 Zhanjun Wang1,2,3
Zhanjun Wang1,2,3 Qianwen Cai1
Qianwen Cai1 Haimeng Xia1Bingqing Han1Minhui Li1
Haimeng Xia1Bingqing Han1Minhui Li1 Yue Wang1Minhui Zhu1
Yue Wang1Minhui Zhu1 Chunyan Jiao1Dandan Wang2Junjie Zhu2Wenya Yuan1Di Zhu1
Chunyan Jiao1Dandan Wang2Junjie Zhu2Wenya Yuan1Di Zhu1 Congcong Xu1
Congcong Xu1 Hongyan Wang1Minghui Zhou1Xie Zhang3
Hongyan Wang1Minghui Zhou1Xie Zhang3 Jisen Shi2
Jisen Shi2 Jinhui Chen2*
Jinhui Chen2*- 1College of Life Sciences, Hefei Normal University, Hefei, China
- 2Key Laboratory of Forest Genetics and Biotechnology, Ministry of Education of China, Co-Innovation Center for the Sustainable Forestry in Southern China, Nanjing Forestry University, Nanjing, China
- 3State Key Laboratory of Utilization of Woody Oil Resource, Hunan Academy of Forestry, Changsha, China
The WUSCHEL-related homeobox (WOX) proteins are widely distributed in plants and play important regulatory roles in growth and development processes such as embryonic development and organ development. Here, series of bioinformatics methods were utilized to unravel the structural basis and genetic hierarchy of WOX genes, followed by regulation of the WOX genes in four Euphorbiaceae species. A genome-wide survey identified 59 WOX genes in Hevea brasiliensis (H. brasiliensis: 20 genes), Jatropha curcas (J. curcas: 10 genes), Manihot esculenta (M. esculenta: 18 genes), and Ricinus communis (R. communis: 11 genes). The phylogenetic analysis revealed that these WOX members could be clustered into three close proximal clades, such as namely ancient, intermediate and modern/WUS clades. In addition, gene structures and conserved motif analyses further validated that the WOX genes were conserved within each phylogenetic clade. These results suggested the relationships among WOX members in the four Euphorbiaceae species. We found that WOX genes in H. brasiliensis and M. esculenta exhibit close genetic relationship with J. curcas and R. communis. Additionally, the presence of various cis-acting regulatory elements in the promoter of J. curcas WOX genes (JcWOXs) reflected distinct functions. These speculations were further validated with the differential expression profiles of various JcWOXs in seeds, reflecting the importance of two JcWOX genes (JcWOX6 and JcWOX13) during plant growth and development. Our quantitative real-time PCR (qRT-PCR) analysis demonstrated that the JcWOX11 gene plays an indispensable role in regulating plant callus. Taken together, the present study reports the comprehensive characteristics and relationships of WOX genes in four Euphorbiaceae species, providing new insights into their characterization.
Introduction
The WUSCHEL-related homeobox (WOX) transcription factors are essential for cell fate determination, cell differentiation, regulation of various developmental processes and plant growth across the plant kingdom (Ueda et al., 2011; Costanzo et al., 2014; He et al., 2019). WOX genes are characterized by a conserved 60–66 residues long DNA-binding homeobox (HB) domain (helix 1-loop-helix 2-turn-helix 3) (Li et al., 2016). In addition, the tail region of the WOX family member are comprised of three specific conserved domains such as the WUS-box (TLXLFPXX), Ethylene-responsive element binding factor-associated Amphiphilic Repression (EAR) domain and acidic domain (Dolzblasz et al., 2016).
Haecker et al. (2004) categorized WOX genes of Arabidopsis thaliana (A. thaliana) into three major phylogenetic clades such as ancient clade (WOX10, WOX13, and WOX14 subfamilies), intermediate clade (WOX8, WOX9, WOX11, and WOX12 subfamilies) and modern/WUS clade (WUS and WOX1-7 subfamilies) (Haecker et al., 2004). In the ancient clade, WOX13 and WOX14 play key roles in regulating flowers and fruits development and conducting tissues (Romera-Branchat et al., 2013; Costanzo et al., 2014). Genes TaWOX8, TaWOX9 and TaWOX12 from the intermediate clade could promote the immature callus proliferation in Triticum aestivum (T. aestivum) embryos (Shi et al., 2021). WOX11, regulated root de novo organogenesis in A. thaliana (Liu et al., 2014), showed high expression of gene ultimately increases nutrient uptake by callus in Oryza Sativa (O. sativa) (Wan Abdullah et al., 2021). In the modern/WUS clade, WOX genes are involved in the regulated development of various types of meristems and are differentially expressed in distinct species (Tvorogova et al., 2021). For instance, WUS, WOX4, and WOX5 exhibit stem cell regulatory functions in the shoot apical meristem (SAM), vascular cambium (VCAM) and root apical meristem (RAM), respectively (Wang et al., 2011; Bueno et al., 2021). CsWOX1 plays a major role in leaf vein morphology, leaf size and cell proliferation (Wang H. et al., 2020). Wu et al. (2007) reported that WOX2 is necessary for the proper development of the embryonic apical region. AtWOX2 positively regulates early embryonic development (Wu et al., 2007). In Picea abies (P. abies), PaWOX2 and PaWOX8/9 are expressed at high levels in the early growth stages of zygotic and somatic embryos (Palovaara et al., 2010). PaWOX3 and OsWOX3 could regulate the hormone expression levels in cells, hence, promoting cell division and development (Yoo et al., 2013; Yang et al., 2021). TaWOX5 may be involved in root formation or development and hormone regulation during somatic embryogenesis in T. aestivum (Zhao et al., 2014). Overall, WOX genes play are not only responsible for the maintenance of stem cells in the apical meristem of shoots, roots, and the VCAM, the development of lateral organs, the formation of floral organs, the dynamic balance of embryonic development and postembryonic development, and the regulation of callus proliferation. To date, WOX gene members have been identified in various plant species, i.e., Sorghum bicolor (11 genes; Zhang et al., 2010), Zea mays (21 genes; Zhang et al., 2010), Solanum lycopersicum (10 genes; Dolzblasz et al., 2016), Salix suchowensis (15 genes; Wang et al., 2018), Ricinus communis (R. communis; http://castorbean.Jcvi. Org/index.php: 11 genes; Han et al., 2019), Jatropha curcas (J. curcas) (12 genes; Tang et al., 2019), and Cucumis sativus (11 genes; Han et al., 2021).
Euphorbiaceae, belongs to dicotyledonous angiosperms, which is widely distributed in tropical and subtropical regions (Webster, 1994). This family is comprised of approximately 300 genera and 800 species. The members of Euphorbiaceae including M. esculenta have extensive medicinal values, including antimicrobial, anti-inflammatory, anticancer and antioxidant activities (Webster, 1994; Santos-Silva et al., 2021). In addition, some Euphorbiaceae species have important economic value because they produce rubber, starch, and other compounds (Li et al., 2019). For example, Hevea brasiliensis (H. brasiliensis) is not only a perennial cross-pollinat tree with a long juvenile stage (Wang Y. et al., 2021) but also the main raw material for many industries, especially the tire industry (Supriya and Priyadarshan, 2019). Due to the high oil contents and adaptability to different environmental conditions, Euphorbiaceae plants such as J. curcas are considered as potential biodiesel sources in response to the current global energy crisis (Debnath and Bisen, 2008; Natarajan and Parani, 2011; Artimo et al., 2012; Maghuly and Laimer, 2013). As the main source of castor oil, R. communis has a high mineral oil accumulation capacity (Rehn et al., 2020). At present, the available whole-genome sequences of H. brasiliensis (Rahman et al., 2013), J. curcas (Hirakawa et al., 2012), M. esculenta (Prochnik et al., 2012) and R. communis (Chan et al., 2010) constitute an important foundation for current research and future molecular exploration. Although members of WOX genes have been identified and studied in many plant species, hitherto a comprehensive research on WOX genes of economically important Euphorbiaceae species is lacking.
In this study, the physicochemical properties, phylogenetic relationships, gene structure, conserved motifs, and codon usage bias of WOX genes in four Euphorbiaceae species were analyzed. Based on this analysis, overall molecular features of WOX genes were further clarified. Additionally, we studied J. curcas to identify the cis-acting elements in its JcWOX gene and determine its expression profile. The results provide further insights into the evolution and genetic relationships of four Euphorbiaceae species, as well as a basis for verifying the function of WOX transcription factors and screening WOX members in Euphorbiaceae species, which might be essential for plant growth and development.
Materials and Methods
Collection of Gene Sequences From Four Euphorbiaceae Species
The genome sequences of J. curcas were retrieved from the Jatropha Genome Database (http://www.kazusa.or.jp/jatropha/; JAT_r4.5; Hirakawa et al., 2012). The genome sequences of H. brasiliensis (Rahman et al., 2013), M. esculenta (Prochnik et al., 2012), and R. communis (Chan et al., 2010) were obtained from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/).
Identification of WOX Genes in Four Euphorbiaceae Species
Redundant sequences were removed from the results obtained using the following two methods described below to identify WOX genes in four Euphorbiaceae species. (1) The hidden Markov model (HMM) file (PF00046) associated with WOX protein family-related domains was downloaded from the Pfam database (http://pfam.xfam.org/) (Cao et al., 2017). Using HMMER 3.0 software to analyze WOX proteins, the HMM file (PF00046) of four species of Euphorbiaceae was screened with an E-value cut off of 0.001. (2) 15 WOX protein sequences from A. thaliana were used as queries for alignment with the total protein sequences of the four Euphorbiaceae species by BLASTP with an E-value of 0.0001 to confirm the accuracy. The candidate sequences of WOX genes were detected using SMART (http://smart.embl-heidelberg.de/) to verify the presence of homeodomains. Ultimately, each WOX gene was assigned a unique name by BLASTP.
