 Poonnada Jiraanont1
Poonnada Jiraanont1 Esther Manor2,3
Esther Manor2,3 Nazi Tabatadze4
Nazi Tabatadze4 Marwa Zafarullah5
Marwa Zafarullah5 Guadalupe Mendoza5
Guadalupe Mendoza5 Gia Melikishvili4
Gia Melikishvili4 Flora Tassone5,6*
Flora Tassone5,6*- 1Faculty of Medicine, King Mongkut’s Institute of Technology Ladkrabang, Bangkok, Thailand
- 2Faculty of Health Sciences, Ben-Gurion University of the Negev, Beersheba, Israel
- 3Genetics Institute, Soroka Medical Center, Beersheba, Israel
- 4Department of Pediatrics, MediClub Georgia Medical Center, Tbilisi, Georgia
- 5Department of Biochemistry and Molecular Medicine, School of Medicine, University of California, Davis, Sacramento, CA, United States
- 6UC Davis MIND Institute, UC Davis Health, Sacramento, CA, United States
Fragile X syndrome (FXS) is the most frequent cause of X-linked inherited intellectual disabilities (ID) and the most frequent monogenic form of autism spectrum disorders. It is caused by an expansion of a CGG trinucleotide repeat located in the 5′UTR of the FMR1 gene, resulting in the absence of the fragile X mental retardation protein, FMRP. Other mechanisms such as deletions or point mutations of the FMR1 gene have been described and account for approximately 1% of individuals with FXS. Here, we report a 7-year-old boy with FXS with a de novo deletion of approximately 1.1 Mb encompassing several genes, including the FMR1 and the ASFMR1 genes, and several miRNAs, whose lack of function could result in the observed proband phenotypes. In addition, we also demonstrate that FMR4 completely overlaps with ASFMR1, and there are no sequencing differences between both transcripts (i.e., ASFMR1/FMR4 throughout the article).
Introduction
Fragile X syndrome (FXS) is the most prevailing cause of X-linked inherited intellectual disability and autism spectrum disorder (Penagarikano et al., 2007; Rogers et al., 2001). It is caused by a CGG trinucleotide repeat expansion within the 5’ UTR region of the fragile X mental retardation 1 (FMR1) gene located on chromosome Xq27.3, which spans approximately 38 kb of genomic sequence (Verkerk et al., 1991; Yu et al., 1991; Fu et al., 1991; Bell et al., 1991). The CGG repeats expand in the female germline during transmission. When the CGG expands to more than 200 repeats, the so-called full mutation, the promoter of FMR1 becomes hypermethylated and transcriptionally silent, leading to gene inactivation and consequently diminished or lack of expression of the FMR1 protein, FMRP (Fu et al., 1991; Tassone et al., 2000) (Figure 1A). Individuals with the fragile X premutation (55-200 CGG repeats) are at risk of developing a number of clinical problems falling under the umbrella of FMR1-associated disorders. Male individuals with FXS have moderate to severe intellectual impairment, and a behavioral phenotype characterized by repetitive behaviors, social difficulties, poor eye contact, excessive shyness, anxiety, aggression, tactile defensiveness, hyperarousal response to sensory stimuli, tantrums, attention deficits, hyperactivity, impulsivity, self‐injury, stereotypic movements including hand flapping, and perseverative speech. Physical manifestations range from the large forehead, prominent ears, hyperextensible finger joints, and macroorchidism (Hagerman and Hagerman, 2002; Hatton et al., 2002; Reiss and Dant, 2003; Hagerman et al., 2009; Symons et al., 2010; Kidd et al., 2014).
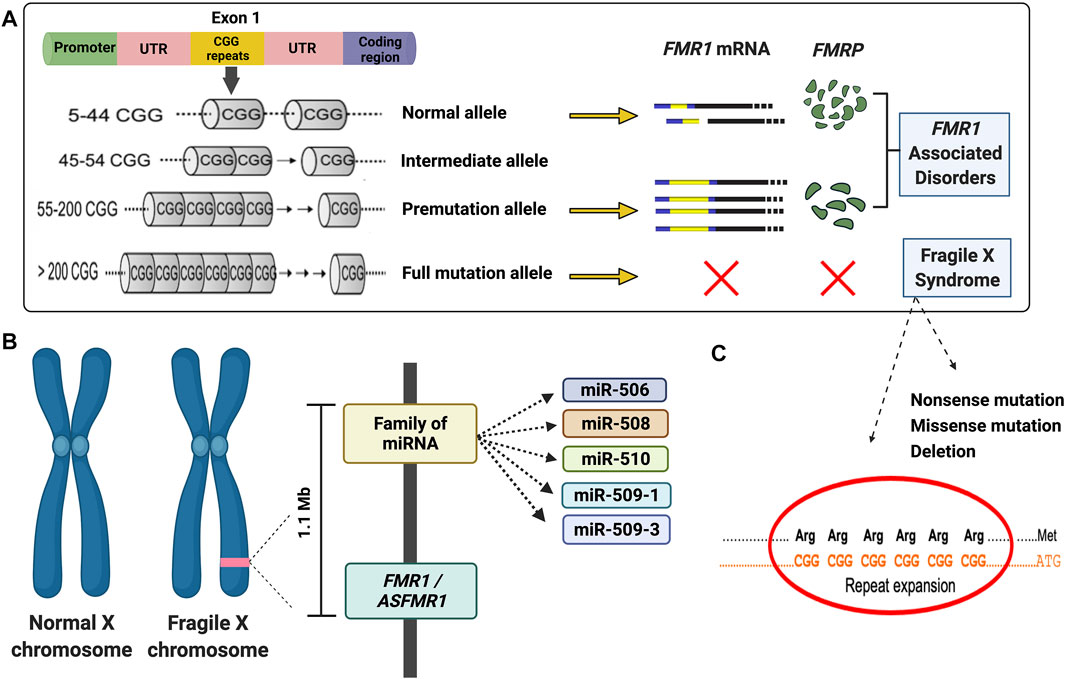
FIGURE 1. Diagram illustrating the main molecular mechanisms leading to fragile X syndrome and FMR1-associated disorders. (A) Schematic of molecular causes of fragile X syndrome and FMR1-associated disorders. (B) Diagram of the deleted region detected in the proband. (C) Majority of the fragile X syndrome cases are due to an expansion of the CGG repeat in the 5′UTR of the FMR1 gene. However, several mutations, including missense, nonsense, and deletion (as demonstrated in the proband) have been reported. A list can be found in the Human Gene Mutation Database for FXS (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FMR1).
Although the CGG repeat expansion is the main underlying cause of FXS, other mechanisms, including point mutations or deletions, can lead to FXS (Handt et al., 2014; Monaghan et al., 2013; Quartier et al., 2017; Sitzmann et al., 2018) (Figures 1B,C). The deletions in FXS individuals commonly range from hundreds to several millions of base pairs and frequently encompass a portion or the entire sequence of the FMR1 gene (Coffee et al., 2008). Several reports during the last 3 decades demonstrated de novo deletions (Tarleton et al., 1993; Gu et al., 1994; Petek et al., 1999; Luo et al., 2014; Zink et al., 2014; Jorge et al., 2018; Myers et al., 2019) spanning the FMR1 or both the FMR1 and FMR2 gene (Clarke et al., 1992; Wolff et al., 1997; Fengler et al., 2002; Probst et al., 2007), resulting in the loss of FMRP and a range of phenotypes including physical, cognitive and behavioral features, intellectual disabilities (ID), seizures, and obesity, similarly observed in individuals with FXS (Coffee et al., 2008).
