 Jiale Xiang
Jiale Xiang Hongfu Zhang
Hongfu Zhang Xiangzhong Sun2
Xiangzhong Sun2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 29 April 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.883617
This article is part of the Research Topic Advancing Whole-Genome Sequencing (WGS) in Clinical Genetic Testing for Human Diseases View all 4 articles
Background: Hearing loss affects approximately two out of every 1,000 newborns. Genetic factors and congenital cytomegalovirus (CMV) infections account for around 90% of the etiology. The purpose of this study was to develop and test a whole genome sequencing (WGS) approach to detect deafness-related genetic variants and CMV infections simultaneously in newborns.
Method: Deafness-related genes causing congenital or childhood hearing loss were curated and selected for newborn screening. Nine dried blood spots from newborns with known genetic variants (n = 6) or CMV infections (n = 3) were employed to develop and validate the WGS testing and analytic pipeline. We then pilot tested the WGS analysis on 51 de-identified clinical samples.
Results: 92 gene-disease pairs were selected for screening hearing loss in newborns. In the validation test, WGS accurately detected all types of genetic variants, including single nucleotide variations, insertions/deletions, and copy number variations in the nuclear or mitochondrial genome. Sequence reads mapping to the CMV reference genome were discovered in CMV infected samples. In the pilot test, WGS identified nine out of 51 (18%) newborns carrying pathogenic variants associated with deafness.
Conclusion: WGS can simultaneously detect genetic variants and CMV infections in dried blood spot specimens from newborns. Our study provides proof of principle that genome sequencing can be a promising alternative for newborn screening of hearing loss.
Hearing loss has an estimated incidence of 0.1–0.3% in newborns, and its causes vary (Korver et al., 2017). Genetic factors are regarded as the primary contributors, accounting for 68% of the causes (Morton and Nance, 2006). Among the genetic factors, variants in both the nuclear and the mitochondrial genome can cause hereditary hearing loss. To date, over 150 genes have been linked with hearing loss in human, and novel associations are still being discovered every year (Azaiez et al., 2018).
Universal hearing screening in newborns has been implemented since the 1990s and has proven remarkably successful for the early identification of neonates with hearing loss, leading to interventions that have improved language development and social interaction (White et al., 2010). However, conventional newborns hearing screening (NHS) is incapable of identifying mild, delayed-onset/progressive or aminoglycoside-induced hearing loss. More importantly, it is unable to uncover the etiology of deafness (Kennedy et al., 2005).
Environmental factors are also important contributors. Congenital cytomegalovirus (CMV) infection is the most prevalent one, accounting for 21% of the prelingual hearing loss (Morton and Nance, 2006). With the advance of molecular technologies, screens for CMV or genetic variants at birth were conducted (Wang et al., 2019; Yamamoto et al., 2019). Newborn CMV screens use a polymerase chain reaction assay that can identify congenitally infected newborns, detecting those who pass auditory screens but would later develop hearing loss (Fowler et al., 2017; Yamamoto et al., 2019). Limited genetic screens using MALDI-TOF mass spectrometry or DNA microarray platforms were clinically adopted in China and were proven to compensate for the limitations of auditory screens by elucidating genetic etiologies and detecting infants who harbored mitochondrial variants and were at risk of aminoglycoside-induced ototoxicity (Dai et al., 2019; Wang et al., 2019; Guo et al., 2020). The feasibility of concurrent genetic and CMV screening in newborns was recently evaluated, and it was shown that the integration of these screening methods could identify the neonates with hearing loss missed by NHS (Lu et al., 2018).
Whole genome sequencing (WGS) is an attractive approach for NHS (Shearer et al., 2019). WGS can detect a wide range of types of genomic variation, including single nucleotide variations (SNV), small insertions and deletions (INDEL), and copy number variations (CNV) (Lindstrand et al., 2019). These variant types are common in genetic hearing loss (Shearer et al., 2014; Sloan-Heggen et al., 2016). In addition, WGS can detect mitochondrial variants which are associated with a predisposition to aminoglycoside ototoxicity and/or late-onset hearing loss (Usami and Nishio, 2004). Moreover, since the viral DNA of CMV is co-isolated with human genomic DNA from blood, WGS could theoretically detect CMV in dried blood specimens (Shearer et al., 2019). However, this approach has not yet been comprehensively evaluated.
In this study, we developed and validated an analytic pipeline using WGS to identify genetic variants and human CMV infections. We then pilot tested this pipeline on 51 de-identified clinical samples. Our study provides proof of principle that genome sequencing can be a promising alternative for newborn screening of hearing loss.
This study was performed with the approval of the Institutional Review Board of BGI (IRB-20016-T2).
Deafness-related genes were curated and selected for WGS analysis in newborn screening. Nine dried blood spot samples were used to develop and validate the WGS analytic pipeline. These six positive samples with known deafness-related genetic variants were specifically selected, including compound heterozygous variants in SLC26A4 (n = 1), compound heterozygous variants in GJB2 (n = 1), homozygous variants in MYO7A (n = 1), a 3.7 Mb CNV (n = 1), and mitochondrial variants in MT-RNR1 (n = 2). The selected samples covered all the variant types associated with hearing loss, including single nucleotide variations, insertions/deletions, and copy number variations, in heterozygous or homozygous statue. Homoplasmy and heteroplasmy of the mitochondrial variants were also covered. The three positive samples with CMV infections were specifically selected with a different level of viral loads. Following this validation analysis, we pilot tested the validated pipeline on samples from 51 newborns. All the dried blood spot samples were collected by heel prick form 3-day-old babies and routinely stored at room temperature.