Physicochemical Properties of WOX Proteins
The properties of WOX proteins, including their molecular weight (MW), isoelectric point (pI), instability index (II), aliphatic index (AI), were computed with ExPASy (https://web.expasy.org/protparam/; Artimo et al., 2012).
Phylogenetic Classification and Gene Structure Analysis
All sequences from the four Euphorbiaceae species were subsequently aligned by DNAMAN software (version 6.0) to visualize the results. Moreover, multiple sequence alignment of all WOX protein sequences was performed by ClustalX 2.0 (Thompson et al., 1997). An interspecific phylogenetic tree containing the species was generated using the Neighbor-Joining (NJ) method with MEGA X software, including 1,000 bootstrap replicates (Kumar et al., 2018). In addition, the exon and intron structures of all WOX genes were obtained from the online Gene Structure Display Server (GSDS; http://gsds.cbi.pku.edu.cn; Hu et al., 2015).
Conserved Motif Analysis
The conserved motifs of WOX proteins from the four Euphorbiaceae species were analyzed by MEME online server (http://meme-suite.org/). The maximum number of identified motifs was set to 25 (E-value = 0.0001; Li et al., 2018).
Codon Usage Bias Analysis
The codon usage bias of WOX genes in the four Euphorbiaceae species was analyzed using CodonW software (version 1.4.2). The relative synonymous codon usage (RSCU) of WOX genes in these species was calculated in such a way that RSCU value >1 represents a codon with positive bias; an RSCU value = 1 indicates no bias (Wang Z. et al., 2021). The relative frequencies of synonymous codons (RFSCs) in the J. curcas WOX genes (JcWOXs) were also calculated. When the RFSC value exceeds 60% or is 0.50 times higher than the average frequency of synonymous codons, the codon is a high-frequency codon (Zhou et al., 2007a). Furthermore, a comparative analysis of the codon usage frequency among WOX genes of the four Euphorbiaceae species and four representative species, A. thaliana, Nicotiana tabacum (N. tabacum), Populus trichocarpa (P. trichocarpa), and O. sativa, was performed. The ratio ranged from 0.50 to 2.00 reflects that the codon usage bias of WOX genes is highly similar to that of the representative plant species (Zhou et al., 2007b).
Cis-Acting Elements of JcWOXs
The genomic sequences located 2000 bp upstream from the initiation codon (ATG) were considered as the promoter fragments. The promoter sequences of JcWOXs were downloaded from the Jatropha Genome Database (Hirakawa et al., 2012). Meanwhile, PlantCARE online sever (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to analyze potential cis-acting elements (Lescot et al., 2002).
Spatial Expression Profiles of JcWOXs
J. curcas RNA transcriptome data from three different tissues (leaves, roots, and seeds) were downloaded from the Sequence Read Archive (SRA) database to investigate the spatial expression characteristics of JcWOXs (Wu et al., 2015; Zou et al., 2016). The spatial expression profiles of each identified JcWOX in three different tissues were determined using three transcriptome datasets. The spatial characteristics of 10 JcWOXs were explored using the R library heatmap, and the trends were ultimately presented as color changes. Detailed information of the three transcriptome datasets was provided in Supplementary Table S1.
Temporal Expression Profiles of JcWOXs
The RNA transcriptome data collected at seven developmental stages (14, 19, 25, 29, 35, 41, and 45 days after pollination-DAP) of seeds were downloaded following the spatial expression profiles of JcWOXs (Jiang et al., 2012). The temporal characteristics of the 10 JcWOXs were explored by generating the heatmap. The detailed data information of the seven transcriptomes is presented in Supplementary Table S1.
Expression Profiles of JcWOXs in Calli
J. curcas calli were selected as experimental materials to test whether WOX gene has functional conservation in regulating callus proliferation. The third leaf from the top of Guangxi J. curcas was selected as the explant. Next, the explants were disinfected by washing with water for 0.50–1.00 min, followed by soaking in 70% ethanol for 15 s and 3% NaClO solution for 12–16 min. Callus induction was performed by inoculating the explant in dorsal contact medium supplemented with MS, BAP (+0.80 mg/L), TDZ (+0.60 mg/L) and NAA (+0.10 mg/L). Explants were cultured in the dark at 25°C. The whole cycle of callus culture was approximately 42 days, which was divided into three stages: 14 days (S1), 28 days (S2), and 42 days (S3), as illustrated in Supplementary Figure S1. With obvious differences in the growth states and callus characteristics were selected as samples. Three biological replicates of each sample were analyzed. All samples were collected and cryo-preserved in liquid nitrogen.
Total RNA was extracted from J. curcas callus was extracted using the FastPure plant total RNA isolation kit (RC401). The RNA concentration was determined using a Nanodrop-2000 spectrophotometer (Thermo, Inc.). The integrity of total RNA was detected by performing gel electrophoresis. First-strand cDNAs were synthesized using the reverse transcriptase method with the HiScriptR III 1st Strand cDNA Synthesis Kit (+ gDNA wiper) (R312-01/02). Additionally, the samples in triplicate were analyzed with qRT-PCR utilizing AceQ qPCR SYBR Green Master Mix (without ROX), as previously described by Wang D. et al. (2020). Four reference genes (JcGAPDH, JcEF1α, JcActin, and JcTUB8) were selected as candidate reference genes according to Zhang et al. (2013), and JcGAPDH and JcActin were determined as reference genes by using semi-quantitative RT-PCR (Liu et al., 2010). Based on the coding DNA sequences (CDSs) of WOX gene from J. curcas, primers were designed using Oligo7 and SnapGene (Supplementary Table S2).
Results
Identification of WOX Genes in Four Euphorbiaceae Species
Both HMMER and local BLAST searches were performed simultaneously with the HMM file (PF00046), and the sequences of 15 members of the WOX family in A. thaliana were used as templates to identify all possible WOX proteins in the four Euphorbiaceae species and confirm the accuracy of the identification. Ultimately, 59 WOX genes were identified in the genomes of the four Euphorbiaceae species such as, 20 in H. brasiliensis, 10 in J. curcas, 18 in M. esculenta, and 11 in R. communis. These WOX genes were named by BLASTP (Supplementary Table S3).
Analysis of Physicochemical Properties
Next, the candidate WOX gene sequences were isolated, followed by analyzing the physicochemical properties of their corresponding proteins utilizing by ExPASy. As shown in Supplementary Table S3, their protein lengths and predicted MW varied little, and few differences in their pI were observed. For H. brasiliensis, the WOX proteins were 185–398 residues long, corresponding MW ranged from 21,346.99 to 44,058.10 Da, and their pI values ranged from 5.63 (HbWOX11b) to 9.51 (HbWOX4b). For J. curcas, the corresponding WOX proteins ranged in length from 190 (JcWOX7) to 392 (JcWOX1) aa, their MW ranged from 21,747.21 to 43,900.70 Da, and their pI values ranged from 5.15 (JcWOX14) to 9.51 (JcWOX4). WOX proteins in M. esculenta were 182 to 391 residues long, with the pI values ranging from 5.61 (MeWUSb) to 9.40 (MeWOX4a). For R. communis, the WOX proteins ranged in length from 192 to 401 residues long, and their pI values ranged from 5.35 (RcWOX14) to 9.42 (RcWOX1a). Therefore, the physicochemical properties of the WOX proteins in the four Euphorbiaceae species were similar. The hydropathicity index represents the hydrophilicity of a protein (Li et al., 2016). The AI ranged from 44.34 to 82.58, indicating that WOX proteins are thermally stable. As shown in Supplementary Table S3, the hydrophilicity index of 59 members of the WOX protein families was negative, suggesting that the members of the four WOX protein families of the Euphorbiaceae species were hydrophilic.
Phylogenetic Classification and Gene Structure Analysis
According to the visualized of sequence alignment (Supplementary Figure S2), the homeodomain of WOX protein of A. thaliana and the four Euphorbiaceae species presented high conserved structures. The number of amino acid residues in WOX homeodomain fluctuated slightly, from 63 to 66. These results proved that the WOX proteins of four Euphorbiaceae species were highly conserved, further indicating that the identified WOX genes were correctly identified.
In order to compare the evolutionary relationships of WOX genes of A. thaliana and the four Euphorbiaceae species, NJ phylogenetic trees of 74 WOX proteins from A. thaliana (15), H. brasiliensis (20), J. curcas (10), M. esculenta (18), and R. communis (11) were constructed with MEGA X software to compare the evolutionary relationships of WOX genes from A. thaliana and the four Euphorbiaceae species. It was observed from Figure 1 that WOX genes from the five species were classified into three major clades, which is consistent with the previously known clades. For instance, ancient clade (WOX10, WOX13, and WOX14 subfamilies), intermediate clade (WOX8, WOX9, and WOX11), and modern/WUS clade contains eight subfamilies (WUS and WOX1-7). The number of WOX genes in the modern/WUS clade (38 genes, 64.4%) was greater than that in the intermediate (12 genes, 20.3%) and the ancient clades (nine genes, 15.3%). Moreover, phylogenetic tree analysis reflected that the homologous genes lies in close proximity, suggesting that the WOX genes in Euphorbiaceae species were highly evolutionarily conserved. Furthermore, members of the WOX transcription factors in H. brasiliensis and M. esculenta were located close to each other in the evolutionary tree.
