 Mutaz Amin1,2
Mutaz Amin1,2 Cedric Vignal3Ahlam A. A. Hamed4Inaam N. Mohammed4Maha A. Elseed4Rayan Abubaker5,6
Cedric Vignal3Ahlam A. A. Hamed4Inaam N. Mohammed4Maha A. Elseed4Rayan Abubaker5,6 Yousuf Bakhit7,8Arwa Babai4
Yousuf Bakhit7,8Arwa Babai4 Eman Elbadi4
Eman Elbadi4 Esraa Eltaraifee4Doua Mustafa4
Esraa Eltaraifee4Doua Mustafa4 Ashraf Yahia4Melka Osman4
Ashraf Yahia4Melka Osman4 Mahmoud Koko5Mohamed Mustafa4
Mahmoud Koko5Mohamed Mustafa4 Mohamed Alsiddig4Sahwah Haroun4Azza Elshafea5
Mohamed Alsiddig4Sahwah Haroun4Azza Elshafea5 Severine Drunat2,3
Severine Drunat2,3 Liena E. O. Elsayed9Ammar E. Ahmed4
Liena E. O. Elsayed9Ammar E. Ahmed4 Odile Boespflug-Tanguy2,10Imen Dorboz2,10*
Odile Boespflug-Tanguy2,10Imen Dorboz2,10*- 1Faculty of Medicine, Al-Neelain University, Khartoum, Sudan
- 2INSERM UMR 1141 PROTECT, Université Paris Diderot-Sorbonne, Paris, France
- 3Unité de Génétique Moleculaire, Departement de Genetique Médicale, APHP, Hopital Robert-Debré, Paris, France
- 4Faculty of Medicine, University of Khartoum, Khartoum, Sudan
- 5Neurogenetics Research group, Faculty of Medicine, University of Khartoum, Khartoum, Sudan
- 6National University Biomedical Research Institute, National University-Sudan, Khartoum, Sudan
- 7Faculty of Dentistry, University of Khartoum, Khartoum, Sudan
- 8Department of Neurobiology, Centre for Neurology, UKB, University of Bonn, Bonn, Germany
- 9Department of Basic Sciences, College of Medicine, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
- 10Neuropediatrics and Metabolic Disorders Department, Reference Center for Leukodystrophies and Rare Leukoencéphalopathies (LEUKOFRANCE), CHU APHP Robert-Debré, Paris, France
Pontocerebellar hypoplasia type 10 (PCH10) is a very rare autosomal recessive neurodegenerative disease characterized by intellectual disability, microcephaly, severe developmental delay, pyramidal signs, mild cerebellar atrophy, and white matter changes in the brain, as shown by magnetic resonance imaging (MRI). The disease has been described in only twenty-one patients from ten Turkish families with a founder missense pathogenic variant in the CLP1 gene involved in tRNA processing and maturation. We analyzed three siblings from a consanguineous Sudanese family who presented with intellectual disability, dysmorphic features, developmental delay, regression of milestones, microcephaly, epilepsy, extrapyramidal signs, mild pontine, and cerebellar atrophy. We identified through whole-exome sequencing the same pathogenic variant (c.419G>A; p(Arg140His) reported before in all Turkish families. Our study extends the phenotypes of PCH10 and reports for the first time cases with PCH10 of non-Turkish origin.
Introduction
Pontocerebellar hypoplasia (PCH) describes a heterogeneous group of rare neurodegenerative disorders characterized by developmental delay, epilepsy, spasticity, and postnatal microcephaly (Rudnik-Schöneborn et al., 2014). Some patients have dysmorphic facial features and axonal sensorimotor neuropathy (Van Dijk et al., 2017; Morton et al., 2018; Somashekar et al., 2021). Brain MRI of patients with PCH reveals the characteristic atrophy of the pons and cerebellum. In most cases, death is invariable within the first decade of life (Van Dijk et al., 2018).