The human genome is extensively transcribed and gives rise to various long non-coding RNAs (lncRNAs), defined as RNAs longer than 200 nucleotides and not translated into functional proteins (Fang et al., 2018). Approximately 40% of mammalian lncRNAs are expressed in the brain (Briggs et al., 2015) and play an essential role in neuronal differentiation and regeneration (Perry et al., 2018). They regulate significant biological roles in DNA damage response and cellular senescence (Tsai et al., 2010; Hung et al., 2011; Kotake et al., 2011; Pastori et al., 2015) through modulation of gene expression regulation. In addition, several lncRNAs play a role in brain development, synaptic signaling mechanisms, differentiation of neural cell lineage, and formation of mature neuronal connections (Mercer et al., 2010; Qureshi et al., 2010). Altered expression of lncRNAs has been associated with neurodevelopmental disorders like Prader–Willi syndrome (Statello et al., 2021) and neurodegenerative disorders such as Parkinson’s disease, Huntington’s disease, lateral amyotrophic sclerosis, or Alzheimer’s disease (Wei et al., 2018).
A comprehensive analysis of the transcriptional landscape of the human FMR1 gene, discovered a decade ago, the ASFMR1 gene, an lncRNA, a unique antisense transcript, overlapping the CGG repeat region at the FMR1 locus. Its expression is determined by two promoters that are flanked by CTCF-binding sites: the FMR1 bidirectional promoter and the other one located in the second intron of the FMR1 gene, which is the major promoter in premutation cells (Ladd et al., 2007). The ASFMR1 mRNA, similarly to the FMR1, is upregulated in lymphoblastoid cells and peripheral blood leukocytes derived from individuals with the premutation, and it is not expressed in subjects with the FMR1 full mutation (Ladd et al., 2007; Loesch et al., 2011). In addition, the ASFMR1 transcript demonstrates premutation-specific alternative splicing suggesting a potential contribution of bidirectional transcription to the clinical phenotypes of the FMR1-associated disorders (Al Olaby et al., 2018; Vittal et al., 2018; Zafarullah et al., 2020). Additional studies have revealed the presence of several lncRNAs within the FMR1 gene, including, FMR4 (Khalil et al., 2008), FMR5, and FMR6 (Pastori et al., 2014). Like the FMR1 mRNA, FMR4, a primate-specific non-coding RNA transcript (∼2.4 kb) residing upstream and sharing a bidirectional promoter with FMR1, is silenced in patients with FXS and upregulated in carriers of an FMR1 premutation allele. It was reported that FMR4 has an antiapoptotic function in HEK293T and HeLa cells but no effect on FMR1 gene regulation, suggesting a potential indirect regulatory transcript for FMR1 (Khalil et al., 2008).
Furthermore, numerous studies have indicated that FMRP acts as a mediator effector of the microRNA (miRNA) pathway by interacting with miRNA and proteins to form RNA-induced silencing complex (RISC) (Caudy et al., 2002; Ishizuka et al., 2002; Jin et al., 2004; Smalheiser and Lugli, 2009; Dionne and Corbin, 2021). Thus, the lack of FMRP in FXS and its role in the miRNA pathway contribute to the abnormal synaptogenesis in FXS and provide one of the mechanisms underlying the fragile X physiopathology (Edbauer et al., 2010; Gong et al., 2015; DeMarco et al., 2019).
Here, we report an FXS case of a seven-year-old boy with a de novo deletion of chromosome X of approximately 1.1 Mb encompassing several genes, including the FMR1 and ASFMR1 genes and the miR-506 family of miRNAs (Figure 1B). Their absence is likely related to neurodevelopment and his FXS phenotypes. Interestingly, we also demonstrated that FMR4 completely overlaps with ASFMR1, and there are no sequencing differences between both transcripts, therefore named in this study as ASFMR1/FMR4 gene. The characterization of the ASFMR1 gene, using SMRT sequencing, identified ∼6-kb ASFMR1 transcript that overlaps with FMR4, demonstrating that FMR4 and ASFMR1 are the same lncRNA (ASFMR1/FMR4).
Materials and Methods
DNA Testing: CGG Sizing
The genomic DNA of the proband, mother, and sisters was isolated from peripheral blood leukocytes (3-5 ml of whole blood) using standard methods (Puregene Kit; Gentra Inc, Minneapolis, MN). PCR and Southern blot analysis were performed to determine the CGG sizing and methylation status. For Southern Blot analysis, 5-10 μg of DNA digested with EcoRI and NruI fixed on a nylon membrane was hybridized with the FMR1 genomic probe StB12.3, labeled with Dig-11-dUTP by PCR (PCR Dig Synthesis Kit; Roche Diagnostics) following the protocol as previously described (Tassone et al., 2008; Filipovic-Sadic et al., 2010).
Chromosomal Microarray
CMA was carried out on the proband using GeneChip auto scan 750 K (Affymetrix Santa Clara, United States) following the manufacturer's instructions. A chromosomal analysis was performed using Chromosome Analysis Suite (ChAS®) software (Affymetrix, Santa Clara, United States). The CNVs found in the patient were analyzed in comparison with public databases, including Database of Genomic Variants (DGV), Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER), and CytoScanHD™ Array Database. Furthermore, CNVs were classified according to their nature, based on (Miller et al., 2010; Battaglia et al., 2013). After the initial Array-CGH result was acquired, informed written consent was obtained from the patient’s mother to draw blood on all available family members for additional investigation. The blood samples were obtained from the patient, his mother, his sisters, and his maternal uncle for further analysis.
Sequencing of the ASFMR1 mRNA
The total RNA was isolated from postmortem brains derived from three normal individuals in a clean and RNA designated area using TRIzol reagent (Invitrogen, Carlsbad, CA), followed by quantification via a Qubit fluorometer (Invitrogen, Waltham, MA, United States) and quality control by measurements of the RNA integrity number utilizing an Agilent 2100 Bioanalyzer system.
The genomic sequence data of 2198-bp FMR4 at locus FJ887036 and 2942-bp ASFMR1 at locus EU048204 were obtained from the UCSC website (http://genome.ucsc.edu/). The ASFMR1 transcripts were identified by PacBio iso-sequencing from isolated total RNA from three brain samples. Then 1 ug of total RNA was reverse transcribed using the Clontech SMARTer cDNA synthesis kit and six samples specific barcoded oligo dT (with PacBio 16mer barcode sequences). Three reverse transcription (RT) reactions were processed in parallel for each sample. PCR optimization was used to determine the optimal amplification cycle number for the downstream large-scale PCRs. A single primer (primer IIA from the Clontech SMARTer kit 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) was used for all PCRs post-RT. Large-scale PCR products were purified separately with 1X AMPure PB beads, and the bioanalyzer was used for QC. An equimolar two pools of 3-plex barcoded cDNA library were input into the probe-based capture with a custom-designed ASFMR1 gene panel. A SMRTBell library was constructed using captured and reamplified cDNA. One SMRT Cell 1M (20 h movie) was sequenced on the PacBio Sequel platform using 2.0 chemistry. The isoform sequencing analysis was performed using the IsoSeq3 application in the PacBio SMRT Analysis v6.0.0.
Results
Clinical History
The patient we present in this study is a 7-year-old boy with moderate intellectual disability (IQ = 41, WISC-V), attention deficit, and hyperactivity. He was a full-term male infant and third child of nonconsanguineous parents, born after a normal pregnancy. His birth weight was 4,500 g, and there were no perinatal problems. His developmental milestones included sitting at 6 months, walking at 21 months, using words at 17 months, and phrases by 3.5 years of age.