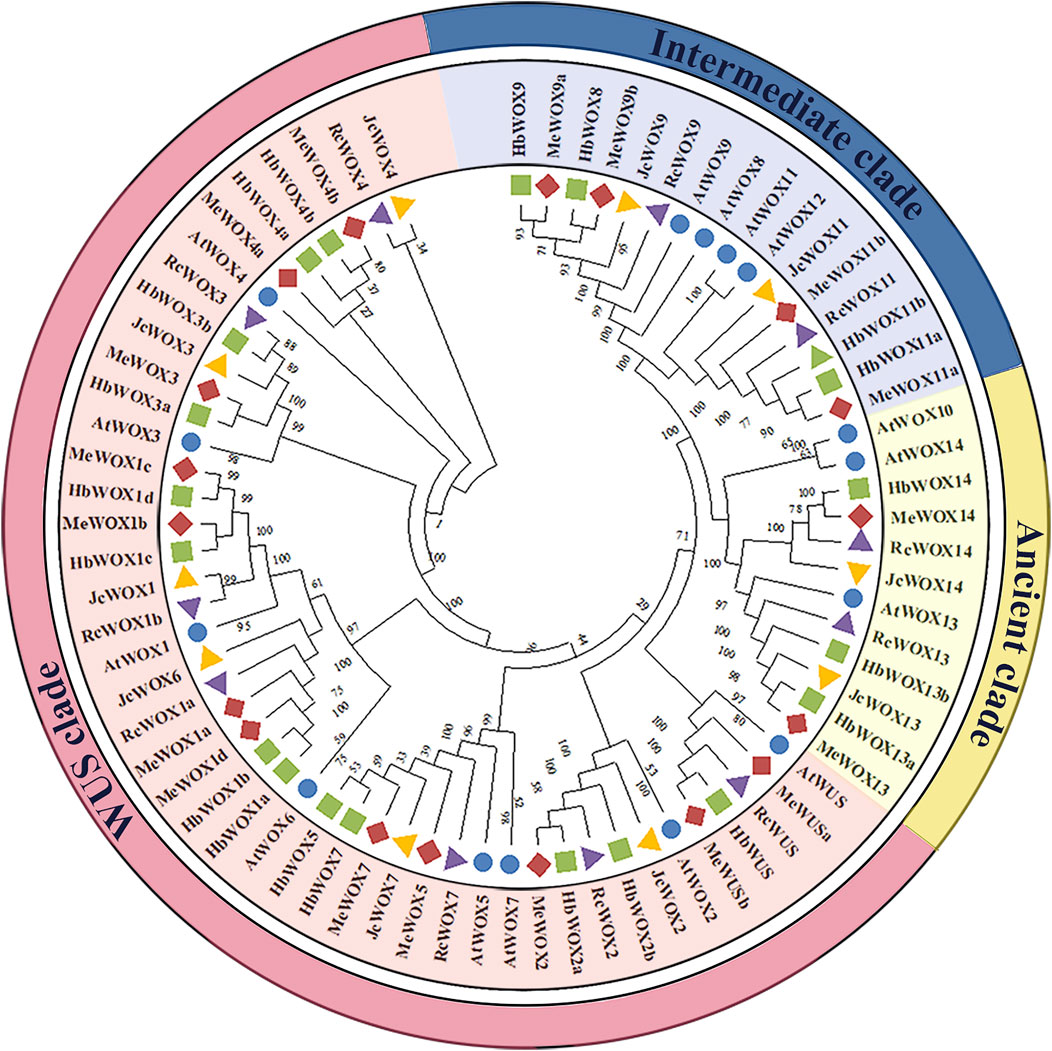
FIGURE 1. Phylogenetic tree of WOX genes from four Euphorbiaceae species and Arabidopsis thaliana. The five plant species that are members of the WOX gene family can be divided into three main clade classes, the ancient clade includes three subfamilies, the intermediate clade includes three subfamilies, and the modern/WUS clade contains eight subfamilies.
Structure analysis of 59 WOX genes was performed with GSDS. WOX gene is composed of exons, introns, and untranslated regions (UTRs), as illustrated in Figure 2. The number of exons in each WOX gene ranged from two to four. The WOX genes members in the same subclade usually presented similar exon–intron patterns. For example, the members of the closely related modern clade i.e., HbWOX7, JcWOX7, HbWOX5, MeWOX5, MeWOX7, and RcWOX7 exhibit similar gene lengths with two exons. In addition, members of the ancient clade had two intron insertion sites, while members of the intermediate and modern/WUS clades exhibited different intron insertion patterns, and the number of introns ranged from one to four. Moreover, five genes (HbWOX9, MeWOX9a, MeWOX9b, HbWOX13b, and MeWOX13) were more than 3 kb. Interestingly, most JcWOX and RcWOX members were located in adjacent regions and had similar gene structures, showing that the similarity between J. curcas and R. communis species is high and the kinship is close. The same phenomenon was also found in most of the HbWOX and MeWOX members. Collectively, these results indicated that similar gene structures also reflect the conservation of WOX gene members and their evolutionary relationships.
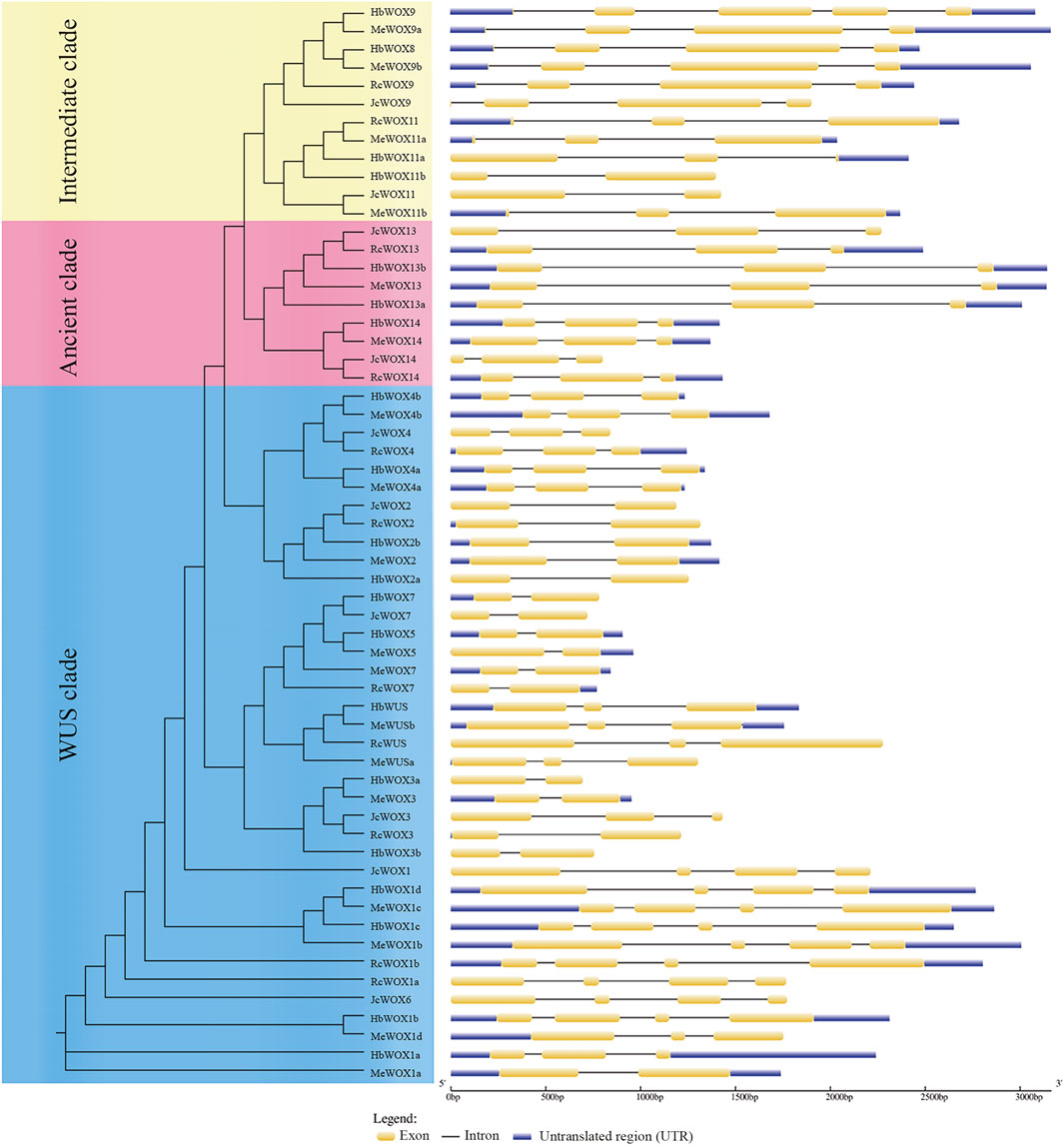
FIGURE 2. WOX gene structure according to the phylogenetic relationship. The composition of the WOX gene includes exons, introns, and untranslated regions (UTRs).
Conserved Motif Analyses
The WOX family typically contains additional conserved motifs that likely have involved in different functions. 25 conserved motifs were identified among the 59 WOX proteins in the four Euphorbiaceae species using the MEME online portal to obtain a comprehensive understanding of the structural features and relationships of the WOX proteins. The phylogenetic tree (Figure 3) was divided into ancient, intermediate, and modern/WUS clades. Among the 25 putative motifs, motif 1 and motif 2 were present in all WOX protein sequences, indicating that motifs 1 and 2 are characteristic domains of the proteins encoded by 59 WOX genes. Structural analysis demonstrated that proteins corresponding HbWOX3a and MeWOX3 exhibit only two motifs while HbWOX1d and MeWOX1c possess 10 motifs. With the exception of JcWOX3, motif 5 was present at a high frequency in nearly all WOX proteins in the modern/WUS clade, but this motif was not present in the intermediate clade or ancient clade, consistent with the distribution of the WUS box (TLXLFPXX). In particular, motif 4 existed only in the ancient clade. Furthermore, the conserved motifs of JcWOX and RcWOX were highly similar, as observed for HbWOX and MeWOX. The WOX members within the same clade had similar motif structures, suggesting that the homologous WOX genes in different plant species are closely related. These results are consistent with previously reported phylogenetic tree analysis and could further strengthens the classification of WOX subfamily members.