There are currently 13 different types of pontocerebellar hypoplasia resulting from mutations in at least 19 different genes (Rüsch et al., 2020). Many of these genes are involved in RNA processing which is essential for neuronal survival (Van Dijk et al., 2018). PCH1 and PCH2 are the most common types; they are caused by mutations in EXOSC3, TSEN54, RARS2, and VRK1 genes (Van Dijk et al., 2018).
Pontocerebellar hypoplasia type 10 (PCH10) is a very rare and recently described type of PCH. It presents with microcephaly, severe developmental delay, pyramidal signs, and very mild cerebellar atrophy and white matter changes in brain MRI (Karaca et al., 2014; Schaffer et al., 2014; Wafik et al., 2018). The disease is caused by a pathogenic variant in the CLP1 gene, which encodes a kinase involved in tRNA processing and maturation (Karaca et al., 2014; Weitzer et al., 2015). Dysfunction of the CLP1 enzyme leads to the accumulation of aberrant tRNA molecules and ultimately to neuronal death (Morisaki et al., 2021).
Only ten families have been reported so far with PCH10 (Karaca et al., 2014; Schaffer et al., 2014; Wafik et al., 2018). All families originated from Turkey, and all shared the same mutation (c.419G>A; p.Arg140His) (Karaca et al., 2014; Schaffer et al., 2014; Wafik et al., 2018). The pathogenicity of the R140H variant has been functionally validated in several studies with the resulting defect in kinase activity (Karaca et al., 2014; Schaffer et al., 2014; Weitzer et al., 2015; Hayne et al., 2020; Monaghan et al., 2021; Morisaki et al., 2021). The associated neurodegenerative phenotype has also been reproduced in both mice (Monaghan et al., 2021; Morisaki et al., 2021) and Zebrafish models (Schaffer et al., 2014).
This study reports for the first time a non-Turkish family with PCH10 from Sudan sharing the same pathogenic variant.
Case Presentation
Three sisters born to first-degree consanguineous parents were studied (Table 1). The first (patient 1) was 8 years old and presented with delayed motor and speech development (sitting at 18 months, crawling at 1 year, disyllabic babbling only). She started to regress at 5 years (lost walking and disyllabic babbling). She had behavioral abnormality (irritability, biting), mild cognitive impairment, and epilepsy since the age of 11 months, controlled with antiepileptic medications. There was no sphincteric control. Patient 2 (7 years) had a severely delayed motor and speech development (sitting at 21 months, no standing or walking, no speech) but no regression. Patient 3 (1 year) had a delayed motor and speech development (head support at 7 months, sitting without support at 8 months, no standing or walking, and no speech). On examination, all patients had mild dysmorphic features, including high arched eyebrows, long palpebral fissures and eyelashes with prominent eyes, microcephaly (<3SD), hypertonia, hyperreflexia, dystonia, extrapyramidal signs (uncontrollable head nodding), poor dentition, and fixed flexion deformity (Table 1). Brain MRI showed mild pontine and cerebellar atrophy, thin corpus callosum, and periventricular white matter changes (Figure 1). The metabolic screening was negative.