On physical examination at age 7 years, his weight was 29 kg (92nd percentile), height was 125 cm (45th percentile), and head circumference was 52.5 cm (64th percentile). The HEENT (head, eyes, ears, nose, and throat) examination was normal. The neurological examination findings were normal. Other characteristics included a long face, prominent forehead, large and protruding ears, normal palate, long palpebral fissures, epicanthal folds, flat nasal bridge, pectus excavatum, macroorchidism, and no joint hypermobility. There are no cardiovascular abnormalities, and echocardiography showed normal results. Overall, the patient is physically a well-developed boy with no significant anomalies.
He hears normally and follows simple commands. He has an acceptable motor delay and deficits in visual analyses and synthesis. He can eat independently regarding daily living skills and uses the toilet but cannot get dressed without help. The child exhibits hand biting and hand flapping when excited, his mood is generally good, and he shows no aggressive behavior toward others but sometimes he is very anxious. The parents noted excessive shyness, and he consistently avoids eye contact by covering his eyes with his hands, although he is exceptionally social and can interact with others. His early behavior included ADHD. He is frequently irritable and hyperactive at home, exhibits concentration problems, short attention span, distractibility, and impulsiveness. He has learning problems, especially difficulty with mathematics.
The proband has a healthy mother and healthy 16 and 14-years old sisters; the maternal uncle has mild to moderate intellectual disability, while the maternal aunt has depressive disorders and ovarian cysts (Figure 2).
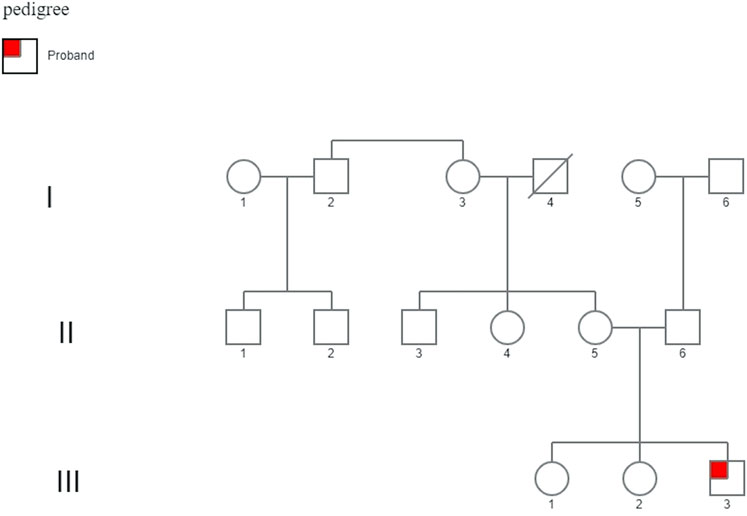
FIGURE 2. Family pedigree. His family history demonstrates two healthy 16- and 14-year-old sisters. The maternal uncle (II-3) displayed mild to moderate intellectual disability, and the maternal aunt (II-4) displayed psychiatric problems (depressive disorder) and ovarian cysts, unrelated to a mutation in the FMR1 gene.
Molecular Testing: CGH Array and FXS Diagnostic Testing
At the age of 7 years, the proband underwent array-CGH on DNA extracted from peripheral blood cells (244 K kit, Agilent Technologies, Santa Clara, CA, United States), which revealed a deletion of approximately 1.1 Mb located on Xq27.3 (NC_000,023.10: g 145,877,075-147,047,871) encompassing several genes and miRNAs, including the entire FMR1 and ASFMR1/FMR4 genes and microRNAs (miRNAs) such miR506, miR508, miR509-1, miR509-3, and miR510. Molecular DNA testing for FXS, carried out by PCR and Southern blot analysis, showed no PCR amplification and lack of hybridization with the FMR1 specific probe in the proband, indicating the presence of a deletion of the FMR1 gene (Supplementary Figure S1). The mother was not a carrier of the FMR1 premutation as determined by Southern Blot and PCR analysis.
The lncRNA, ASFMR1, and FMR4 Are the Same Transcripts
The sequence identity of ASFMR1/FMR4 using the Integrative Genomic Viewer (IGV) was obtained by blasting the transcriptomic sequences reported in the UCSC human genome database. Interestingly, we identify the ASFMR1 transcript in our library data set, and by sequencing comparison, we confirmed that no genetic sequence differences exist between the two transcripts (Figure 3A), as one of the hypotheses, proposed by Khalil et al. (2008). The complete sequence overlaps between the two genes (ASFMR1/FMR4) are shown inFigure 3B.
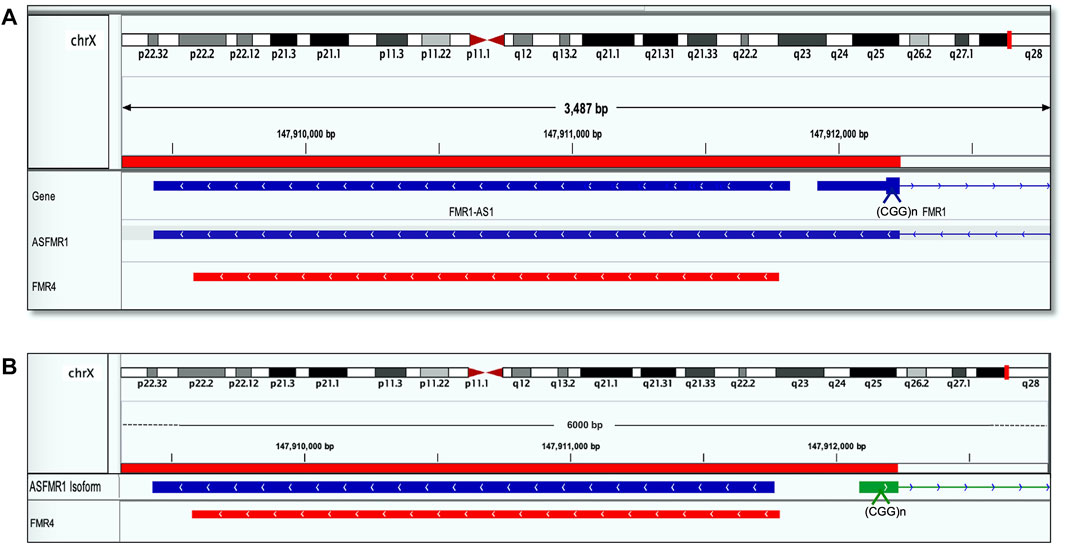
FIGURE 3. FMR4 overlaps with ASFMR1. The figure shows a visualization of sequence alignment in Integrative Genomic Viewer (IGV) at 3487 base-pair resolution on chromosome X in human genome assembly GRCh38 (hg38). (A) ASFMR1 and FMR1 genes from the human genome assembly are shown (the top of the three tracks are highlighted in blue). The middle track is the ASFMR1 transcript reported by (Ladd et al., 2007) overlapping with the FMR1 gene (going in the opposite direction). For the FMR1 gene, only exon 1 (containing the CGG repeat) and intron 1 (thin blue line) are shown. The bottom track (highlighted in red) shows the sequence of the FMR4 reported by (Khalil et al., 2008). The location of the CGG repeat in the FMR1 is indicated (CCG in the antisense direction). (B) Diagram shows the overlap between the two transcripts ASFMR1 (highlighted in blue) and FMR4 (highlighted in red) on a zoom image of the 6-kb ASFMR1 identified transcripts. The overlap demonstrates the sequencing identity of the two transcripts. The location of the CGG repeat in the FMR1 is indicated (CCG in the antisense direction).
Discussion
We report a case of FXS caused by a de novo deletion spanning a 1.1 Mb of DNA comprising the FMR1 and the ASFMR1 genes, and the miR-506 family of miRNAs. Importantly, we verified the overlap of the ASFMR1/FMR4 genes, previously identified which are therefore the same transcripts.