A total of 174 gene-disease pairs and their associations were retrieved from clinical testing panels in the Genetic Testing Registry that had been curated by the ClinGen Hearing Loss Working Group (DiStefano et al., 2020). The exclusion criteria are: 1) the gene-disease association is lower than “strong” (Ceyhan-Birsoy et al., 2017); 2) hearing loss is not a presenting feature of the disease (DiStefano et al., 2019); or 3) the age of onset is greater than 18 years (Ceyhan-Birsoy et al., 2017). The youngest age at which individuals with pathogenic variants in a gene presented with disease was curated from the OMIM database. If the information was not available in the OMIM database, we integrated age of onset from the literature. Finally, two mitochondrial genes (MT-RNR1 and MT-TS1) were added because MT-RNR1 is definitively associated with maternally-inherited, aminoglycoside-induced hearing loss and MT-TS1 is definitively associated with mitochondrial nonsyndromic hearing loss (DiStefano et al., 2019). Newborns who harbor mitochondrial variants generally pass the auditory screens because they are asymptomatic before they receive external stimulation such as aminoglycoside drugs (Wang et al., 2019).
DNA was extracted from dried blood spots using a QIAamp DNA Mini Kit (QIAGEN, Germany). Briefly, two dried blood spots were punched into a 1.5 ml Eppendorf tube with 360 μl Buffer ATL and incubated at 85°C for 10 min. A total of 40 μl proteinase K was added with incubation at 56°C for 60 min, followed by addition of 400 μl Buffer AL with incubation at 70°C for 10 min. Then, 400 μl ethanol was added and the sample was mixed by pulse-vortexing. The mixture was then transferred to a QIAamp Mini spin column and centrifuged at 6,000 × g for 1 min. 500 μl Buffer AW1 was added, followed by centrifuging at 8,000 × g for 1 min, and then 500 μl Buffer AW2 was added and the samples were centrifuged at 20,000 × g for 3 min. The QIAamp Mini spin column was placed in a clean 1.5 ml microcentrifuge tube with 80 µl Buffer TE and incubated at room temperature for 1 min. Finally, the tube was centrifuged at 12,000 × g for 1 min to collect the DNA.
The genomic DNA was fragmented using covaris S220 (Covaris, Brighton, UK). DNA fragments between 100 bp and 300 bp were selected using AMPure XP beads (AGENCOURT). The selected DNA fragments were then repaired to obtain blunt ends and modified at the 3’ end to add dATP as a sticky end. A dTTP-tailed adapter sequence was ligated to both ends of the DNA fragments. The ligation product was heat-denatured together with splint oligos that were reverse-complementary to a specific strand of the product, and the single-strand molecule was ligated using DNA ligase. The remaining linear molecule was digested with exonuclease, obtaining a single-strand circular DNA library. The library was subjected to rolling circle amplification to generate DNA nanospheres. Whole-genome sequencing was then performed on an MGISEQ-2000 with 100-bp paired-end fragments (Figure 1).
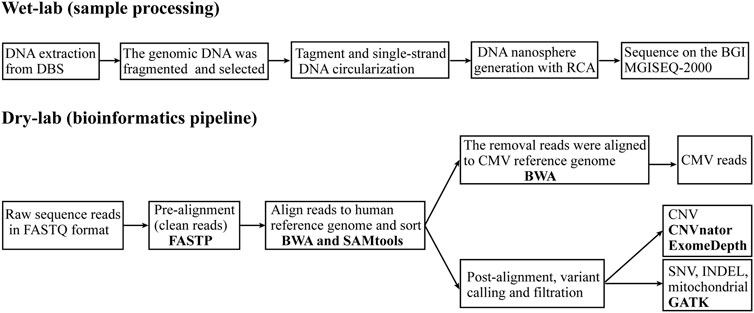
FIGURE 1. Overview of the whole genome sequencing and analytics pipeline. The WGS pipeline includes two sections: wet-lab (sample processing), which starts with DNA extraction from dried blood spots (DBS) and continues to generate a whole genome library that is sequenced on an MGISEQ-2000 sequencer; and dry-lab (bioinformatics pipeline), which starts with raw sequence reads in FASTQ format and continues with analysis using a series of software tools to identify genetic variants and reads from CMV. RCA, rolling circle amplification; BWA, Burrows-Wheeler Aligner; GATK, Genome Analysis Toolkit; CNV, copy number variant; SNV, single nucleotide variant; INDEL, small insertions and deletion; CMV, cytomegalovirus.