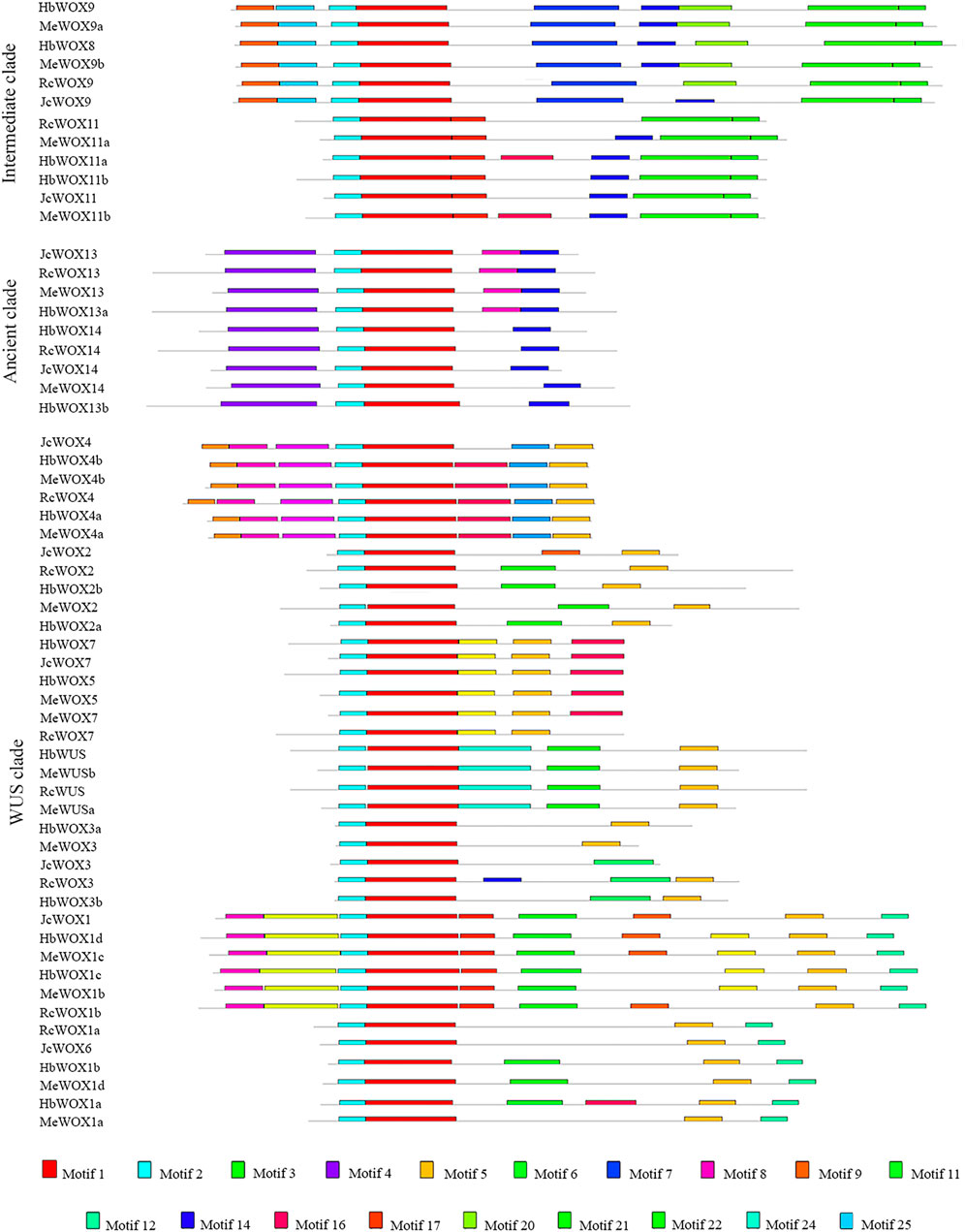
FIGURE 3. Distribution of conserved motifs identified among four Euphorbiaceae species. 25 conserved motifs identified in 59 WOX proteins, which can also be divided into ancient, intermediate, and modern/WUS clades according to the phylogenetic tree.
Codon Usage Bias Analysis
CodonW software was used to study the codon usage bias in the 59 WOX genes from the four Euphorbiaceae species. As shown in Table 1, the codon usage of WOX genes across the Euphorbiaceae species was quite conserved. According to the RSCU values, strong commonalities were detected in WOX genes from the four Euphorbiaceae species. 19, 23, and two identical codons displayed positive bias, negative bias and no bias, respectively; the remaining 20 codons differed in bias. The strongest positive bias in all species presents same codons (AGA), however, the codons with the strongest negative bias were different such as J. curcas (CGC) and M. esculenta (CGC), H. brasiliensis (GCG) and R. communis (ACG). An analysis of the RFSCs of the JcWOXs revealed four identical high-frequency codons from WOX genes of the four Euphorbiaceae species: GCA, GAU, CCA, and AGA. However, some differences in the high-frequency codons were observed. For instance, CUU, CAA, and ACU were high-frequency codons in M. esculenta but not in the other three species. Additionally, the codon usage frequency ratios between the four Euphorbiaceae species and four model organisms were analyzed (Table 2). The ratio of six codons (GCU, CGC, UCG, UAA, UAG, and UGA; GCU, GAU, CGC, UAA, UAG, and UGA) were greater than 2.00 and lower than 0.50 in the JcWOX genes than in the A. thaliana and P. trichocarpa genes, respectively. Comparative analysis reflected, five similar codons (GCC, CGG, UAA, UAG, and UGA) in RcWOX genes and P. trichocarpa genes, five codons (GCC, CGC, UAA, UAG, and UGA) showed differences in the comparison between the MeWOX genes and N. tabacum genes, and six codons (GCG, UUG, CGG, UAA, UAG, and UGA) showed differences in the comparison between the HbWOX genes and P. trichocarpa genes. Hence, we considered P. trichocarpa to be the optimal choice for comparing exogenous WOX genes in J. curcas, R. communis, and H. brasiliensis and N. tabacum the optimal choice for comparing exogenous WOX genes in M. esculenta.
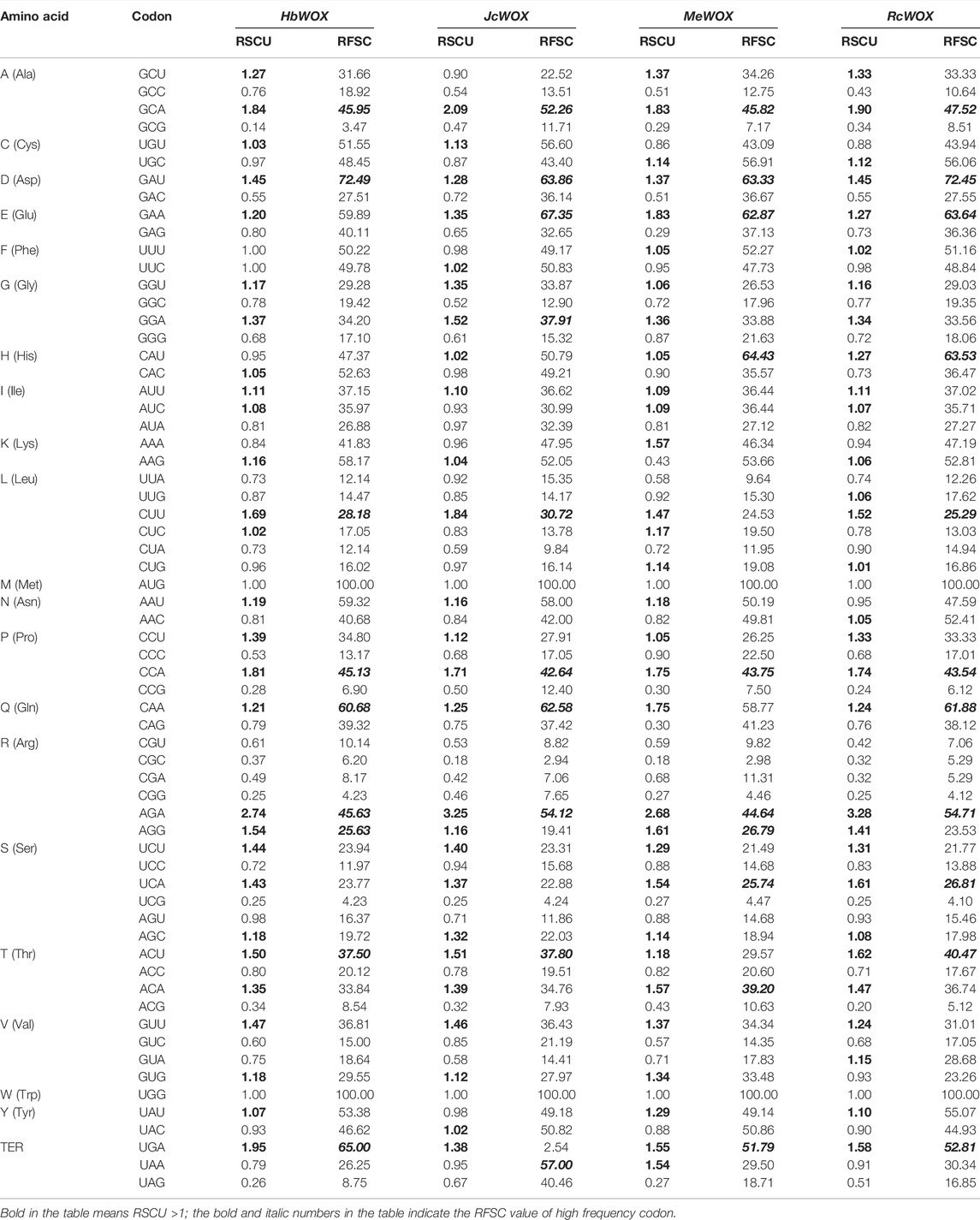
TABLE 1. RSCU and RFSC of codons in WOX genes of four Euphorbiaceae species.