In 2007, 1 year prior to the discovery of the FMR4 lncRNA, Ladd (Ladd et al., 2007) identified the novel ASFMR1 (antisense fragile X mental retardation 1) gene, highly expressed in the brain and kidney but hardly noticeable in the heart. Similarly, to the FMR1 gene, the novel ASFMR1 transcript was upregulated in premutation alleles but repressed in full mutations and exhibited premutation-specific alternative splicing (Ladd et al., 2007; Zafarullah et al., 2020). One of the alternative splicing isoforms, ASFMR1 mRNAs Iso131 bp, positively correlates with the CGG repeat number and could distinguish between carriers of the premutation and controls, and its expression levels in premutation carriers, with and without FXTAS, were significantly different from controls (Vittal et al., 2018; Zafarullah et al., 2020).
In 2008, Khalil and his colleagues reported on the identification of FMR4, a novel 2.4-kb lncRNA, which transcribed in an antisense direction upstream of the FMR1 gene. The authors showed that the FMR4 transcript was highly expressed in the heart and kidney of human fetal tissues and in human adult brains including the hippocampus, frontal cortex, and cerebellum. Thus, similarly to the FMR1 gene, the ASFMR1/FMR4 mRNA expression is silenced in the brain of full mutations but highly expressed in premutation carriers. In addition, ASFMR1/FMR4 demonstrated an antiapoptotic function in vitro suggesting it may promote human cell proliferation (Khalil et al., 2008). Further study revealed that the ASFMR1/FMR4 transcript suppresses MBD4 (methyl-CpG-binding domain protein 4), which is a translational repressor, pivotal for DNA mismatch repair, inhibition of transcription, and regulation of apoptosis, in trans-activity (Yakovlev et al., 2017). Interestingly, ASFMR1/FMR4 expression is significantly reduced, while both FMR1 and MBD4 expressions are increased, in differentiating human neural precursor cells proposing a role for ASFMR1/FMR4 as a gene-regulatory lncRNA in normal development (Peschansky et al., 2015). In addition, overexpression and silencing of ASFMR1/FMR4 can induce genome-wide alteration in histone methylation of several genes implicating developmental or neurophysiological roles (Peschansky et al., 2016). These findings suggest that ASFMR1/FMR4 may regulate neurodevelopment, and its level of expression could influence the pathogenesis of FMR1-associated disorders.
In this study, we confirmed the complete sequence overlaps between the two transcripts, the ASFMR1 mRNA and the FMR4 mRNA, which is not surprising given the similar characteristics and qualities. In addition to the different reported lengths, the ASFMR1/FMR4 transcript is polyadenylated, and expressed highly in the brains of premutation carriers but suppressed in full mutation individuals. Notably, evidence demonstrated that the ASFMR1/FMR4 gene plays a role in neurodevelopment as it is involved in early neural differentiation and emerged as a promising biomarker for FXTAS, a neurodegenerative disorder (Peschansky et al., 2016; Vittal et al., 2018). Taken together, the deletion encompassing the ASFMR1/FMR4 region may influence the neurodevelopment and neuropathogenesis observed in the proband.
Furthermore, the deletion present in the proband also included a panel of miR-506 family miRNAs consisting of miR506, miR508, miR509-1, miR509-3, and miR510, all belonging to the “fragile-X miRNA (FX-MIR)” cluster (Ramaiah et al., 2019). miRNAs are crucial for various critical functions of cellular differentiation and homeostasis, as they regulate the expression of hundreds of mRNAs resulting in diverse pathologies, including cancer, neural disorders, and infertility (Greenberg and Soreq, 2014; Khazaie and Nasr Esfahani, 2014; Bracken et al., 2016). miRNAs are approximately 22-nucleotide-long small RNAs that regulate the gene expression at post-transcriptional levels by binding with target mRNAs like FMRP and Argonaute protein incorporated into the RNA-induced silencing complex (RISC) to silence their target mRNAs (Filipowicz et al., 2008; Edbauer et al., 2010; Santhekadur and Kumar, 2020). The FX-MIR is a member of an X chromosome-linked miRNA cluster initially determined in primate testis and found well conserved among placental mammals (Bentwich et al., 2005; Zhang et al., 2007; Li et al., 2010). The FX-MIR consists of 22 miRNAs, located and transcribed from five large clusters on the X chromosome and encompassing approximately 62-kb region. One of these clusters is adjacent to FMR1, which suggests the possibility that the FX-MIR miRNAs might target the FMR1 (Fromm et al., 2015; Wang et al., 2020). Most of them are primarily expressed in the human testis and brain, in which FMR1, their target, is also highly expressed in these two organs. Accordingly, the FX-MIR cluster may play a role in gene expression in both tissues since it can lead to silencing and the consequent absence of FMRP (Tamanini et al., 1997; Peier et al., 2000). Furthermore, the loss of function of FMRP demonstrated deterioration of synaptic formation and plasticity in the central nervous system and macroorchidism by overproduction of sertoli cells in the testes (Slegtenhorst-Eegdeman et al., 1998; Terracciano et al., 2005). A subset of the FX-MIR cluster has increased expression levels significantly in FXS NPCs and neurons, in which miR-509-3p and miR-890 were upregulated by approximately 78-fold and 106-fold, respectively, compared to the control. Thus, dysregulation of specific miRNAs may play a vital role in FXS (Ramaiah et al., 2019). Consistently with these findings, our proband exhibited moderate ID and ADHD, and macroorchidism, partly due to deletion of the FX-MIR cluster.
Albeit the prevalent mechanism of FXS is the presence of hypermethylation of the CGG repeat tract and the upstream CpG island in the promoter region leading to transcriptional silencing (Bell et al., 1991; Verkerk et al., 1991), several reports involving point mutations and deletions, either small or large, have been described (Sitzmann et al., 2018; Saldarriaga et al., 2020; Erbs et al., 2021). The deletions affecting the FMR1 gene account for the second most common cause of FXS, although they are rare (Coffee et al., 2009). Small deletions (<10 kb) are consistently concomitant with full mutation (>200 CGG repeats) or premutation (55-200 CGG repeats) alleles and happen during the transmission of a premutation allele from mother to offspring (Hammond et al., 1997; Han et al., 2006). Rather, large deletions (>10 kb) result in meiotic or mitotic ectopic recombination in which crossing over occurs at non-homologous loci (Coffee et al., 2008). Individuals with large deletions affecting the FMR1 gene usually display typical features of FXS, including seizure and obesity and if the deletions span over adjacent genes, they may present with additional manifestations (Moore et al., 1999).
A review of the current literature found six male individuals with de novo large deletion (>10 kb) as their mothers did not carry either a premutation or a full mutation allele, or a deleted allele and index cases did not have an expansion of CGG repeat but only large deletions harboring the FMR1 gene and contiguous DNA sequences (Wöhrle et al., 1992; Tarleton et al., 1993; Gu et al., 1994; Hirst et al., 1995; Nagamani et al., 2012; Jiraanont et al., 2016). The reported deletions encompassing the FMR1 gene range from 35 kb to 3 Mb; they all have typical features of FXS involving ID from moderate to severe and developmental delay (DD) including motor and language development at various severities. Regarding physical anomalies, four out of seven cases, including our case, had dysmorphic features including macroorchidism and shawl scrotum, and two cases presented with macrocephaly. Interestingly, three of them had vision-related problems including epicanthus inversus, esotropia, and our case presents with mild deficits in visual-related cognitive abilities. Three cases had neurodevelopmental disorders, including autistic behaviors and attention and concentration problems. This evidence strongly supports that the development of typical functions of cognition and behavior are regulated by FMR1.