The WGS data were analyzed using our in-house WGS analysis pipeline following Genome Analysis Toolkit (GATK) best practices (DePristo et al., 2011). Briefly, clean reads were generated from the raw sequence reads in FASTQ format after quality control with FASTP (Chen et al., 2018). These were aligned to the human nuclear reference genome (GRCh37/hg19) and mitochondrial reference genome (GenBank:NC_012920) with the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009) and then sorted with SAMtools to create binary alignment map (BAM) files (Li et al., 2009). Unmatched reads were mapped to the cytomegalovirus reference genome (NCBI Accession NC_006273.2) with BWA. CNV were analyzed using CNVnator (Abyzov et al., 2011) and ExomeDepth (Plagnol et al., 2012). The GATK was used to call SNV, INDEL and mitochondrial variants (Van der Auwera et al., 2013). The GATK Depth of Coverage tool was employed to obtain coverage statistics of genomic positions. The “well-coverage” category was defined as 20x coverage for nuclear variants (Sheppard et al., 2018) and 2000x coverage for mitochondrial variants (Schon et al., 2020). Using the ANNOVAR bioinformatics toolset to functionally annotate the genetic variants (Yang and Wang, 2015). Sequence variants were interpreted based on the expert specifications of variant interpretation guidelines for genetic hearing loss (Oza et al., 2018).
A total of 174 gene-disease pairs were retrieved from the ClinGen Hearing Loss Expert Panel. Of these, 74 were excluded because their associations were lower than strong. Eight gene-disease pairs were excluded because hearing loss is not a presenting feature of the disease: namely ALMS1/alstrom syndrome, ATP6V1B1/DRTA2, BTD/biotinidase deficiency, COL4A5/alport syndrome, GJB3/erythrokeratodermia variabilis, DSPP/dentinogenesis imperfecta, GJB6/clouston syndrome, and TCOF1/treacher-collins syndrome. Two gene-disease pairs (COCH/DFNA9 and DNMT1/ADCADN) characterized by progressive sensorineural hearing loss were excluded because of the disease’s age of onset. The age of onset for DFNA9 is approximate 20 years old (Robertson et al., 2006). The age of onset for ADCADN is usually from thirties to forties (Winkelmann et al., 2012). Two mitochondrial genes (MT-RNR1 and MT-TS1) were manually added. MT-RNR1 is definitively associated with maternally inherited, aminoglycoside-induced hearing loss, and MT-TS1 is definitively associated with mitochondrial nonsyndromic hearing loss. Both gene-disease pairs were previously approved by the ClinGen Hearing Loss Expert Panel but were not listed on the ClinGen website (DiStefano et al., 2019).
Based on this, a total of 84 genes (92 gene-disease pairs) were finally included in the WGS analytic pipeline for screening hearing loss in newborns. The 84 genes included some of the genes most strongly linked with deafness and recommended for newborn screening, namely GJB2, STRC, SLC26A4, MYO7A, TECTA, MYO15A, CDH23, USH2A, ADGRV1, WFS1, TMPRSS3, OTOF, and TMC1 (Shearer et al., 2019).
Of these, 48 gene-disease pairs were associated with nonsyndromic hearing loss and 44 with syndromic hearing loss (Figure 2 and Supplementary Table S1). Autosomal recessive, autosomal dominant, X-linked and mitochondrial inheritance patterns accounted for 61 (66%), 24 (26%), 5 (6%), and 2 (2%) of the diseases, respectively.
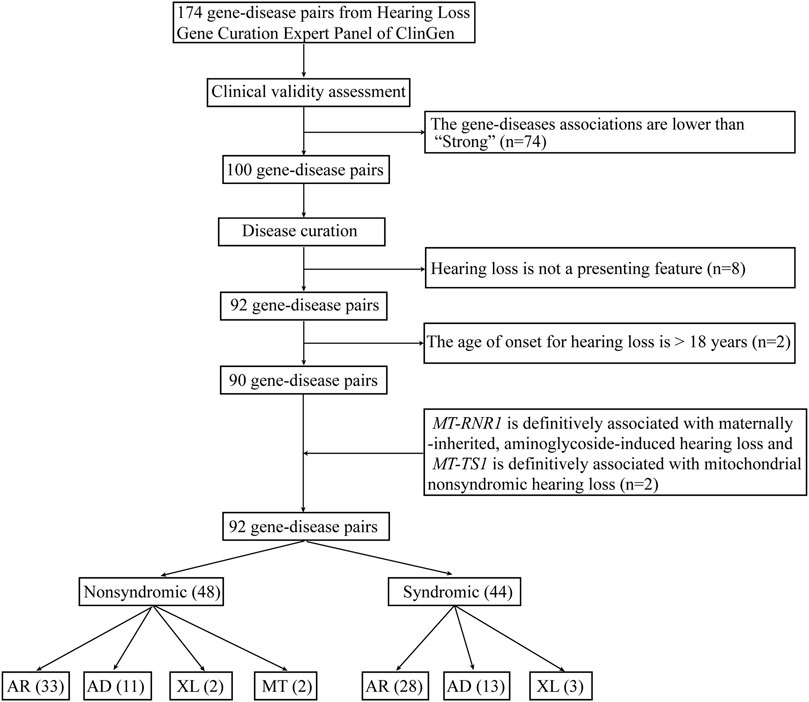
FIGURE 2. The workflow of gene curation for hearing loss. AR, autosomal recessive; AD, autosomal dominant; XL, X-linked; MT, mitochondrial.
To develop and validate a WGS pipeline for detecting genetic variants and CMV infections, WGS data from nine samples with known causes of deafness were analyzed. Overall, our sequencing produced a median of 99.54% mappable bases with coverage of at least 20X in the human nuclear genome and 98.92% mappable bases with coverage of at least 2000X in the mitochondrial genome (Supplementary Table S2). These metrics demonstrated that WGS data was of high quality and could be used to detect both nuclear and mitochondrial variants.