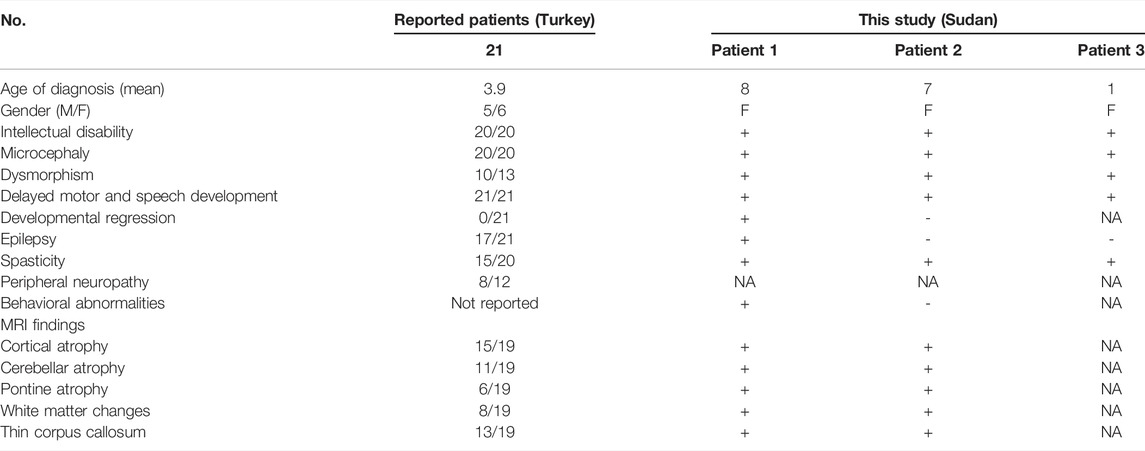
TABLE 1. Clinical features of patients with CLP1 mutation.
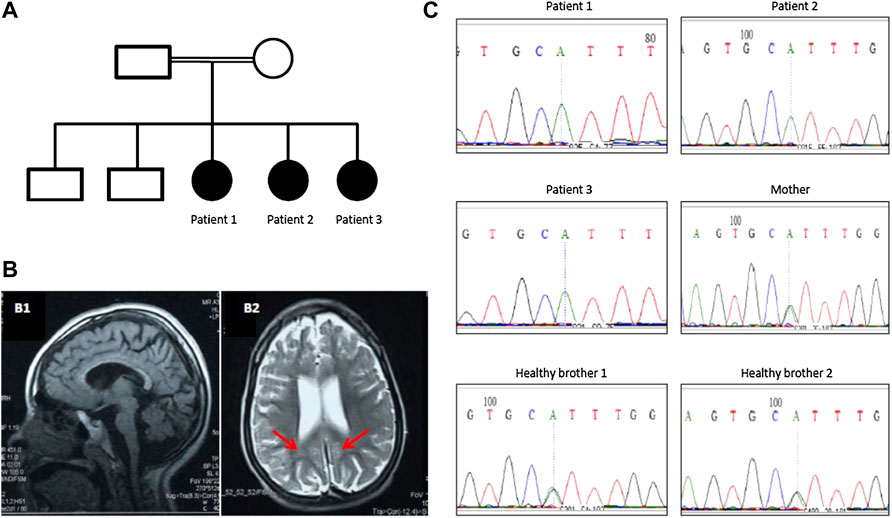
FIGURE 1. (A) Family pedigree of a Sudanese family with PCH10. (B) Brain MRI of patient 1 showing mild pontine and cerebellar atrophy and thin corpus callosum (B1) and periventricular white matter changes (red arrows in B2). (C) Segregation of CLP1 (c.419G>A; p.Arg140His) pathogenic variant in the family with pontocerebellar hypoplasia 10.
Genetic Testing
DNA Extraction
Two ml of saliva was collected from patients and other healthy family members using DNA Oragene Saliva kits (DNA Genotek Inc., Ottawa, ON, Canada). DNA extraction was done according to the prepIT.L2P manual protocol provided by the manufacturer.
Whole Exome Sequencing
Whole exome sequencing was performed using the Hiseq property of the NextSeq-500 sequencer (Illumina®, San Diego, United States) available in the Bioinformatics Department in the Institute of Brain and Spinal cord (ICM) in Paris, France. High-quality paired-end reads (minimum coverage 100x) were aligned to human reference genome hg19 using the Burrows–Wheeler (BWA-MEM) algorithm v0.7.12 (Li and Durbin, 2009) with default parameters. The resulting BAM files were processed (removal of PCR duplicates, sorting, and indexing) using samtools v1.7 (Li et al., 2009). Variant calling was performed using Freebayes v1.0.0 (Garrison and Marth, 2012) with default parameters, and the resulting VCF files were filtered according to depth (>20) and genotype quality (>20). The VCF file was annotated using SnpEff v4.3t (Cingolani et al., 2012), loaded to Gemini v0.17 (Paila et al., 2013), and then filtered for rare (ExAC MAF <0.01) potentially pathogenic variants that follow an autosomal recessive inheritance pattern.