Our patient manifested typical FXS characteristics with distinct facial features, macroorchidism, and moderate ID with ADHD. However, he is a well-developed physical boy with good social interaction and delay in fine motor and subtle visual deficits. To date, there are only two studies mentioning de novo FXS cases with gain and loss CNV of the ASFMR1/FMR4 gene (Nagamani et al., 2012; Vengoechea et al., 2012). Our colleagues reported a 4-year-old boy with 86-kb microduplication of the FMR1 and of the ASFMR1/FMR4 genes, together with a 363-kb duplication on 1q44 and a 168-kb deletion on 4p15.31 inherited from a healthy father. The boy presented with myoclonic seizures and later developed absence seizures, persistent speech, and fine motor delay. He also had signs and symptoms of hyperactivity (Vengoechea et al., 2012). (Nagamani et al., 2012) reported a 5-year-old boy with a deletion genomic region containing the FMR1 and the ASFMR1/FMR4 genes. This boy had relatively macrocephaly and large ears, epicanthus inversus, and bilateral trigones of the lateral ventricles. He could not speak at 4-years of age indicating significant language delay. Our case phenotypes are more like a boy with microduplication of the FMR1 and of the ASFMR1/FMR4 genes with a relatively milder phenotype, fine motor delay and ADHD although this boy did not have distinctive facial features and macroorchidism, as in our case. The author suggested that either a loss or a gain copy number of the FMR1 gene, which is tightly regulated, is essential for the normal development of neurocognitive structures and functions, and can lead to neurodevelopmental disorders (Nagamani et al., 2012). Finally, we report that the deletion observed in the proband is due to a de novo deletion, as the two probands’ sisters, the mother and maternal uncle do not have the deletion (Supplementary Figure 1). However, although unlikely, given the two probands’ normal sisters, we cannot completely exclude the possibility of germinal mosaicism in his mother.
Importantly, this study, suggested a complex transcription within the FMR1 locus, and further advanced investigation would be needed to determine the exact nature and function of all the transcripts, and their relevance to the FMR1-associated disorders. An Mb deletion encompassing the FMR1, ASFMR1/FMR4 genes, and FX-MIR cluster detected in the proband may provoke the observed clinical phenotype including intellectual disability, attention-deficit hyperactivity disorder, distinct facial features, and macroorchidism, which are typical characteristics exhibited in individuals with FXS. Importantly, we verified that the ASFMR1/FMR4 is the same genes.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Request to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by UC Davis Institutional Review Board. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FT, PJ, NT, and EM conceptualized the manuscript. FT and NT discussed and designed the study. PJ, EM, MZ, and FT conducted the methods and data analysis. PJ drafted the manuscript, and all authors edited and revised the manuscript.
Funding
This study was funded by the National Institute of Health HD024356.
Conflict of Interest
FT received funds from Azrieli Foundation and Zynerba for studies on Fragile X syndrome.
The remaining authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to this family for their willingness and enthusiasm to participate in our investigation. This article is dedicated to the memory of Matteo. We thank Hiu-Tung Tang for assistance with figures, generated using Adobe Photoshop CS2. Figure 2 was generated by Biorender, and Figure 3 was generated by MZ using Integrative Genomics Viewer (IGV) software.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.884424/full#supplementary-material
Supplementary Figure S1 | Southern blot analysis of genomic DNA isolated from a negative control (lane 1, normal female) and from a positive control (lane 5, full mutation male). Lane 2 shows lack of hybridization to the FMR1-specific probe Stb 12.3 in the proband’s DNA indicating the presence of a deletion. Lanes 2 and 3 show a normal band patter in the proband’s mother and in the maternal uncle, respectively. One-kilobyte ladder is shown in lane M. The normal unmethylated band (2.8 kb) and normal methylated band (5.2 kb) in a female individual are shown on the left side. PCR analysis using FMR1 specific primers did not yield any amplicons (data not showed).
References
Al Olaby, R. R., Tang, H.-T., Durbin-Johnson, B., Schneider, A., Hessl, D., Rivera, S. M., et al. (2018). Assessment of Molecular Measures in Non-FXTAS Male Premutation Carriers. Front. Genet. 9, 302. doi:10.3389/fgene.2018.00302
Battaglia, A., Doccini, V., Bernardini, L., Novelli, A., Loddo, S., Capalbo, A., et al. (2013). Confirmation of Chromosomal Microarray as a First-Tier Clinical Diagnostic Test for Individuals with Developmental Delay, Intellectual Disability, Autism Spectrum Disorders and Dysmorphic Features. Eur. J. Paediatric Neurol. 17, 589–599. doi:10.1016/j.ejpn.2013.04.010
Bell, M. V., Hirst, M. C., Nakahori, Y., MacKinnon, R. N., Roche, A., Flint, T. J., et al. (1991). Physical Mapping across the Fragile X: Hypermethylation and Clinical Expression of the Fragile X Syndrome. Cell 64, 861–866. doi:10.1016/0092-8674(91)90514-y
Bentwich, I., Avniel, A., Karov, Y., Aharonov, R., Gilad, S., Barad, O., et al. (2005). Identification of Hundreds of Conserved and Nonconserved Human microRNAs. Nat. Genet. 37, 766–770. doi:10.1038/ng1590
Bracken, C. P., Scott, H. S., and Goodall, G. J. (2016). A Network-Biology Perspective of microRNA Function and Dysfunction in Cancer. Nat. Rev. Genet. 17, 719–732. doi:10.1038/nrg.2016.134
Briggs, J. A., Wolvetang, E. J., Mattick, J. S., Rinn, J. L., and Barry, G. (2015). Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 88, 861–877. doi:10.1016/j.neuron.2015.09.045
Caudy, A. A., Myers, M., Hannon, G. J., and Hammond, S. M. (2002). Fragile X-Related Protein and VIG Associate with the RNA Interference Machinery. Genes Dev. 16, 2491–2496. doi:10.1101/gad.1025202
Clarke, J. T., Wilson, P. J., Morris, C. P., Hopwood, J. J., Richards, R. I., Sutherland, G. R., et al. (1992). Characterization of a Deletion at Xq27-Q28 Associated with Unbalanced Inactivation of the Nonmutant X Chromosome. Am. J. Hum. Genet. 51, 316–322.