Most of the 84 deafness-related genes in our study well covered, with coverage of 90% to nearly 100% of the corresponding gene regions. Only two genes had less than 90% of their regions covered at 20X. They were OTOA (85.82%) and STRC (64.42%) (Figure 3). These two genes have homologous pseudogenes that are nearly identical to their sequence, explaining the low coverage in our sequencing data.
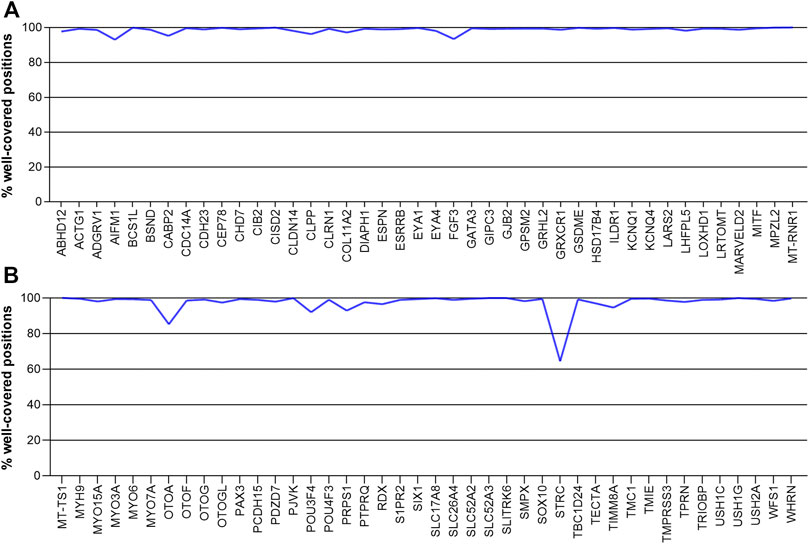
FIGURE 3. The coverage of deafness-related genes in our sequencing, showing the percentage of gene positions that are well covered. Genes are arranged in alphabetical order. (A) ABHD12- MT-RNR1 (B) MT-TS1-WHRN.
WGS identified all of the types of known variants associated with hearing loss in the six samples, including single nucleotide variations, insertions/deletions, and copy number variations in the nuclear or mitochondrial genome. More specifically, samples HL-V1, HL-V2 and HL-V3 had heterozygous or homozygous single nucleotide variations and insertions/deletions, while a chromosomal deletion (chr17:g.16670884-20391194, 3.7 Mb) leading to Smith-Magenis syndrome was found in HL-V4. In HL-V5 and HL-V6, homoplasmy and heteroplasmy of the mitochondrial variant m.1555A>G in the MT-RNR1 gene was detected (Table 1).
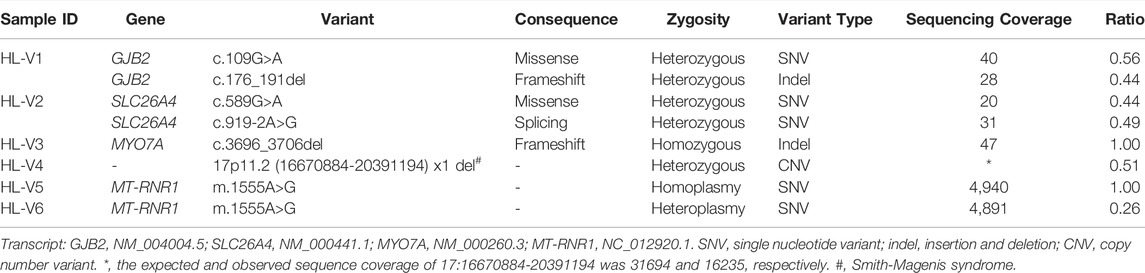
TABLE 1. The results of whole genome sequencing for six samples with known genetic variation.
The other three samples (HL-V7, HL-V8 and HL-V9) were known to have CMV infections. The viral load, confirmed by qPCR, differed between the samples. Sequencing reads mapping to CMV reference genome increased substantially with the increase in viral load, with 4,551, 28,924, and 20,876 reads in the samples, respectively (Table 2).
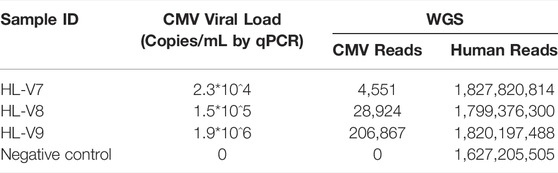
TABLE 2. The results of WGS for three samples with known human CMV infection.
The WGS analytic pipeline was pilot tested on 51 de-identified dried blood spots from newborns. Of these, nine (18%) were found to carry pathogenic variants in one of the 84 deafness-related genes (Supplementary Table S3). NM_004004.5:c.109G > A in the GJB2 gene was detected in five individuals and was the most prevalent in the population. CMV infection was not detected in any of these 51 samples. All pathogenic variants detected by WGS in the pilot study were validated by Sanger sequencing (Supplementary Figure S1).