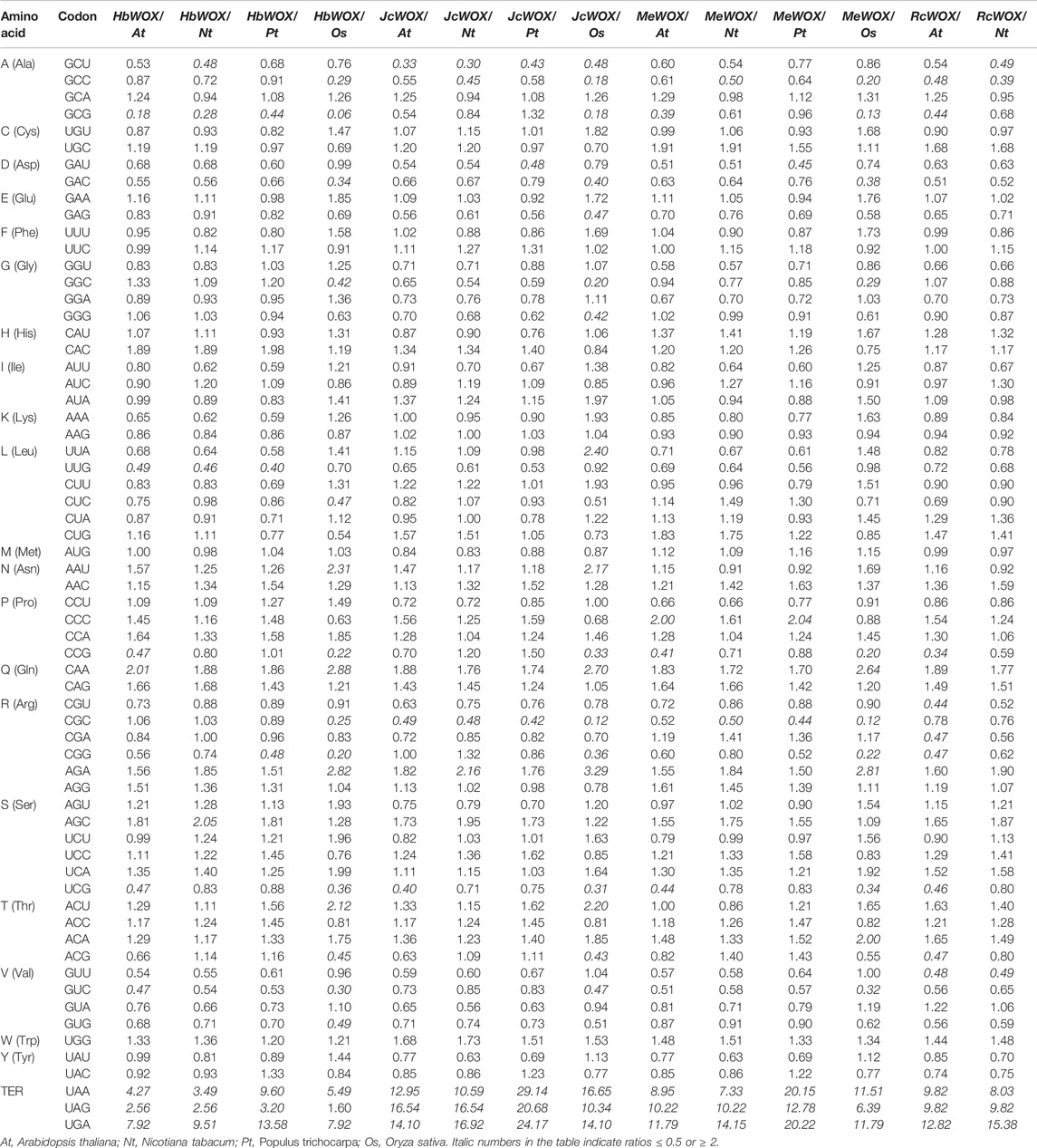
TABLE 2. Comparison of codon usage frequency of WOX genes of four Euphorbiaceae species and four representative plant genomes.
Analysis of Cis-Acting Elements of the JcWOXs
As shown in Figure 4, the cis-acting elements identified in this study were divided into three types based on their functions such as stress responses, hormone responses, and growth and development. The largest group of cis-acting elements was related to stress at 104 among the JcWOXs. For most of JcWOXs, the number of identical cis-acting elements usually ranged from one to three. A large number of cis-acting elements were associated with growth and development—10. The most abundant cis-acting elements were MYCs (related to abscisic acid (ABA) induction), of which 26 existed in the promoters of nine JcWOX genes, excluding JcWOX6. Moreover, AREs (related to the anaerobic stress response) were the most widespread cis-acting elements in the JcWOXs (present within all 10 JcWOX gene members), suggesting that JcWOXs are widely involved in reactions related to anaerobic stress. In addition, JcWOX14 contained the most stress-related cis-acting elements, four MYB elements (related to the drought stress response) and four LTR elements (JcWOXs related to the low-temperature stress responses), which reflected that JcWOX14 plays an indispensable role in the drought stress response and low-temperature response. With respect to hormone-related promoter elements, JcWOX1 contains the largest number six types and 11 elements—suggesting that JcWOX1 is likely involved in hormone-related responses. ABREs (related to the ABA reaction element) were the most widespread cis-acting elements in the JcWOXs (present within eight JcWOX gene members, excluding JcWOX2 and JcWOX7), which demonstrates that JcWOXs are widely involved in reactions related to ABA-related reactions. In terms of cis-acting elements related to growth and development, JcWOX1 and JcWOX4 contained the most—seven types and 10 elements. In addition, the G-box (related to the light response element) was present within seven JcWOX gene members, excluding JcWOX2, JcWOX7, and JcWOX11. Interestingly, JcWOX13 contains four GT1 motifs that participate in the light response.
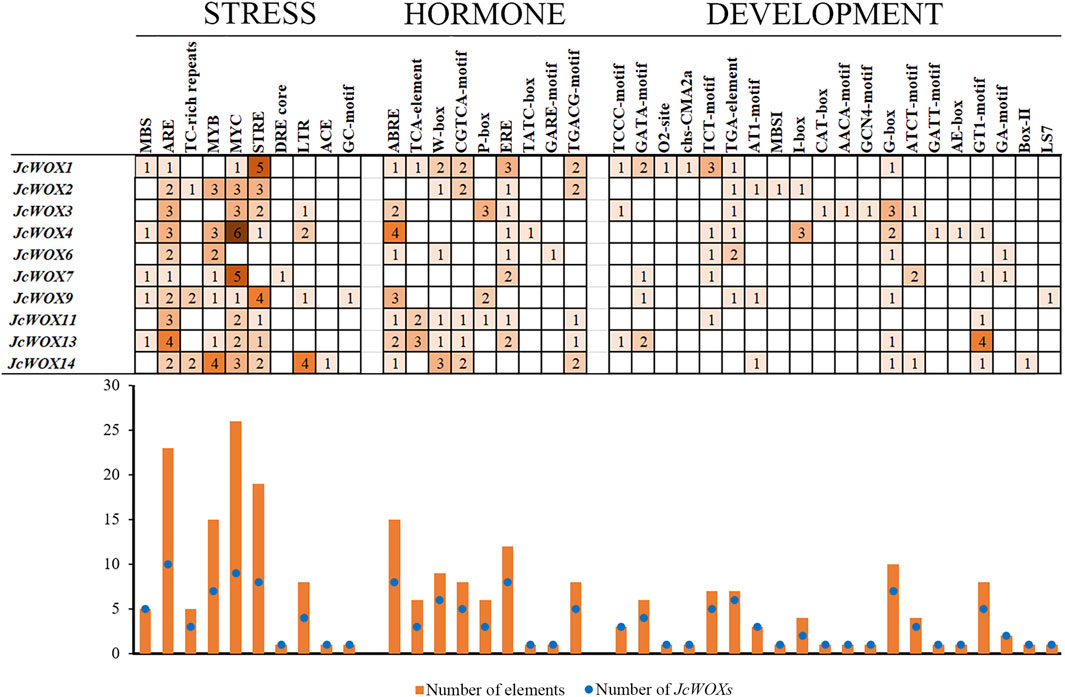
FIGURE 4. Cis-acting elements in the promoter regions of 10 JcWOX genes. The number of cis-acting elements related to stress was the largest.
JcWOXs are Specifically Involved in Spatial Expression
We investigated previously published transcriptome profiles of various tissue types to determine the critical role of WOX genes in the development of J. curcas. Based on a heatmap of these data, we found that four genes (JcWOX4, JcWOX7, JcWOX9, and JcWOX11) in the roots and five genes (JcWOX1, JcWOX2, JcWOX6, JcWOX13, and JcWOX14) in the seeds. Nevertheless, WOX genes in J. curcas were expressed at low levels in the leaves (Figure 5). Based on these results, the JcWOXs might play more significant roles in the growth and development of roots and seeds than in leaves. In summary, the expression patterns of JcWOXs in different tissues respectively are conducive to identifying functional genes expressed in J. curcas.
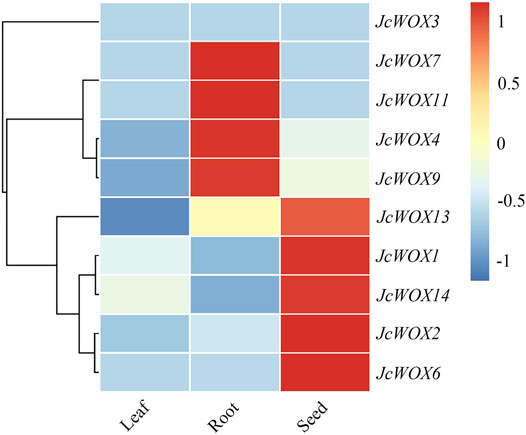
FIGURE 5. Tissue-specific expression patterns of 10 JcWOX genes. The role of JcWOX family members in the growth and development of roots and seeds may be more important than that of leaves.