Coffee, B., Ikeda, M., Budimirovic, D. B., Hjelm, L. N., Kaufmann, W. E., and Warren, S. T. (2008). MosaicFMR1 Deletion Causes Fragile X Syndrome and Can lead to Molecular Misdiagnosis: A Case Report and Review of the Literature. Am. J. Med. Genet. 146A, 1358–1367. doi:10.1002/ajmg.a.32261
Coffee, B., Keith, K., Albizua, I., Malone, T., Mowrey, J., Sherman, S. L., et al. (2009). Incidence of Fragile X Syndrome by Newborn Screening for Methylated FMR1 DNA. Am. J. Hum. Genet. 85, 503–514. doi:10.1016/j.ajhg.2009.09.007
DeMarco, B., Stefanovic, S., Williams, A., Moss, K. R., Anderson, B. R., Bassell, G. J., et al. (2019). FMRP - G-Quadruplex mRNA - miR-125a Interactions: Implications for miR-125a Mediated Translation Regulation of PSD-95 mRNA. PLoS One 14, e0217275. doi:10.1371/journal.pone.0217275
Dionne, O., and Corbin, F. (2021). An "Omic" Overview of Fragile X Syndrome. Biology 10, 433. doi:10.3390/biology10050433
Edbauer, D., Neilson, J. R., Foster, K. A., Wang, C.-F., Seeburg, D. P., Batterton, M. N., et al. (2010). Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs miR-125b and miR-132. Neuron 65, 373–384. doi:10.1016/j.neuron.2010.01.005
Erbs, E., Fenger-Grøn, J., Jacobsen, C. M., Lildballe, D. L., and Rasmussen, M. (2021). Spontaneous rescue of a FMR1 Repeat Expansion and Review of Deletions in the FMR1 Non-coding Region. Eur. J. Med. Genet. 64, 104244. doi:10.1016/j.ejmg.2021.104244
Fang, S., Zhang, L., Guo, J., Niu, Y., Wu, Y., Li, H., et al. (2018). NONCODEV5: a Comprehensive Annotation Database for Long Non-coding RNAs. Nucleic Acids Res. 46, D308–d314. doi:10.1093/nar/gkx1107
Fengler, S., Fuchs, S., König, R., and Arnemann, J. (2002). Mosaicism for FMR1 and FMR2 Deletion: a New Case. J. Med. Genet. 39, 200–201. doi:10.1136/jmg.39.3.200
Filipovic-Sadic, S., Sah, S., Chen, L., Krosting, J., Sekinger, E., Zhang, W., et al. (2010). A Novel FMR1 PCR Method for the Routine Detection of Low Abundance Expanded Alleles and Full Mutations in Fragile X Syndrome. Clin. Chem. 56, 399–408. doi:10.1373/clinchem.2009.136101
Filipowicz, W., Bhattacharyya, S. N., and Sonenberg, N. (2008). Mechanisms of post-transcriptional Regulation by microRNAs: Are the Answers in Sight? Nat. Rev. Genet. 9, 102–114. doi:10.1038/nrg2290
Fromm, B., Billipp, T., Peck, L. E., Johansen, M., Tarver, J. E., King, B. L., et al. (2015). A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 49, 213–242. doi:10.1146/annurev-genet-120213-092023
Fu, Y. H., Kuhl, D. P., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG Repeat at the Fragile X Site Results in Genetic Instability: Resolution of the Sherman Paradox. Cell 67, 1047–1058. doi:10.1016/0092-8674(91)90283-5
Gong, X., Wang, Y., Zeng, J., Li, S., and Luo, Y. (2015). Computational Identification and Experimental Validation of MicroRNAs Binding to the Fragile X Syndrome Gene Fmr1. Neurochem. Res. 40, 109–117. doi:10.1007/s11064-014-1471-3
Greenberg, D., and Soreq, H. (2014). MicroRNA Therapeutics in Neurological Disease. Cpd 20, 6022–6027. doi:10.2174/1381612820666140314151924
Gu, Y., Lugenbeel, K. A., Vockley, J. G., Grody, W. W., and Nelson, D. L. (1994). A De Novo Deletion in FMR1 in a Patient with Developmental Delay. Hum. Mol. Genet. 3, 1705–1706. doi:10.1093/hmg/3.9.1705
Hagerman, R. J., Berry-Kravis, E., Kaufmann, W. E., Ono, M. Y., Tartaglia, N., Lachiewicz, A., et al. (2009). Advances in the Treatment of Fragile X Syndrome. Pediatrics 123, 378–390. doi:10.1542/peds.2008-0317
Hagerman, R. J., and Hagerman, P. J. (2002). The Fragile X Premutation: into the Phenotypic Fold. Curr. Opin. Genet. Develop. 12, 278–283. doi:10.1016/s0959-437x(02)00299-x
Hammond, L. S., Macias, M. M., Tarleton, J. C., and Pai, G. S. (1997). Fragile X Syndrome and Deletions in FMR1: New Case and Review of the Literature. Am. J. Med. Genet. 72, 430–434. doi:10.1002/(sici)1096-8628(19971112)72:4<430:aid-ajmg11>3.0.co;2-s
Han, X. D., Powell, B. R., Phalin, J. L., and Chehab, F. F. (2006). Mosaicism for a Full Mutation, Premutation, and Deletion of the CGG Repeats Results in 22% FMRP and Elevated FMR1 mRNA Levels in a High-Functioning Fragile X Male. Am. J. Med. Genet. A. 140, 1463–1471. doi:10.1002/ajmg.a.31291
Handt, M., Epplen, A., Hoffjan, S., Mese, K., Epplen, J. T., and Dekomien, G. (2014). Point Mutation Frequency in the FMR1 Gene as Revealed by Fragile X Syndrome Screening. Mol. Cell Probes 28, 279–283. doi:10.1016/j.mcp.2014.08.003
Hatton, D. D., Hooper, S. R., Bailey, D. B., Skinner, M. L., Sullivan, K. M., and Wheeler, A. (2002). Problem Behavior in Boys with Fragile X Syndrome. Am. J. Med. Genet. 108, 105–116. doi:10.1002/ajmg.10216
Hirst, M., Grewal, P., Flannery, A., Slatter, R., Maher, E., Barton, D., et al. (1995). Two New Cases of FMR1 Deletion Associated with Mental Impairment. Am. J. Hum. Genet. 56, 67–74.
Hung, T., Wang, Y., Lin, M. F., Koegel, A. K., Kotake, Y., Grant, G. D., et al. (2011). Extensive and Coordinated Transcription of Noncoding RNAs within Cell-Cycle Promoters. Nat. Genet. 43, 621–629. doi:10.1038/ng.848
Ishizuka, A., Siomi, M. C., and Siomi, H. (2002). A Drosophila Fragile X Protein Interacts with Components of RNAi and Ribosomal Proteins. Genes Dev. 16, 2497–2508. doi:10.1101/gad.1022002
Jin, P., Zarnescu, D. C., Ceman, S., Nakamoto, M., Mowrey, J., Jongens, T. A., et al. (2004). Biochemical and Genetic Interaction between the Fragile X Mental Retardation Protein and the microRNA Pathway. Nat. Neurosci. 7, 113–117. doi:10.1038/nn1174
Jiraanont, P., Hagerman, R. J., Neri, G., Zollino, M., Murdolo, M., and Tassone, F. (2016). Germinal Mosaicism for a Deletion of the FMR1 Gene Leading to Fragile X Syndrome. Eur. J. Med. Genet. 59, 459–462. doi:10.1016/j.ejmg.2016.08.009
Jorge, P., Garcia, E., Gonçalves, A., Marques, I., Maia, N., Rodrigues, B., et al. (2018). Classical Fragile-X Phenotype in a Female Infant Disclosed by Comprehensive Genomic Studies. BMC Med. Genet. 19, 74. doi:10.1186/s12881-018-0589-6
Khalil, A. M., Faghihi, M. A., Modarresi, F., Brothers, S. P., and Wahlestedt, C. (2008). A Novel RNA Transcript with Antiapoptotic Function Is Silenced in Fragile X Syndrome. PLoS One 3, e1486. doi:10.1371/journal.pone.0001486
Khazaie, Y., and Nasr Esfahani, M. H. (2014). MicroRNA and Male Infertility: A Potential for Diagnosis. Int. J. Fertil. Steril 8, 113–118.