To facilitate the integration of WGS into newborn screening, we curated and selected 84 deafness-related genes (92 gene-disease pairs) using criteria established by expert groups (Ceyhan-Birsoy et al., 2017; DiStefano et al., 2019). We then validated our WGS analytics pipeline in samples harboring the most common types of deafness-related variants in nuclear and mitochondrial genomes. We revealed that WGS can not only detect these variants, but also detect CMV infection in specimens with different viral loads.
Genetic factors and congenital CMV infection are two major causes of congenital and childhood hearing loss (Shearer et al., 2019). A comprehensive newborn hearing screen that includes physiologic, genetic, and CMV screening would have multiple clinical benefits for newborns (Shearer et al., 2019). Incorporating limited genetic screening into an auditory screening has been widely adopted in large population screens in China (Dai et al., 2019; Wang et al., 2019; Guo et al., 2020). However, CMV screening in newborns has been less well investigated. Recently, Yamamoto and co-authors reported a prospective study of CMV screening in 11900 newborns in Brazil (Yamamoto et al., 2019). These studies demonstrated that either genetic or CMV screening can identify affected newborns who passed the auditory screens. As such, WGS could be a promising supplement to a traditional auditory newborn screening program since it can identify both genetic variants and CMV infections in parallel.
WGS has several advantages for newborn hearing screening. First, limited genetic screens only include a small number of pre-selected variants. This presents an inherent ethnic bias, which is an important drawback in a population with racial and ethnic diversity (Shearer et al., 2019). WGS, however, can analyze the full sequence of selected genes. More importantly, WGS is more powerful than whole-exome sequencing for detecting exome variants (Belkadi et al., 2015). Second, WGS can detect both chromosomal and exon-level CNVs, which are common causes of genetic hearing loss (Shearer et al., 2014). Third, WGS can simultaneously detect genomic variants and CMV infections, as we demonstrated in this study. These features highlight the advantages of WGS over limited genetic screens or WES.
It should be noted that WGS is limited in its ability to detect variants in genes with high homology. Two deafness-related genes (STRC and OTOA) have homologous regions in the human genome, which explains their low coverage in our WGS data. The STRC gene encodes stereocilin, a large extracellular structural protein found in the stereocilia of outer hair cells in the inner ear (Verpy et al., 2001). It is the second most common etiology in cases of mild-to-moderate hearing loss, after the GJB2 gene (Yokota et al., 2019). STRC is highly homologous to pseudogene pSTRC (99.6% of coding regions and 98.9% including introns) (Francey et al., 2012). OTOA encodes otoancorin, a protein that is important for conditioning proper stimulation of the inner hair cells (Zwaenepoel et al., 2002). It has 99% sequence similarity with exons 22–28 of the pseudogene OTOAP1, which generally leads to non-allelic homologous recombination-mediated deletions (Laurent et al., 2021). Considering the high prevalence of these two genes in hearing loss patients and the limitation of WGS, the residual risk should be calculated and the newborns’ guardians should be informed prior to testing.
Allelic heterogeneity, which leads to variable phenotypes, poses challenges to clinical counseling. For example, we identified two cases carrying c.2802T>G (p.Cys934Trp) in the USH2A gene (Supplementary Table S3). Although this variant is associated with both Usher syndrome and retinitis pigmentosa, most patients present with nonsyndromic retinitis pigmentosa with normal hearing (Meng et al., 2021; Zhu et al., 2021). To obtain an accurate clinical diagnosis, comprehensively ophthalmic examinations and audiological assessments are required. More importantly, clinical follow-up is needed.
Another challenge of implementing WGS as a screening test in newborns is the burden of variant interpretation (Narravula et al., 2017). To overcome this issue, our group recently developed a semi-automated variant interpretation platform for genetic hearing loss (Peng et al., 2021). This tool semi-automates variant interpretation based on the rules recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Peng et al., 2021). It facilitates and standardizes the interpretation of genetic hearing loss variants.
One important caveat that we wish to stress is that the viral load of CMV might be low in peripheral blood specimens. This led to low test sensitivity when dried blood spots from newborns’ heel were used (Boppana et al., 2010). Samples of saliva or umbilical cord blood might be a better alternative. Several studies have demonstrated that saliva specimens are reliable for screening newborns for CMV, offering high sensitivity and specificity (Boppana et al., 2011; Sun and Reichenberger, 2014). Umbilical cord blood, however, has received less investigation. Tagawa and co-authors reported that dried umbilical cord blood samples can be used to identify congenital CMV infections (Tagawa et al., 2009). In addition, Reyes et al. (2020) concluded that dried umbilical cord blood is an alternative to dried blood spots from heels and has better sensitivity.
It is also worth noting that the association between CMV infections and hearing loss relies on the virus loads. Boppana et al. revealed that the risk of hearing loss was low when the viral DNA is less than 5 × 103 pfu/ml in urine or low than 1 × 104 copies/mL in urine (Boppana et al., 2005). In addition, Ross et al. reported that a viral load of <3,500 genomic equivalents per milliliter is associated with a lower risk of hearing loss in children born with asymptomatic congenital infection (Ross et al., 2009). That is, when the CMV infection is revealed by WGS, a quantitative test (i.e., qPCR) is warranted to evaluate the risk.