JcWOXs are Specifically Involved in Temporal Expression
Based on the analysis of the expression of WOX genes and their variation trends across seven J. curcas seed development stages (Figure 6), all JcWOXs were expressed at relatively high level during the initial stage (19 DAP, 25 DAP, and 29 DAP) and at relatively low level during the fast oil accumulation stage (35 DAP, 41 DAP, and 45 DAP). The expression of four WOX genes (JcWOX3, JcWOX6, JcWOX11, and JcWOX13) markedly decreased between 29 and 35 DAP. Interestingly, the expression of two WOX genes (JcWOX6 and JcWOX13) decreased continuously during all seven stages. In general, the expression of WOX genes tended to decrease during the seven stages, hence, we deduced that JcWOX6 and JcWOX13 play a significant anti-regulatory role in seed development.
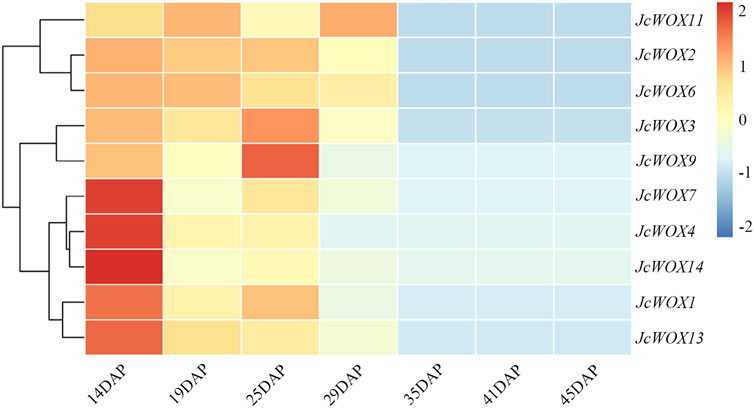
FIGURE 6. Expression patterns of 10 JcWOX genes during seven seed developmental stages.
qRT-PCR Analysis of JcWOXs Expression in Calli
The expression levels of JcWOXs were analyzed in calli collected at three stages (S1, S2, and S3) via qRT-PCR to better understand their functions. We performed qRT-PCR on WOX genes of J. curcas; the findings for some genes with polar expression levels were discarded (JcWOX1, JcWOX2, JcWOX3, JcWOX6, and JcWOX9). The results of qRT-PCR experiments (Figure 7) showed that among the five WOX genes of J. curcas, the expression of four WOX genes (JcWOX4, JcWOX7, JcWOX13, and JcWOX14) did not display obvious variation in the three biological repetitions. The expression of the JcWOX11 gene was significantly upregulated at S1. Moreover, the gene variation in the expression of JcWOX11 was most obvious. These findings provide new insights and a comprehensive understanding of the characteristics of JcWOXs for functional validation in the future.
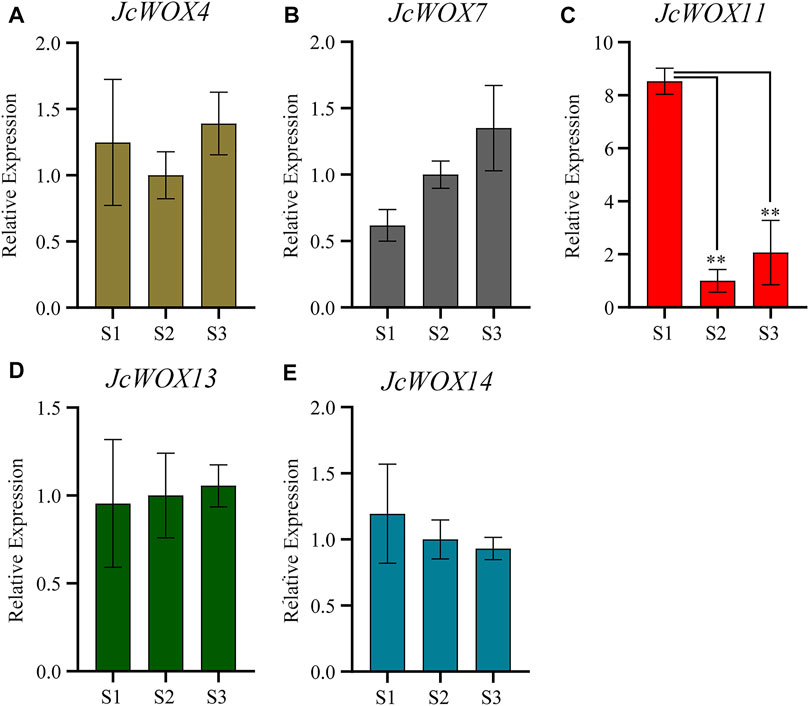
FIGURE 7. Expression of five JcWOX genes in callus in qRT-PCR experiments (Student’s t-test: **p < 0.01). (A) JcWOX4; (B) JcWOX7; (C) JcWOX11; (D) JcWOX 13; (E) JcWOX14.
Discussion
In the recent study, WOX genes in four Euphorbiaceae species (H. brasiliensis (20 members), J. curcas (10 members), M. esculenta (18 members) and R. communis (11 members) ) were identified. Through the ExPASy analysis, we found that their protein lengths, predicted MW, and pI values did not differ substantially, and we concluded that the physicochemical properties of WOX proteins from the four Euphorbiaceae species were thermostable and hydrophilic, with very similar physicochemical properties. As plant-specific transcription factors, WOX family transcription factors are widely involved in the regulation of plant meristems and play distinct roles in the development of different tissues (Haecker et al., 2004). The function of WOX genes is conserved between A. thaliana and the four Euphorbiaceae species, and we speculated that WOX family transcription factors play a significant role in plant growth and development by regulating the subsistence of plants (Vandenbussche et al., 2009; Zhang and Tadege, 2015).
Our evolutionary tree revealed that 59 WOX genes were divided into three major clades. Phylogenetic analysis of S. lycopersicum, O. sativa, A. thaliana, and Petunia hybrida have also divided WOX genes into three separate clades: the ancient, intermediate, and modern/WUS clades (Figure 1). The ancient clade includes WOX13 and WOX14 (Lin et al., 2013; Dolzblasz et al., 2016), consistent with the evolutionary relationship between WOX genes in Rosaceae species (Cao et al., 2017); the intermediate clade contain WOX8, WOX9, and WOX11; and the modern/WUS clade consists of WUS and WOX1-7. Our results were consistent with the representative taxonomic results from phylogenetic analysis of P. abies (Hedman et al., 2013). In addition, in the ancient clade, AtWOX13 promotes fruit embryonic development (Romera-Branchat et al., 2013); in the intermediate clade, OsWOX11 is expressed specifically in the cambium and promotes adventitious root formation (Zhao et al., 2009); in the modern/WUS clade, AtWUS could maintain stem cell population stability in SAMs; and AtWOX5 has similar roles in the apical meristem (Wang et al., 2018). According to the evolutionary results of orthologous genes, JcWOX13 may be involved in fruit embryonic development, and JcWOX11 may participate in root growth.
Moreover, the genetic relationships of WOX members in different species were very close and members of the WOX families in M. esculenta and H. brasiliensis were evolutionarily close to each other in the phylogenetic tree, suggesting that these two species were more closely related than J. curcas and R. communis. These results were further supported by conserved motif analysis and gene structure analysis. The gene structure, motif number and location of each WOX subfamily were highly similar, which further indicated that the structure of WOX genes is quite conserved. The conserved motif analysis revealed that all proteins encoded by 59 WOX genes contained both motif 1 and motif 2, showing that these motifs are homeodomains of WOX proteins (Figure 3). We found that each clade contained specific conserved motifs, implying that these specific motifs are likely required for specific functions among the different subfamily members. For instance, motif 5 exists only in the modern/WUS clade of WOX genes, and the sequence represented by motif 5 is consistent with that of the WUS-box (TLXLFPXX), revealing that motif 5 is the specific WOX structural domain WUS-box, which is consistent with previous studies (Zhang et al., 2010; Cao et al., 2017; Wang et al., 2018).
Analysis of RSCU and RFSC values based on the codon bias demonstrated similarities in their codon usage of the four Euphorbiaceae species (Table 1), we mainly found that there were similarities in their codon usage, that is, there were commonalities between the four plants. We also realized that the codon usage of WOX genes of the Euphorbiaceae species was quite conserved. Based on the RSCU values, the four Euphorbiaceae species had 19 identical codons with positive bias. Moreover, the RFSC values reflected four identical high-frequency codons in WOX genes of the four Euphorbiaceae species: GCA, GAU, CCA, and AGA. After comparing the codon usage frequencies, we considered P. trichocarpa to be the optimal choice for comparison of exogenous WOX genes in J. curcas, R. communis, and H. brasiliensis; N. tabacum considered as the optimal choice for exogenous WOX genes in M. esculenta. Interestingly, N. tabacum to be was considered the optimal choice for comparison with the whole J. curcas whole genome in a previous study (Wang Z. et al., 2021), which is different from the conclusion in the present study. This result further shows the significance and value of our study of WOX genes and provides a direction for future research. At the same time, this finding indicates that the expression patterns receptors of exogenous genes differ for different genes even although they are from the same species.
We identified three major categories of cis-acting elements among JcWOXs: stress-related, hormone-related, and growth-related elements (Figure 4). Among them, cis-acting elements related to growth and development were the most abundant, with 22 types were identified. G-boxes constituted the greatest number of cis-acting elements associated with growth and development, which demonstrates that WOX genes are actively involved in light response regulation. JcWOX14 contained the most stress-related promoters with seven types across 18 different promoters, proving that JcWOX14 is likely involved in stress-related responses (Figure 5). This echoes a previous study of JcWOX5 in transgenic rice in which the gene increased its sensitivity to the drought stress (Tang et al., 2020). These results confirmed the hypothesis that JcWOXs played a potential role in the response to abiotic stress. Additionally, JcWOX1 contained the most hormone-related elements, with six types across 10 different promoters, suggesting that JcWOX1 is likely involved in hormone-related responses.