Sanger Sequencing
Sanger sequencing was used to analyze the segregation pattern of candidate variants and confirm the genotypes in affected individuals. Primer3 online tool (Untergasser et al., 2012) was used to design forward and reverse primers. Multiple sequence alignment was performed using Bioedit software v7.2.5 (Hall, 1999) with default parameters.
Ethical Consideration
Written informed consent was obtained from each family member (or parents/legal guardians in case of minors) before participation in the study according to the LEUKOFRANCE research program for undetermined leukodystrophies (authorization CPP AU788; CNIL 1406552; AFSSAPS B90298-60).
Results
Exome sequencing revealed a homozygous missense pathogenic variant (NM_006831.3:c.419G>A; p.Arg140His) in the CLP1 gene. The segregation of the variant in the family was consistent with autosomal recessive inheritance (Figure 1). The variant has a very low allele frequency (<0.0001) in the ExAC and gnomAD databases and was predicted to be pathogenic using 11 different pathogenicity prediction tools: SIFT (Ng and Henikoff, 2003), PolyPhen (Adzhubei et al., 2013), MutationTaster (Schwarz et al., 2010), MutationAssessor (Reva et al., 2011), BayesDel_addAF (Feng, 2017), DANN (Quang et al., 2015), EIGEN (Ionita-Laza et al., 2016), FATHMM-MKL (Rogers et al., 2018), LIST-S2 (Malhis et al., 2020), M-CAP (Jagadeesh et al., 2016), and PrimateAI (Sundaram et al., 2018). The wild-type amino acid is highly conserved (Schaffer et al., 2014). Structural differences between the wild type and mutant residues are shown in Figure 2. This pathogenic variant has been reported before in Turkish families with the same clinical presentation (Schaffer et al., 2014) and has been shown to impair CLP1 protein, which is involved in tRNA and mRNA processing leading to the accumulation of unspliced pre-tRNAs (Schaffer et al., 2014). The mutant CLP1 enzyme loses its kinase activity and causes a neurodegenerative phenotype in clp1 null zebrafish (Schaffer et al., 2014), and dysfunction of CLP1 in mice resulted in a similar phenotype of microcephaly, brain atrophy, and neurodegeneration (Karaca et al., 2014).
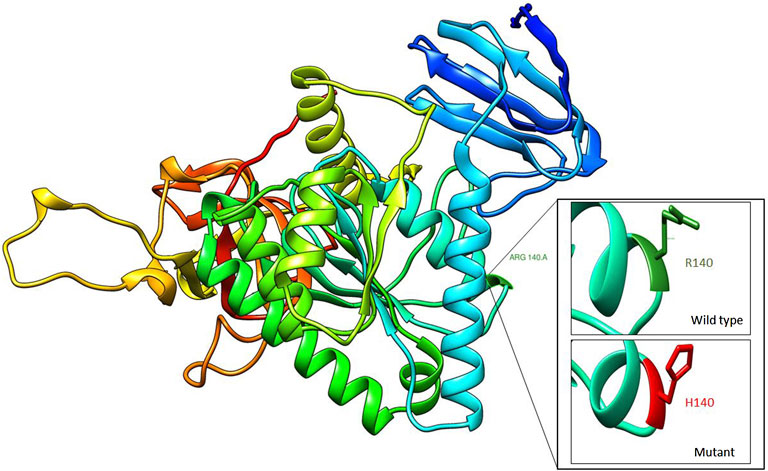
FIGURE 2. Homology modeling of CLP1 protein showing structural differences between wild type (R140) and mutant residue (H140).