In summary, WGS can simultaneously detect genetic variants and CMV infections in dried blood spot specimens from newborns. Our study provides proof of principle that genome sequencing can be a promising alternative for newborn screening of hearing loss.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author. The data reported in this study are also available in the CNGB Nucleotide Sequence Archive (CNSA: https://db.cngb.org/cnsa; accession number CNP0002697).
The studies involving human participants were reviewed and approved by Institutional Review Board of BGI. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JX, JS, and ZP conceived and designed the study. JX, XS, and HZ drafted and revised the manuscript. JX, JS, and ZP reviewed the analyses and edited the manuscript. XS and HZ performed data analysis. All authors read and approved the final manuscript.
JX, HZ, XS, JZ, ZX, JS, and ZP were employed by BGI
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.883617/full#supplementary-material
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: an Approach to Discover, Genotype, and Characterize Typical and Atypical CNVs from Family and Population Genome Sequencing. Genome Res. 21 (6), 974–984. doi:10.1101/gr.114876.110
Azaiez, H., Booth, K. T., Ephraim, S. S., Crone, B., Black-Ziegelbein, E. A., Marini, R. J., et al. (2018). Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 103 (4), 484–497. doi:10.1016/j.ajhg.2018.08.006
Belkadi, A., Bolze, A., Itan, Y., Cobat, A., Vincent, Q. B., Antipenko, A., et al. (2015). Whole-genome Sequencing Is More Powerful Than Whole-Exome Sequencing for Detecting Exome Variants. Proc. Natl. Acad. Sci. U.S.A. 112 (17), 5473–5478. doi:10.1073/pnas.1418631112
Boppana, S. B., Fowler, K. B., Pass, R. F., Rivera, L. B., Bradford, R. D., Lakeman, F. D., et al. (2005). Congenital Cytomegalovirus Infection: Association between Virus burden in Infancy and Hearing Loss. J. Pediatr. 146 (6), 817–823. doi:10.1016/j.jpeds.2005.01.059
Boppana, S. B., Ross, S. A., Novak, Z., Shimamura, M., Tolan, R. W., Palmer, A. L., et al. (2010). Dried Blood Spot Real-Time Polymerase Chain Reaction Assays to Screen Newborns for Congenital Cytomegalovirus Infection. JAMA 303 (14), 1375–1382. doi:10.1001/jama.2010.423
Boppana, S. B., Ross, S. A., Shimamura, M., Palmer, A. L., Ahmed, A., Michaels, M. G., et al. (2011). Saliva Polymerase-Chain-Reaction Assay for Cytomegalovirus Screening in Newborns. N. Engl. J. Med. 364 (22), 2111–2118. doi:10.1056/NEJMoa1006561
Ceyhan-Birsoy, O., Machini, K., Lebo, M. S., Yu, T. W., Agrawal, P. B., Parad, R. B., et al. (2017). A Curated Gene List for Reporting Results of Newborn Genomic Sequencing. Genet. Med. 19 (7), 809–818. doi:10.1038/gim.2016.193
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Dai, P., Huang, L.-H., Wang, G.-J., Gao, X., Qu, C.-Y., Chen, X.-W., et al. (2019). Concurrent Hearing and Genetic Screening of 180,469 Neonates with Follow-Up in Beijing, China. Am. J. Hum. Genet. 105 (4), 803–812. doi:10.1016/j.ajhg.2019.09.003
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 43 (5), 491–498. doi:10.1038/ng.806
DiStefano, M. T., Hemphill, S. E., Oza, A. M., Siegert, R. K., Grant, A. R., Hughes, M. Y., et al. (2019). ClinGen Expert Clinical Validity Curation of 164 Hearing Loss Gene-Disease Pairs. Genet. Med. 21 (10), 2239–2247. doi:10.1038/s41436-019-0487-0
DiStefano, M. T., Hughes, M. Y., Patel, M. J., Wilcox, E. H., and Oza, A. M. (2020). Expert Interpretation of Genes and Variants in Hereditary Hearing Loss. Medizinische Genetik 32(2), 109–115. doi:10.1515/medgen-2020-2018
Fowler, K. B., McCollister, F. P., Sabo, D. L., Shoup, A. G., Owen, K. E., Woodruff, J. L., et al. (2017). A Targeted Approach for Congenital Cytomegalovirus Screening within Newborn Hearing Screening. Pediatrics 139 (2), e20162128. doi:10.1542/peds.2016-2128
Francey, L. J., Conlin, L. K., Kadesch, H. E., Clark, D., Berrodin, D., Sun, Y., et al. (2012). Genome-wide SNP Genotyping Identifies the Stereocilin (STRC) Gene as a Major Contributor to Pediatric Bilateral Sensorineural Hearing Impairment. Am. J. Med. Genet. 158a (2), 298–308. doi:10.1002/ajmg.a.34391
Guo, L., Xiang, J., Sun, L., Yan, X., Yang, J., Wu, H., et al. (2020). Concurrent Hearing and Genetic Screening in a General Newborn Population. Hum. Genet. 139 (4), 521–530. doi:10.1007/s00439-020-02118-6
Kennedy, C., McCann, D., Campbell, M. J., Kimm, L., and Thornton, R. (2005). Universal Newborn Screening for Permanent Childhood Hearing Impairment: an 8-year Follow-Up of a Controlled Trial. Lancet 366 (9486), 660–662. doi:10.1016/s0140-6736(05)67138-3