Based on our expression profile, we propose that these genes may play a major part in the growth and development of roots (JcWOX4, JcWOX7, JcWOX9, and JcWOX11) or seeds (JcWOX1, JcWOX2, JcWOX6, JcWOX13, and JcWOX14). According to the expression analysis of WOX genes and their variation trends across seven J. curcas seed development stages, these genes play an anti-regulatory role during seed development (Figure 6). Furthermore, the expression of four WOX genes (JcWOX3, JcWOX6, JcWOX11, and JcWOX13) showed a significant downward trend between 29 and 35 DAP. In addition, JcWOX6 and JcWOX13 decreased continuously during all seven stages. Numerous studies have shown that WOX genes play different roles in the development of rice roots, stems, and leaves (Li et al., 2012; Wang et al., 2017; Zhou et al., 2017). The WOX6 gene plays a significant role in the regulation of seed development, especially for the growth and development of seeds under water-deficient conditions (Shafique Khan et al., 2021). In particular, WOX6 gene in rice regulates the asymmetric expression of auxin, resulting in the appearance of rice tiller horns (Zhang et al., 2018). WOX13 is expressed in plant pods, flowers and seeds, with the most prominent expression in roots (Han et al., 2019). In addition, WOX13 could be also significantly expressed in reproductive organs and the developing embryo in cotton (Yang et al., 2017). The AtWOX13 mutant remained defective after grafting, suggesting that the WOX13 gene is essential for tissue repair in seed plants (Ikeuchi et al., 2022). The qRT-PCR results indicated that the expression of JcWOX11 in the callus had the most obvious change. Our results demonstrated that JcWOX11 had the highest expression level in roots, and the expression of JcWOX4, JcWOX7, JcWOX13, and JcWOX14 showed no obvious variations (Figure 7). AtWUS is expressed in organizing center cells of the SAM and regulates the maintenance of shoot stem cells (Dolzblasz et al., 2016). The feedback regulation mechanisms in the SAM and RAM are similar, while the expression pattern of SAM may have similarities in root apex (Wang et al., 2011). Further analysis of the shoot-containing SAM provides a new direction for our further research. In A. thaliana, lateral root formation can be promoted through an AtWOX11-mediated pathway, thus further promoting callus initiation (Kong et al., 2016; Guo et al., 2018). The high expression of OsWOX11 enhanced the formation of adventitious roots and finally increased the uptake of nutrients by callus (Wan Abdullah et al., 2021). OsWOX11 can integrate auxin and cytokinin signals, thereby promoting cell division during crown root development and playing a crucial role in the regulation of root development (Zhao et al., 2009). This study lays a foundation for further research in the field related to WOX genes and provides important reference value.
Conclusions
In the present study, 59 WOX genes from four Euphorbiaceae species were identified and comprehensively analyzed to clarify their overall and molecular characteristics. Moreover, cis-acting elements and expression patterns of JcWOXs were determined under different spatiotemporal conditions. The results showed that the structures and genetic relationships of WOX genes in H. brasiliensis, J. curcas, M. esculenta, and R. communis providing a foundation for the functional verification of functional genes in WOX genes. Moreover, analyses of the spatial and temporal expression pattern analysis of JcWOXs in different tissues and at variety of stages of seed development indicated that two JcWOXs (JcWOX6 and JcWOX13) may be involved in plant growth and development. Furthermore, qRT-PCR proved that JcWOX11 was particularly worthy of further functional analysis in promoting the callus proliferation. Overall, our study lays a foundation for future research in exploring the molecular mechanisms through which WOX genes drive development in Euphorbiaceae species and in other species.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
Conceptualization, ZW, JS and JC; methodology, ZW, JS and JC; software, CJ, WY and DZ; validation, ML, YW and MhZ; formal analysis, ZW., QC, HX, BH, CJ, DW and JZ; investigation, MgZ; resources, XZ; data curation, CX and HW; writing original draft preparation, ZW, QC, HX and BH; writing review and editing, ZW, ML, YW and MhZ; visualization, CJ; supervision, JS; project administration, JC; funding acquisition, ZW, JC, MgZ. and CX All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant No. 32071784), Foundation of State Key Laboratory of Utilization of Woody Oil Resource (grant No. GZKF202103), Open Fund of State Key Laboratory of Tea Plant Biology and Utilization (grant No. SKLTOF20200129), Outstanding Young Talents in Colleges and universities of Anhui Province (Grant No. gxyq2020040), Special Scientific Research Project of Graduate Tutor of Hefei Normal University (grant No. DSKY17), Key Project of Natural Science Foundation of Universities in Anhui Province (Grant No. KJ2019A0728), and Priority Academic Program Development of Jiangsu Higher Education Institutions (Grant No. PAPD), the College Students’ Innovation and Entrepreneurship Project in Anhui Province (grant No. 14098006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.878554/full#supplementary-material
References
Artimo, P., Jonnalagedda, M., Arnold, K., Baratin, D., Csardi, G., de Castro, E., et al. (2012). ExPASy: SIB Bioinformatics Resource Portal. Nucleic Acids Res. 40, W597–W603. doi:10.1093/nar/gks400
Bueno, N., Cuesta, C., Centeno, M. L., Ordás, R. J., and Alvarez, J. M. (2021). In Vitro Plant Regeneration in Conifers: The Role of WOX and KNOX Gene Families. Genes 12, 438. doi:10.3390/genes12030438
Cao, Y., Han, Y., Meng, D., Li, G., Li, D., Abdullah, M., et al. (2017). Genome-Wide Analysis Suggests the Relaxed Purifying Selection Affect the Evolution of WOX Genes in Pyrus Bretschneideri, Prunus Persica, Prunus Mume, and Fragaria Vesca. Front. Genet. 8, 78. doi:10.3389/fgene.2017.00078
Chan, A. P., Crabtree, J., Zhao, Q., Lorenzi, H., Orvis, J., Puiu, D., et al. (2010). Draft Genome Sequence of the Oilseed Species Ricinus communis. Nat. Biotechnol. 28, 951–956. doi:10.1038/nbt.1674
Costanzo, E., Trehin, C., and Vandenbussche, M. (2014). The Role of WOX Genes in Flower Development. Ann. Bot. 114, 1545–1553. doi:10.1093/aob/mcu123
Debnath, M., and Bisen, P. S. (2008). Jatropha Curcas L., a Multipurpose Stress Resistant Plant with a Potential for Ethnomedicine and Renewable Energy. Curr. Pharm. Biotechnol. 9, 288–306. doi:10.2174/138920108785161541
Dolzblasz, A., Nardmann, J., Clerici, E., Causier, B., van der Graaff, E., Chen, J., et al. (2016). Stem Cell Regulation by Arabidopsis WOX Genes. Mol. Plant 9, 1028–1039. doi:10.1016/j.molp.2016.04.007
Guo, F., Zhang, H., Liu, W., Hu, X., Han, N., Qian, Q., et al. (2018). Callus Initiation from Root Explants Employs Different Strategies in Rice and Arabidopsis. Plant Cell Physiol. 59, 1782–1789. doi:10.1093/pcp/pcy095
Haecker, A., Gross-Hardt, R., Geiges, B., Sarkar, A., Breuninger, H., Herrmann, M., et al. (2004). Expression Dynamics of WOX Genes Mark Cell Fate Decisions during Early Embryonic Patterning in Arabidopsis thaliana. Development 131, 657–668. doi:10.1242/dev.00963
Han, N., Tang, R., Chen, X., Xu, Z., Ren, Z., and Wang, L. (2021). Genome-Wide Identification and Characterization of WOX Genes in Cucumis Sativus. Genome 64 (8), 761–776. doi:10.1139/gen-2020-0029
Han, W., Li, G., Feng, L., Yan, X., Bai, Y., Li, M., et al. (2019). Genome-Wide Characterization Analysis of WOX Transcription Factors and Response to Abiotic Stresses in Ricinus communis L. J. Agric. Sci. 33 (10), 1921–1927. doi:10.11869/j.issn.100-8551.2019.10.1921
He, P., Zhang, Y., Liu, H., Yuan, Y., Wang, C., Yu, J., et al. (2019). Comprehensive Analysis of WOX Genes Uncovers that WOX13 is Involved in Phytohormone-Mediated Fiber Development in Cotton. BMC Plant Biol. 19, 312. doi:10.1186/s12870-019-1892-x
Hedman, H., Zhu, T., von Arnold, S., and Sohlberg, J. J. (2013). Analysis of the WUSCHEL-RELATED HOMEOBOX Gene Family in the Conifer Picea Abiesreveals Extensive Conservation as well as Dynamic Patterns. BMC Plant Biol. 13, 89. doi:10.1186/1471-2229-13-89