Discussion
One of the main distinguishing features of PCH10 is the ethnic origin of the family (Turkish origin) (Rüsch et al., 2020) because PCH10 has not been reported so far from outside Turkey. The disease was first described in 2014 by two independent Turkish studies (Karaca et al., 2014; Schaffer et al., 2014), who reported nine different families with a distinct form of pontocerebellar hypoplasia. Later, another study (Wafik et al., 2018) reported another Turkish family with the same disease. All these families shared the same mutation, a homozygous missense pathogenic variant in the CLP1 gene, which is involved in the splicing and processing of tRNAs. This study reports a Sudanese family with PCH10 with the same reported variant. All patients with PCH10, including the patients in our study, presented with intellectual disability, microcephaly, and delayed developmental milestones. The patients in our study presented with similar clinical and radiological features reported in the Turkish studies (Table 1). However, the eldest patient in our study presented with a more severe phenotype, including behavioral abnormalities (biting and irritability) and developmental regression. She ultimately succumbed to the disease at the age of 8.5 years following an acute respiratory tract infection. These severe features have not been reported before in patients with PCH10 (Table 1).
All previously reported families originated from Eastern Turkey. Therefore, a founder effect was suggested (Karaca et al., 2014; Schaffer et al., 2014). Schaffer et al. (2014) analyzed the haplotypes of the four Turkish families in their study and estimated from the expected mutation rate and the number of haploblocks in Middle Eastern cohorts that the most recent common ancestor has probably lived around 16 generations ago (+/− 8.7), which coincides with the time of the Ottoman’s expansion. It is possible that the R140H pathogenic variant has been transferred to or from Turkey at this time. However, it is more likely that this mutation arose independently in the Sudanese family, considering the lack of shared genotypes in the region surrounding the mutation (Supplementary Figure S1).
Our study extends the phenotypes of PCH10 and reports for the first time cases with PCH10 of non-Turkish origin.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the LEUKOFRANCE research program for undetermined leukodystrophies (authorization CPP AU788; CNIL 1406552; AFSSAPS B90298-60). Written informed consent to participate in this study was provided by the participants’ legal guardians/next of kin.
Author Contributions
MuA, ID, and OB-T made substantial contributions to the conception and the design of the study. AH, IM, and ME recruited the patients and did the clinical interpretation. CV and SD did the laboratory work. Fieldwork, data collection, data analysis, and data interpretation were done by RA, YB, AB, EmE, EsE, DM, AY, MO, MK, MM, MoS, SH, and AE. MuA drafted the manuscript. ME, MK, LE, and AA critically revised it. ID gave final approval of the version to be published. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.883211/full#supplementary-material
References
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting Functional Effect of Human Missense Mutations Using PolyPhen‐2. Curr. Protoc. Hum. Genet. 76. Chapter 7Unit7.20. doi:10.1002/0471142905.hg0720s76
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 6, 80–92. iso-2; iso-3. doi:10.4161/FLY.19695
Feng, B.-J. (2017). PERCH: A Unified Framework for Disease Gene Prioritization. Hum. Mutat. 38, 243–251. doi:10.1002/HUMU.23158
Garrison, E., and Marth, G. (2012). Haplotype-based Variant Detection from Short-Read Sequencing. Quant. Biol. Genomics 1, 3907. doi:10.48550/arxiv.1207.3907
Hall, T. (1999). BioEdit: a User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98. citeulike-article-id:691774.