Korver, A. M. H., Smith, R. J. H., Van Camp, G., Schleiss, M. R., Bitner-Glindzicz, M. A. K., Lustig, L. R., et al. (2017). Congenital Hearing Loss. Nat. Rev. Dis. Primers 3, 16094. doi:10.1038/nrdp.2016.94
Laurent, S., Gehrig, C., Nouspikel, T., Amr, S. S., Oza, A., Murphy, E., et al. (2021). Molecular Characterization of Pathogenic OTOA Gene Conversions in Hearing Loss Patients. Hum. Mutat. 42 (4), 373–377. doi:10.1002/humu.24167
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Lindstrand, A., Eisfeldt, J., Pettersson, M., Carvalho, C. M. B., Kvarnung, M., Grigelioniene, G., et al. (2019). From Cytogenetics to Cytogenomics: Whole-Genome Sequencing as a First-Line Test Comprehensively Captures the Diverse Spectrum of Disease-Causing Genetic Variation Underlying Intellectual Disability. Genome Med. 11 (1), 68. doi:10.1186/s13073-019-0675-1
Lu, C.-Y., Tsao, P.-N., Ke, Y.-Y., Lin, Y.-H., Lin, Y.-H., Hung, C.-C., et al. (2018). Concurrent Hearing, Genetic, and Cytomegalovirus Screening in Newborns, Taiwan. J. Pediatr. 199, 144–150. doi:10.1016/j.jpeds.2018.02.064
Meng, X., Liu, X., Li, Y., Guo, T., and Yang, L. (2021). Correlation between Genotype and Phenotype in 69 Chinese Patients with USH2A Mutations: A Comparative Study of the Patients with Usher Syndrome and Nonsyndromic Retinitis Pigmentosa. Acta Ophthalmol. 99 (4), e447–e460. doi:10.1111/aos.14626
Morton, C. C., and Nance, W. E. (2006). Newborn Hearing Screening - A Silent Revolution. N. Engl. J. Med. 354 (20), 2151–2164. doi:10.1056/NEJMra050700
Narravula, A., Garber, K. B., Askree, S. H., Hegde, M., and Hall, P. L. (2017). Variants of Uncertain Significance in Newborn Screening Disorders: Implications for Large-Scale Genomic Sequencing. Genet. Med. 19 (1), 77–82. doi:10.1038/gim.2016.67
Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., Siegert, R. K., et al. (2018). Expert Specification of the ACMG/AMP Variant Interpretation Guidelines for Genetic Hearing Loss. Hum. Mutat. 39 (11), 1593–1613. doi:10.1002/humu.23630
Peng, J., Xiang, J., Jin, X., Meng, J., Song, N., Chen, L., et al. (2021). VIP‐HL: Semi‐automated ACMG/AMP Variant Interpretation Platform for Genetic Hearing Loss. Hum. Mutat. 42 (12), 1567–1575. doi:10.1002/humu.24277
Plagnol, V., Curtis, J., Epstein, M., Mok, K. Y., Stebbings, E., Grigoriadou, S., et al. (2012). A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics 28 (21), 2747–2754. doi:10.1093/bioinformatics/bts526
Reyes, A., Taravillo, I., Moral, N., Moraleda, C., Blázquez-Gamero, D., and Folgueira, L. (2020). Feasible Alternatives to DBS in the Retrospective Diagnosis of Congenital Cytomegalovirus Infection. J. Clin. Virol. 129, 104504. doi:10.1016/j.jcv.2020.104504
Robertson, N. G., Cremers, C. W. R. J., Huygen, P. L. M., Ikezono, T., Krastins, B., Kremer, H., et al. (2006). Cochlin Immunostaining of Inner Ear Pathologic Deposits and Proteomic Analysis in DFNA9 Deafness and Vestibular Dysfunction. Hum. Mol. Genet. 15 (7), 1071–1085. doi:10.1093/hmg/ddl022
Ross, S. A., Novak, Z., Fowler, K. B., Arora, N., Britt, W. J., and Boppana, S. B. (2009). Cytomegalovirus Blood Viral Load and Hearing Loss in Young Children with Congenital Infection. Pediatr. Infect. Dis. J. 28 (7), 588–592. doi:10.1097/INF.0b013e3181979a27
Schon, K. R., Ratnaike, T., van den Ameele, J., Horvath, R., and Chinnery, P. F. (2020). Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 36 (9), 702–717. doi:10.1016/j.tig.2020.06.009
Shearer, A., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E., et al. (2014). Copy Number Variants Are a Common Cause of Non-syndromic Hearing Loss. Genome Med. 6 (5), 37. doi:10.1186/gm554
Shearer, A. E., Shen, J., Amr, S., Morton, C. C., and Smith, R. J. (2019). Newborn Hearing Screening Working Group of the National Coordinating Center for the Regional Genetics, NA Proposal for Comprehensive Newborn Hearing Screening to Improve Identification of Deaf and Hard-Of-Hearing Children. Genet. Med. 21 (11), 2614–2630. doi:10.1038/s41436-019-0563-5
Sheppard, S., Biswas, S., Li, M. H., Jayaraman, V., Slack, I., Romasko, E. J., et al. (2018). Utility and Limitations of Exome Sequencing as a Genetic Diagnostic Tool for Children with Hearing Loss. Genet. Med. 20 (12), 1663–1676. doi:10.1038/s41436-018-0004-x
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive Genetic Testing in the Clinical Evaluation of 1119 Patients with Hearing Loss. Hum. Genet. 135 (4), 441–450. doi:10.1007/s00439-016-1648-8
Sun, F., and Reichenberger, E. J. (2014). Saliva as a Source of Genomic DNA for Genetic Studies: Review of Current Methods and Applications. Oral Health Dent Manag. 13 (2), 217–222.