Kidd, S. A., Lachiewicz, A., Barbouth, D., Blitz, R. K., Delahunty, C., McBrien, D., et al. (2014). Fragile X Syndrome: a Review of Associated Medical Problems. Pediatrics 134, 995–1005. doi:10.1542/peds.2013-4301
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., et al. (2011). Long Non-coding RNA ANRIL Is Required for the PRC2 Recruitment to and Silencing of p15INK4B Tumor Suppressor Gene. Oncogene 30, 1956–1962. doi:10.1038/onc.2010.568
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., et al. (2007). An Antisense Transcript Spanning the CGG Repeat Region of FMR1 Is Upregulated in Premutation Carriers but Silenced in Full Mutation Individuals. Hum. Mol. Genet. 16, 3174–3187. doi:10.1093/hmg/ddm293
Li, J., Liu, Y., Dong, D., and Zhang, Z. (2010). Evolution of an X-Linked Primate-specific Micro RNA Cluster. Mol. Biol. Evol. 27, 671–683. doi:10.1093/molbev/msp284
Loesch, D. Z., Godler, D. E., Evans, A., Bui, Q. M., Gehling, F., Kotschet, K. E., et al. (2011). Evidence for the Toxicity of Bidirectional Transcripts and Mitochondrial Dysfunction in Blood Associated with Small CGG Expansions in the FMR1 Gene in Patients with Parkinsonism. Genet. Med. 13, 392–399. doi:10.1097/GIM.0b013e3182064362
Luo, S., Huang, W., Xia, Q., Xia, Y., Du, Q., Wu, L., et al. (2014). Cryptic FMR1 Mosaic Deletion in a Phenotypically normal Mother of a Boy with Fragile X Syndrome: Case Report. BMC Med. Genet. 15, 125. doi:10.1186/s12881-014-0125-2
Mercer, T. R., Qureshi, I. A., Gokhan, S., Dinger, M. E., Li, G., Mattick, J. S., et al. (2010). Long Noncoding RNAs in Neuronal-Glial Fate Specification and Oligodendrocyte Lineage Maturation. BMC Neurosci. 11, 14. doi:10.1186/1471-2202-11-14
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 86, 749–764. doi:10.1016/j.ajhg.2010.04.006
Monaghan, K. G., Lyon, E., and Spector, E. B. (2013). ACMG Standards and Guidelines for Fragile X Testing: a Revision to the Disease-specific Supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet. Med. 15, 575–586. doi:10.1038/gim.2013.61
Moore, S. J., Strain, L., Cole, G. F., Miedzybrodzka, Z., Kelly, K. F., and Dean, J. C. (1999). Fragile X Syndrome with FMR1 and FMR2 Deletion. J. Med. Genet. 36, 565–566. doi:10.1136/jmg.36.7.565
Myers, K. A., van 't Hof, F. N. G., Sadleir, L. G., Legault, G., Simard-Tremblay, E., Amor, D. J., et al. (2019). Fragile Females: Case Series of Epilepsy in Girls with FMR1 Disruption. Pediatrics 144, e20190599. doi:10.1542/peds.2019-0599
Nagamani, S. C. S., Erez, A., Probst, F. J., Bader, P., Evans, P., Baker, L. A., et al. (2012). Small Genomic Rearrangements Involving FMR1 Support the Importance of its Gene Dosage for normal Neurocognitive Function. Neurogenetics 13, 333–339. doi:10.1007/s10048-012-0340-y
Pastori, C., Kapranov, P., Penas, C., Peschansky, V., Volmar, C.-H., Sarkaria, J. N., et al. (2015). The Bromodomain Protein BRD4 Controls HOTAIR, a Long Noncoding RNA Essential for Glioblastoma Proliferation. Proc. Natl. Acad. Sci. U.S.A. 112, 8326–8331. doi:10.1073/pnas.1424220112
Pastori, C., Peschansky, V. J., Barbouth, D., Mehta, A., Silva, J. P., and Wahlestedt, C. (2014). Comprehensive Analysis of the Transcriptional Landscape of the Human FMR1 Gene Reveals Two New Long Noncoding RNAs Differentially Expressed in Fragile X Syndrome and Fragile X-Associated Tremor/ataxia Syndrome. Hum. Genet. 133, 59–67. doi:10.1007/s00439-013-1356-6
Peier, A. M., McIlwain, K. L., Kenneson, A., Warren, S. T., Paylor, R., Nelson, D. L., et al. (2000). (Over)Correction of FMR1 Deficiency With YAC Transgenics: Behavioral and Physical Features. Hum. Mol. Genet. 9 (8), 1145–1159. doi:10.1093/hmg/9.8.1145
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The Pathophysiology of Fragile X Syndrome. Annu. Rev. Genom. Hum. Genet. 8, 109–129. doi:10.1146/annurev.genom.8.080706.092249
Perry, R. B.-T., Hezroni, H., Goldrich, M. J., and Ulitsky, I. (2018). Regulation of Neuroregeneration by Long Noncoding RNAs. Mol. Cel 72, 553–567. e555. doi:10.1016/j.molcel.2018.09.021
Peschansky, V. J., Pastori, C., Zeier, Z., Motti, D., Wentzel, K., Velmeshev, D., et al. (2015). Changes in Expression of the Long Non-coding RNA FMR4 Associate with Altered Gene Expression during Differentiation of Human Neural Precursor Cells. Front. Genet. 6, 263. doi:10.3389/fgene.2015.00263
Peschansky, V. J., Pastori, C., Zeier, Z., Wentzel, K., Velmeshev, D., Magistri, M., et al. (2016). The Long Non-coding RNA FMR4 Promotes Proliferation of Human Neural Precursor Cells and Epigenetic Regulation of Gene Expression in Trans. Mol. Cell Neurosci. 74, 49–57. doi:10.1016/j.mcn.2016.03.008
Petek, E., Kroisel, P. M., Schuster, M., Zierler, H., and Wagner, K. (1999). Mosaicism in a Fragile X Male Including a De Novo Deletion in theFMR1 Gene. Am. J. Med. Genet. 84, 229–232. doi:10.1002/(sici)1096-8628(19990528)84:3<229:aid-ajmg13>3.0.co;2-t
Probst, F. J., Roeder, E. R., Enciso, V. B., Ou, Z., Cooper, M. L., Eng, P., et al. (2007). Chromosomal Microarray Analysis (CMA) Detects a Large X Chromosome Deletion includingFMR1,FMR2, andIDS in a Female Patient with Mental Retardation. Am. J. Med. Genet. 143A, 1358–1365. doi:10.1002/ajmg.a.31781
Quartier, A., Poquet, H., Gilbert-Dussardier, B., Rossi, M., Casteleyn, A.-S., Portes, V. d., et al. (2017). Intragenic FMR1 Disease-Causing Variants: a Significant Mutational Mechanism Leading to Fragile-X Syndrome. Eur. J. Hum. Genet. 25, 423–431. doi:10.1038/ejhg.2016.204
Qureshi, I. A., Mattick, J. S., and Mehler, M. F. (2010). Long Non-coding RNAs in Nervous System Function and Disease. Brain Res. 1338, 20–35. doi:10.1016/j.brainres.2010.03.110
Ramaiah, M., Tan, K., Plank, T. D. M., Song, H. W., Dumdie, J. N., Jones, S., et al. (2019). A Micro RNA Cluster in the Fragile‐X Region Expressed during Spermatogenesis Targets FMR 1. EMBO Rep. 20. doi:10.15252/embr.201846566
Reiss, A. L., and Dant, C. C. (2003). The Behavioral Neurogenetics of Fragile X Syndrome: Analyzing Gene-Brain-Behavior Relationships in Child Developmental Psychopathologies. Dev. Psychopathol. 15, 927–968. doi:10.1017/s0954579403000464