Hayne, C. K., Schmidt, C. A., Haque, M. I., Matera, A. G., and Stanley, R. E. (2020). Reconstitution of the Human tRNA Splicing Endonuclease Complex: Insight into the Regulation of Pre-tRNA Cleavage. Nucleic Acids Res. 48, 7609–7622. doi:10.1093/NAR/GKAA438
Ionita-Laza, I., Mccallum, K., Xu, B., and Buxbaum, J. D. (2016). A Spectral Approach Integrating Functional Genomic Annotations for Coding and Noncoding Variants. Nat. Genet. 48, 214–220. doi:10.1038/NG.3477
Jagadeesh, K. A., Wenger, A. M., Berger, M. J., Guturu, H., Stenson, P. D., Cooper, D. N., et al. (2016). M-CAP Eliminates a Majority of Variants of Uncertain Significance in Clinical Exomes at High Sensitivity. Nat. Genet. 48, 1581–1586. doi:10.1038/NG.3703
Karaca, E., Weitzer, S., Pehlivan, D., Shiraishi, H., Gogakos, T., Hanada, T., et al. (2014). Human CLP1 Mutations Alter tRNA Biogenesis, Affecting Both Peripheral and Central Nervous System Function. Cell 157, 636–650. doi:10.1016/J.CELL.2014.02.058
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/BIOINFORMATICS/BTP324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/BIOINFORMATICS/BTP352
Malhis, N., Jacobson, M., Jones, S. J. M., and Gsponer, J. (2020). LIST-S2: Taxonomy Based Sorting of Deleterious Missense Mutations across Species. Nucleic Acids Res. 48, W154–W161. doi:10.1093/NAR/GKAA288
Monaghan, C. E., Adamson, S. I., Kapur, M., Chuang, J. H., and Ackerman, S. L. (2021). The Clp1 R140H Mutation Alters tRNA Metabolism and mRNA 3′ Processing in Mouse Models of Pontocerebellar Hypoplasia. Proc. Natl. Acad. Sci. U.S.A. 118. doi:10.1073/PNAS.2110730118
Morisaki, I., Shiraishi, H., Fujinami, H., Shimizu, N., Hikida, T., Arai, Y., et al. (2021). Modeling a Human CLP1 Mutation in Mouse Identifies an Accumulation of Tyrosine Pre-tRNA Fragments Causing Pontocerebellar Hypoplasia Type 10. Biochem. Biophysical Res. Commun. 570, 60–66. doi:10.1016/J.BBRC.2021.07.036
Morton, D. J., Kuiper, E. G., Jones, S. K., Leung, S. W., Corbett, A. H., and Fasken, M. B. (2018). The RNA Exosome and RNA Exosome-Linked Disease. RNA 24, 127–142. doi:10.1261/RNA.064626.117
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting Amino Acid Changes that Affect Protein Function. Nucleic Acids Res. 31, 3812–3814. doi:10.1093/nar/gkg509
Paila, U., Chapman, B. A., Kirchner, R., and Quinlan, A. R. (2013). GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLoS Comput. Biol. 9, e1003153. doi:10.1371/JOURNAL.PCBI.1003153
Quang, D., Chen, Y., and Xie, X. (2015). DANN: a Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 31, 761–763. doi:10.1093/BIOINFORMATICS/BTU703
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 39, e118. doi:10.1093/NAR/GKR407
Rogers, M. F., Shihab, H. A., Mort, M., Cooper, D. N., Gaunt, T. R., and Campbell, C. (2018). FATHMM-XF: Accurate Prediction of Pathogenic Point Mutations via Extended Features. Bioinformatics 34, 511–513. doi:10.1093/BIOINFORMATICS/BTX536
Rudnik-Schöneborn, S., Barth, P. G., and Zerres, K. (2014). Pontocerebellar Hypoplasia. Am. J. Med. Genet. 166, 173–183. doi:10.1002/AJMG.C.31403