Tagawa, M., Tanaka, H., Moriuchi, M., and Moriuchi, H. (2009). Retrospective Diagnosis of Congenital Cytomegalovirus Infection at a School for the Deaf by Using Preserved Dried Umbilical Cord. J. Pediatr. 155 (5), 749–751. doi:10.1016/j.jpeds.2009.04.033
Usami, S.-i., and Nishio, S.-y. (2004). “Nonsyndromic Hearing Loss and Deafness, Mitochondrial,” in GeneReviews® [Internet]. Editors M. P. Adam, H. H. Ardinger, and R. A. Pagon (Seattle (WA): University of Washington, Seattle). Available at https://www.ncbi.nlm.nih.gov/books/NBK1422/.
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ Data to High Confidence Variant Calls: the Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics 43 (1110), 11–33. doi:10.1110.3310.1002/0471250953.bi1110s4310.1002/0471250953.bi1110s43
Verpy, E., Masmoudi, S., Zwaenepoel, I., Leibovici, M., Hutchin, T. P., Del Castillo, I., et al. (2001). Mutations in a New Gene Encoding a Protein of the Hair Bundle Cause Non-syndromic Deafness at the DFNB16 Locus. Nat. Genet. 29 (3), 345–349. doi:10.1038/ng726
Wang, Q., Xiang, J., Sun, J., Yang, Y., Guan, J., Wang, D., et al. (2019). Nationwide Population Genetic Screening Improves Outcomes of Newborn Screening for Hearing Loss in China. Genet. Med. 21 (10), 2231–2238. doi:10.1038/s41436-019-0481-6
White, K. R., Forsman, I., Eichwald, J., and Munoz, K. (2010). The Evolution of Early Hearing Detection and Intervention Programs in the United States. Semin. Perinatol. 34 (2), 170–179. doi:10.1053/j.semperi.2009.12.009
Winkelmann, J., Lin, L., Schormair, B., Kornum, B. R., Faraco, J., Plazzi, G., et al. (2012). Mutations in DNMT1 Cause Autosomal Dominant Cerebellar Ataxia, Deafness and Narcolepsy. Hum. Mol. Genet. 21 (10), 2205–2210. doi:10.1093/hmg/dds035
Yamamoto, A. Y., Anastasio, A. R. T., Massuda, E. T., Isaac, M. L., Manfredi, A. K. S., Cavalcante, J. M. S., et al. (2019). Contribution of Congenital Cytomegalovirus Infection to Permanent Hearing Loss in a Highly Seropositive Population: The Brazilian Cytomegalovirus Hearing and Maternal Secondary Infection Study. Clin. Infect. Dis. 70, 1379–1384. doi:10.1093/cid/ciz413
Yang, H., and Wang, K. (2015). Genomic Variant Annotation and Prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10 (10), 1556–1566. doi:10.1038/nprot.2015.105
Yokota, Y., Moteki, H., Nishio, S.-y., Yamaguchi, T., Wakui, K., Kobayashi, Y., et al. (2019). Frequency and Clinical Features of Hearing Loss Caused by STRC Deletions. Sci. Rep. 9 (1), 4408. doi:10.1038/s41598-019-40586-7
Zhu, T., Chen, D.-F., Wang, L., Wu, S., Wei, X., Li, H., et al. (2021). USH2A Variants in Chinese Patients with Usher Syndrome Type II and Non-syndromic Retinitis Pigmentosa. Br. J. Ophthalmol. 105 (5), 694–703. doi:10.1136/bjophthalmol-2019-315786
Zwaenepoel, I., Mustapha, M., Leibovici, M., Verpy, E., Goodyear, R., Liu, X. Z., et al. (2002). Otoancorin, an Inner Ear Protein Restricted to the Interface between the Apical Surface of Sensory Epithelia and Their Overlying Acellular Gels, Is Defective in Autosomal Recessive Deafness DFNB22. Proc. Natl. Acad. Sci. U.S.A. 99 (9), 6240–6245. doi:10.1073/pnas.082515999
Keywords: WGS, hearing loss, newborns, genetic variation, cytomegalovirus infection
Citation: Xiang J, Zhang H, Sun X, Zhang J, Xu Z, Sun J and Peng Z (2022) Utility of Whole Genome Sequencing for Population Screening of Deafness-Related Genetic Variants and Cytomegalovirus Infection in Newborns. Front. Genet. 13:883617. doi: 10.3389/fgene.2022.883617
Received: 25 February 2022; Accepted: 07 April 2022;
Published: 29 April 2022.
Edited by:
Fengxiao Bu, Sichuan University, ChinaCopyright © 2022 Xiang, Zhang, Sun, Zhang, Xu, Sun and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyu Peng, cGVuZ3poaXl1QGJnaS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.