Rogers, S. J., Wehner, E. A., and Hagerman, R. (2001). The Behavioral Phenotype in Fragile X: Symptoms of Autism in Very Young Children with Fragile X Syndrome, Idiopathic Autism, and Other Developmental Disorders. J. Develop. Behav. Pediatr. 22, 409–417. doi:10.1097/00004703-200112000-00008
Saldarriaga, W., Payán-Gómez, C., González-Teshima, L. Y., Rosa, L., Tassone, F., and Hagerman, R. J. (2020). Double Genetic Hit: Fragile X Syndrome and Partial Deletion of Protein Patched Homolog 1 Antisense as Cause of Severe Autism Spectrum Disorder. J. Dev. Behav. Pediatr. 41, 724–728. doi:10.1097/dbp.0000000000000850
Santhekadur, P. K., and Kumar, D. P. (2020). RISC Assembly and post-transcriptional Gene Regulation in Hepatocellular Carcinoma. Genes Dis. 7, 199–204. doi:10.1016/j.gendis.2019.09.009
Sitzmann, A. F., Hagelstrom, R. T., Tassone, F., Hagerman, R. J., and Butler, M. G. (2018). Rare FMR1 Gene Mutations Causing Fragile X Syndrome: A Review. Am. J. Med. Genet. 176, 11–18. doi:10.1002/ajmg.a.38504
Slegtenhorst-Eegdeman, K. E., de Rooij, D. G., Verhoef-Post, M., van de Kant, H. J. G., Bakker, C. E., Oostra, B. A., et al. (1998). Macroorchidism in FMR1 Knockout Mice Is Caused by Increased Sertoli Cell Proliferation during Testicular Development*. Endocrinology 139, 156–162. doi:10.1210/endo.139.1.5706
Smalheiser, N. R., and Lugli, G. (2009). microRNA Regulation of Synaptic Plasticity. Neuromol Med. 11, 133–140. doi:10.1007/s12017-009-8065-2
Statello, L., Guo, C. J., Chen, L. L., and Huarte, M. (2021). Gene Regulation by Long Non-coding RNAs and its Biological Functions. Nat. Rev. Mol. Cel Biol. 22, 96–118. doi:10.1038/s41580-020-00315-9
Symons, F. J., Byiers, B. J., Raspa, M., Bishop, E., and Bailey, D. B. (2010). Self-injurious Behavior and Fragile X Syndrome: Findings from the National Fragile X Survey. Am. J. Intellect. Dev. Disabil. 115, 473–481. doi:10.1352/1944-7558-115.6.473
Tamanini, F., Willemsen, R., van Unen, L., Bontekoe, C., Galjaard, H., Oostra, B. A., et al. (1997). Differential Expression of FMR1, FXR1 and FXR2 Proteins in Human Brain and Testis. Hum. Mol. Genet. 6 (8), 1315–1322. doi:10.1093/hmg/6.8.1315
Tarleton, J., Richle, R., Schwarsz, C., Rao, K., Aylsworth, A. S., and Lachlewicz, A. (1993). An Extensive De Novo Deletion Removing FMR1 in a Patient with Mental Retardation and the Fragile X Syndrome Phenotype. Hum. Mol. Genet. 2, 1973–1974. doi:10.1093/hmg/2.11.1973
Tassone, F., Hagerman, R. J., Chamberlain, W. D., and Hagerman, P. J. (2000). Transcription of the FMR1 Gene in Individuals with Fragile X Syndrome. Am. J. Med. Genet. 97, 195–203. doi:10.1002/1096-8628(200023)97:3<195:AID-AJMG1037>3.0.CO;2-R
Tassone, F., Pan, R., Amiri, K., Taylor, A. K., and Hagerman, P. J. (2008). A Rapid Polymerase Chain Reaction-Based Screening Method for Identification of All Expanded Alleles of the Fragile X (FMR1) Gene in Newborn and High-Risk Populations. J. Mol. Diagn. 10, 43–49. doi:10.2353/jmoldx.2008.070073
Terracciano, A., Chiurazzi, P., and Neri, G. (2005). Fragile X Syndrome. Am. J. Med. Genet. 137c, 32–37. doi:10.1002/ajmg.c.30062
Tsai, M.-C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 329, 689–693. doi:10.1126/science.1192002
Vengoechea, J., Parikh, A. S., Zhang, S., and Tassone, F. (2012). De Novo microduplication of the FMR1 Gene in a Patient with Developmental Delay, Epilepsy and Hyperactivity. Eur. J. Hum. Genet. 20, 1197–1200. doi:10.1038/ejhg.2012.78
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 65, 905–914. doi:10.1016/0092-8674(91)90397-h
Vittal, P., Pandya, S., Sharp, K., Berry-Kravis, E., Zhou, L., Ouyang, B., et al. (2018). ASFMR1splice Variant. Neurol. Genet. 4, e246. doi:10.1212/NXG.0000000000000246
Wang, Z., Xie, Y., Wang, Y., Morris, D., Wang, S., Oliver, D., et al. (2020). X‐linked miR‐506 Family miRNAs Promote FMRP Expression in Mouse Spermatogonia. EMBO Rep. 21, e49024. doi:10.15252/embr.201949024
Wei, C.-W., Luo, T., Zou, S.-S., and Wu, A.-S. (2018). The Role of Long Noncoding RNAs in Central Nervous System and Neurodegenerative Diseases. Front. Behav. Neurosci. 12, 175. doi:10.3389/fnbeh.2018.00175
Wöhrle, D., Kotzot, M. C., Hirst, A., Manca, B., Korn, A., Schmidt, G., et al. (1992). A Microdeletion of Less Than 250 Kb, Including the Proximal Part of the FMR-I Gene and the Fragile-X Site, in a Male with the Clinical Phenotype of Fragile-X Syndrome. Am. J. Hum. Genet. 51 (2), 299–306.
Wolff, D. J., Gustashaw, K. M., Zurcher, V., Ko, L., White, W., Weiss, L., et al. (1997). Deletions in Xq26.3-q27.3 Including FMR1 Result in a Severe Phenotype in a Male and Variable Phenotypes in Females Depending upon the X Inactivation Pattern. Hum. Genet. 100, 256–262. doi:10.1007/s004390050501
Yakovlev, D. A., Kuznetsova, A. A., Fedorova, O. S., and Kuznetsov, N. A. (2017). Search for Modified DNA Sites with the Human Methyl-CpG-Binding Enzyme MBD4. Acta Naturae 9, 88–98. doi:10.32607/20758251-2017-9-1-88-98
Yu, S., Pritchard, M., Kremer, E., Lynch, M., Nancarrow, J., Baker, E., et al. (1991). Fragile X Genotype Characterized by an Unstable Region of DNA. Science 252, 1179–1181. doi:10.1126/science.252.5009.1179
Zafarullah, M., Tang, H.-T., Durbin-Johnson, B., Fourie, E., Hessl, D., Rivera, S. M., et al. (2020). FMR1 Locus Isoforms: Potential Biomarker Candidates in Fragile X-Associated Tremor/ataxia Syndrome (FXTAS). Sci. Rep. 10, 11099. doi:10.1038/s41598-020-67946-y
Zhang, R., Peng, Y., Wang, W., and Su, B. (2007). Rapid Evolution of an X-Linked microRNA Cluster in Primates. Genome Res. 17, 612–617. doi:10.1101/gr.6146507
Keywords: fragile X syndrome, FMR1 gene, miRNA, ASFMR1/FMR4 gene, large deletion
Citation: Jiraanont P, Manor E, Tabatadze N, Zafarullah M, Mendoza G, Melikishvili G and Tassone F (2022) De Novo Large Deletion Leading to Fragile X Syndrome. Front. Genet. 13:884424. doi: 10.3389/fgene.2022.884424
Received: 26 February 2022; Accepted: 13 April 2022;
Published: 11 May 2022.
Edited by:
Corrado Romano, University of Catania, ItalyReviewed by:
Daniele Di Marino, Polytechnic University of Marche, ItalyDaman Kumari, National Institutes of Health, United States
Copyright © 2022 Jiraanont, Manor, Tabatadze, Zafarullah, Mendoza, Melikishvili and Tassone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flora Tassone, ZnRhc3NvbmVAdWNkYXZpcy5lZHU=