Rüsch, C. T., Bölsterli, B. K., Kottke, R., Steinfeld, R., and Boltshauser, E. (2020). Pontocerebellar Hypoplasia: a Pattern Recognition Approach. Cerebellum 19, 569–582. doi:10.1007/S12311-020-01135-5
Schaffer, A. E., Eggens, V. R. C., Caglayan, A. O., Reuter, M. S., Scott, E., Coufal, N. G., et al. (2014). CLP1 Founder Mutation Links tRNA Splicing and Maturation to Cerebellar Development and Neurodegeneration. Cell 157, 651–663. doi:10.1016/J.CELL.2014.03.049
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster Evaluates Disease-Causing Potential of Sequence Alterations. Nat. Methods 7, 575–576. doi:10.1038/nmeth0810-575
Somashekar, P. H., Kaur, P., Stephen, J., Guleria, V. S., Kadavigere, R., Girisha, K. M., et al. (2021). Bi‐allelic Missense Variant, P. Ser35Leu in EXOSC1 Is Associated with Pontocerebellar hypoplasiaSer35Leu in EXOSC1 Is Associated with Pontocerebellar Hypoplasia. Clin. Genet. 99, 594–600. doi:10.1111/CGE.13928
Sundaram, L., Gao, H., Padigepati, S. R., McRae, J. F., Li, Y., Kosmicki, J. A., et al. (20182018). Predicting the Clinical Impact of Human Mutation with Deep Neural Networks. Nat. Genet. 50 (50), 1161–1170. doi:10.1038/s41588-018-0167-z
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new Capabilities and Interfaces. Nucleic Acids Res. 40, e115. doi:10.1093/nar/gks596
Van Dijk, T., Baas, F., Barth, P. G., and Poll-The, B. T. (2018). What's New in Pontocerebellar Hypoplasia? an Update on Genes and Subtypes. Orphanet J. Rare Dis. 13. doi:10.1186/S13023-018-0826-2
Van Dijk, T., Rudnik-Schöneborn, S., Senderek, J., Hajmousa, G., Mei, H., Dusl, M., et al. (2017). Pontocerebellar Hypoplasia with Spinal Muscular Atrophy (PCH1): Identification of SLC25A46 Mutations in the Original Dutch PCH1 Family. Brain 140, e46. doi:10.1093/BRAIN/AWX147
Wafik, M., Taylor, J., Lester, T., Gibbons, R. J., and Shears, D. J. (2018). 2 New Cases of Pontocerebellar Hypoplasia Type 10 Identified by Whole Exome Sequencing in a Turkish Family. Eur. J. Med. Genet. 61, 273–279. doi:10.1016/J.EJMG.2018.01.002
Keywords: pontocerebellar hypoplasia 10, CLP1, Sudan, pontocerebellar hypoplasia, family
Citation: Amin M, Vignal C, Hamed AAA, Mohammed IN, Elseed MA, Abubaker R, Bakhit Y, Babai A, Elbadi E, Eltaraifee E, Mustafa D, Yahia A, Osman M, Koko M, Mustafa M, Alsiddig M, Haroun S, Elshafea A, Drunat S, Elsayed LEO, Ahmed AE, Boespflug-Tanguy O and Dorboz I (2022) Case Report: A New Family With Pontocerebellar Hypoplasia 10 From Sudan. Front. Genet. 13:883211. doi: 10.3389/fgene.2022.883211
Received: 24 February 2022; Accepted: 29 April 2022;
Published: 02 June 2022.
Edited by:
Stephen J. Bush, University of Oxford, United KingdomReviewed by:
Gaetan Lesca, Université Claude Bernard Lyon 1, FrancePeter Natesan Pushparaj, King Abdulaziz University, Saudi Arabia
Muzammil Ahmad Khan, Gomal University, Pakistan
Copyright © 2022 Amin, Vignal, Hamed, Mohammed, Elseed, Abubaker, Bakhit, Babai, Elbadi, Eltaraifee, Mustafa, Yahia, Osman, Koko, Mustafa, Alsiddig, Haroun, Elshafea, Drunat, Elsayed, Ahmed, Boespflug-Tanguy and Dorboz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Imen Dorboz, aW1lbi5kb3Jib3pAYXBocC5mcg==