cystatin C levels, CKD, and risk of incident cardiovascular disease and mortality. Am. J. Kidney Dis. 63 (1), 16–22. doi:10.1053/j.ajkd.2013.06.015
O’Seaghdha, C. M., and Fox, C. S. (2011). Genome-wide association studies of chronic kidney disease: What have we learned? Nat. Rev. Nephrol. 8, 89–99. doi:10.1038/nrneph.2011.189
Philippe, A., Nevo, F., Esquivel, E. L., Reklaityte, D., Gribouval, O., Tete, M. J., et al. (2008). Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 19 (10), 1871–1878. doi:10.1681/ASN.2008010059
Prokop, J. W., Yeo, N. C., Ottmann, C., Chhetri, S. B., Florus, K. L., Ross, E. J., et al. (2018). Characterization of coding/noncoding variants for SHROOM3 in patients with CKD. J. Am. Soc. Nephrol. 29, 1525–1535. doi:10.1681/ASN.2017080856
Riedhammer, K. M., Braunisch, M. C., Günthner, R., Wagner, M., Hemmer, C., Strom, T. M., et al. (2020). Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. Am. J. Kidney Dis. 76, 460. doi:10.1053/j.ajkd.2019.12.008
Romagnani, P., Remuzzi, G., Glassock, R., Levin, A., Jager, K. J., Tonelli, M., et al. (2017). Chronic kidney disease. Nat. Rev. Dis. Prim. 3, 17088. doi:10.1038/nrdp.2017.88
Santín, S., Bullich, G., Tazón-Vega, B., Garcia-Maset, R., Gimenez, I., Silva, I., et al. (2011). Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 6 (5), 1139–1148. doi:10.2215/CJN.05260610
Satko, S. G., and Freedman, B. I. (2005). The familial clustering of renal disease and related phenotypes. Med. Clin. North Am. 89, 447–456. doi:10.1016/J.MCNA.2004.11.011
Sveinbjornsson, G., Mikaelsdottir, E., Palsson, R., Indridason, O. S., Holm, H., Jonasdottir, A., et al. (2014). Rare mutations associating with serum creatinine and chronic kidney disease. Hum. Mol. Genet. 23, 6935–6943. doi:10.1093/hmg/ddu399
Thomas, B., Matsushita, K., Abate, K. H., Al-Aly, Z., Arnlov, J., Asayama, K., et al. (2017). Global cardiovascular and renal outcomes of reduced GFR. J. Am. Soc. Nephrol. 28 (7), 2167–2179. doi:10.1681/ASN.2016050562
Keywords: chronic kidney disease, next-generation sequencing, renal failure, panel sequencing, novel variants
Citation: Alaamery M, Alghamdi J, Massadeh S, Alsawaji M, Aljawini N, Albesher N, Alghamdi B, Almutairi M, Hejaili F, Alfadhel M, Baz B, Almuzzaini B, Almutairi AF, Abdullah M, Quintana FJ and Sayyari A (2022) Analysis of chronic kidney disease patients by targeted next-generation sequencing identifies novel variants in kidney-related genes. Front. Genet. 13:886038. doi: 10.3389/fgene.2022.886038
Received: 28 February 2022; Accepted: 29 June 2022;
Published: 11 August 2022.
Edited by:
Sadeq Vallian, University of Isfahan, IranReviewed by:
Katarina Trebušak Podkrajšek, University of Ljubljana, SloveniaCopyright © 2022 Alaamery, Alghamdi, Massadeh, Alsawaji, Aljawini, Albesher, Alghamdi, Almutairi, Hejaili, Alfadhel, Baz, Almuzzaini, Almutairi, Abdullah, Quintana and Sayyari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manal Alaamery, YWxhYW1lcnltYUBuZ2hhLm1lZC5zYQ==; Abdullah Sayyari, c2F5eWFyaWFiQG5naGEubWVkLnNh
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.