Hirakawa, H., Tsuchimoto, S., Sakai, H., Nakayama, S., Fujishiro, T., Kishida, Y., et al. (2012). Upgraded Genomic Information of Jatropha Curcas L. Plant Biotechnol. 29, 123–130. doi:10.5511/plantbiotechnology.12.0515a
Hu, B., Jin, J., Guo, A., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 31, 1296–1297. doi:10.1093/bioinformatics/btu817
Ikeuchi, M., Iwase, A., Ito, T., Tanaka, H., Favero, D. S., Kawamura, A., et al. (2022). Wound-Inducible WUSCHEL-RELATED HOMEOBOX 13 is Required for Callus Growth and Organ Reconnection. Plant Physiol. 188, 425–441. doi:10.1093/plphys/kiab510
Jiang, H., Wu, P., Zhang, S., Song, C., Chen, Y., Li, M., et al. (2012). Global Analysis of Gene Expression Profiles in Developing Physic Nut (Jatropha Curcas L.) Seeds. PLoS One 7, e36522. doi:10.1371/journal.pone.0036522
Kong, D., Hao, Y., and Cui, H. (2016). The WUSCHEL Related Homeobox Protein WOX7 Regulates the Sugar Response of Lateral Root Development in Arabidopsis thaliana. Mol. Plant 9, 261–270. doi:10.1016/j.molp.2015.11.006
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi:10.1093/molbev/msy096
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a Database of Plant Cis-Acting Regulatory Elements and a Portal to Tools for in Silico Analysis of Promoter Sequences. Nucleic Acids Res. 30, 325–327. doi:10.1093/nar/30.1.325
Li, J., Gao, X., Sang, S., and Liu, C. (2019). Genome-Wide Identification, Phylogeny, and Expression Analysis of the SBP-Box Gene Family in Euphorbiaceae. BMC Genomics 20, 912. doi:10.1186/s12864-019-6319-4
Li, J., Yuan, Y., Lu, Z., Yang, L., Gao, R., Lu, J., et al. (2012). Glabrous Rice 1, Encoding a Homeodomain Protein, Regulates Trichome Development in Rice. Rice 5, 1–10. doi:10.1186/1939-8433-5-32
Li, X., Hamyat, M., Liu, C., Ahmad, S., Gao, X., Guo, C., et al. (2018). Identification and Characterization of the WOX Family Genes in Five Solanaceae Species Reveal Their Conserved Roles in Peptide Signaling. Genes 9, 260. doi:10.3390/genes9050260
Li, X., Liu, C., Li, W., Zhang, Z., Gao, X., Zhou, H., et al. (2016). Genome-Wide Identification, Phylogenetic Analysis and Expression Profiling of the WOX Family Genes in Solanum lycopersicum. Hereditas. 38, 444–460. doi:10.16288/j.yczz.15-499
Lin, H., Niu, L., McHale, N. A., Ohme-Takagi, M., Mysore, K. S., and Tadege, M. (2013). Evolutionarily Conserved Repressive Activity of WOX Proteins Mediates Leaf Blade Outgrowth and Floral Organ Development in Plants. Proc. Natl. Acad. Sci. U.S.A. 110, 366–371. doi:10.1073/pnas.1215376110
Liu, B., Wang, W., Gao, J., Chen, F., Wang, S., Xu, Y., et al. (2010). Molecular Cloning and Characterization of a Jasmonate Biosynthetic Pathway Gene for Allene Oxide Cyclase from Jatropha Curcas. Acta Physiol. Plant. 32, 531–539. doi:10.1007/s11738-009-0430-0
Liu, J., Sheng, L., Xu, Y., Li, J., Yang, Z., Huang, H., et al. (2014). WOX11 and 12 are Involved in the First-Step Cell Fate Transition during de Novo Root Organogenesis in Arabidopsis. Plant Cell 26 (3), 1081–1093. doi:10.1105/tpc.114.122887
Maghuly, F., and Laimer, M. (2013). Jatropha Curcas, a Biofuel Crop: Functional Genomics for Understanding Metabolic Pathways and Genetic Improvement. Biotechnol. J. 8, 1172–1182. doi:10.1002/biot.201300231
Natarajan, P., and Parani, M. (2011). De Novo Assembly and Transcriptome Analysis of Five Major Tissues of Jatropha Curcas L. Using GS FLX Titanium Platform of 454 Pyrosequencing. BMC Genomics 12, 191. doi:10.1186/1471-2164-12-191
Palovaara, J., Hallberg, H., Stasolla, C., and Hakman, I. (2010). Comparative Expression Pattern Analysis of WUSCHEL-Related Homeobox 2 (WOX2) and WOX8/9 in Developing Seeds and Somatic Embryos of the Gymnosperm Picea Abies. New Phytol. 188, 122–135. doi:10.1111/j.1469-8137.2010.03336.x
Prochnik, S., Marri, P. R., Desany, B., Rabinowicz, P. D., Kodira, C., Mohiuddin, M., et al. (2012). The Cassava Genome: Current Progress, Future Directions. Trop. Plant Biol. 5, 88–94. doi:10.1007/s12042-011-9088-z
Rahman, A. Y. A., Usharraj, A. O., Misra, B. B., Thottathil, G. P., Jayasekaran, K., Feng, Y., et al. (2013). Draft Genome Sequence of the Rubber Tree Hevea Brasiliensis. BMC Genomics. 14, 75. doi:10.1186/1471-2164-14-75
Rehn, L. S., Rodrigues, A. A., Vasconcelos-Filho, S. C., Rodrigues, D. A., de Freitas Moura, L. M., Costa, A. C., et al. (2020). Ricinus communis as a Phytoremediator of Soil Mineral Oil: Morphoanatomical and Physiological Traits. Ecotoxicology 29, 129–139. doi:10.1007/s10646-019-02147-6
Romera-Branchat, M., Ripoll, J. J., Yanofsky, M. F., and Pelaz, S. (2013). The WOX13 Homeobox Gene Promotes Replum Formation in the Arabidopsis Thaliana Fruit. Plant J. 73, 37–49. doi:10.1111/tpj.12010
Santos-Silva, C. A. D., Vilela, L. M. B., Oliveira-Silva, R. L. d., Silva, J. B. D., Machado, A. R., Bezerra-Neto, J. P., et al. (2021). Cassava (Manihot Esculenta) Defensins: Prospection, Structural Analysis and Tissue-Specific Expression under Biotic/Abiotic Stresses. Biochimie 186, 1–12. doi:10.1016/j.biochi.2021.03.012
Shafique Khan, F., Zeng, R., Gan, Z., Zhang, J., and Hu, C. (2021). Genome-Wide Identification and Expression Profiling of the WOX Gene Family in Citrus Sinensis and Functional Analysis of a CsWUS Member. Int. J. Mol. Sci. 22, 4919. doi:10.3390/ijms22094919
Shi, L., Wang, K., Du, L., Song, Y., Li, H., and Ye, X. (2021). Genome-Wide Identification and Expression Profiling Analysis of WOX Family Protein-Encoded Genes in Triticeae Species. Int. J. Mol. Sci. 22, 9325. doi:10.3390/ijms22179325
Supriya, R., and Priyadarshan, P. M. (2019). Genomic Technologies for Hevea Breeding. Adv. Genet. 104, 1–73. doi:10.1016/bs.adgen.2019.04.001
Tang, Y., Bao, X., Jian, W., Lou, H., Feng, Y., Jie, T., et al. (2019). Genome-Wide Identification and Expression Analysis of the WOX Gene Family in Physic Nut. Mol. Plant Breed. 17, 1154–1162. doi:10.13271/j.mpb.017.001154
Tang, Y., Li, H., Guan, Y., Li, S., Xun, C., Dong, Y., et al. (2020). Genome-Wide Identification of the Physic Nut WUSCHEL-Related Homeobox Gene Family and Functional Analysis of the Abiotic Stress Responsive Gene JcWOX5. Front. Genet. 11, 670. doi:10.3389/fgene.2020.00670
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic acids. Res. 25, 4876–4882. doi:10.1093/nar/25.24.4876
Tvorogova, V. E., Krasnoperova, E. Y., Potsenkovskaia, E. A., Kudriashov, A. A., Dodueva, I. E., and Lutova, L. A. (2021). What Does the WOX Say? Review of Regulators, Targets, Partners. Mol. Biol. Mosk. 55, 362–391. doi:10.31857/S0026898421030174
Ueda, M., Zhang, Z., and Laux, T. (2011). Transcriptional Activation of Arabidopsis Axis Patterning Genes WOX8/9 Links Zygote Polarity to Embryo Development. Dev. Cell 20, 264–270. doi:10.1016/j.devcel.2011.01.009
Vandenbussche, M., Horstman, A., Zethof, J., Koes, R., Rijpkema, A. S., and Gerats, T. (2009). Differential Recruitment of WOX Transcription Factors for Lateral Development and Organ Fusion in Petunia and Arabidopsis. Plant Cell 21 (8), 2269–2283. doi:10.1105/tpc.109.065862
Wan Abdullah, W. M. A. N., Tan, N. P., Low, L. Y., Loh, J. Y., Wee, C. Y., Md Taib, A. Z., et al. (2021). Calcium Lignosulfonate Improves Proliferation of Recalcitrant Indica Rice Callus via Modulation of Auxin Biosynthesis and Enhancement of Nutrient Absorption. Plant Physiology Biochem. 161, 131–142. doi:10.1016/j.plaphy.2021.01.046
Wang, D., Hao, Z., Long, X., Wang, Z., Zheng, X., Ye, D., et al. (2020). The Transcriptome of Cunninghamia Lanceolata Male/Female Cone Reveal the Association between MIKC MADS-Box Genes and Reproductive Organs Development. BMC Plant Biol. 20, 508. doi:10.1186/s12870-020-02634-7
Wang, H., Niu, H., Li, C., Shen, G., Liu, X., Weng, Y., et al. (2020). WUSCHEL-Related Homeobox1 (WOX1) Regulates Vein Patterning and Leaf Size in Cucumis Sativus. Hortic. Res. 7, 182. doi:10.1038/s41438-020-00404-y
Wang, X., Bi, C., Wang, C., Ye, Q., Yin, T., and Ye, N. (2018). Genome-Wide Identification and Characterization of WUSCHEL-Related Homeobox (WOX) Genes in Salix Suchowensis. J. For. Res. 30, 1811–1822. doi:10.1007/s11676-018-0734-2
Wang, Y., Li, H., Zhou, Y., Guo, D., Zhu, J., and Peng, S. (2021). Transcriptomes Analysis Reveals Novel Insight into the Molecular Mechanisms of Somatic Embryogenesis in Hevea Brasiliensis. BMC Genomics 22, 183. doi:10.1186/s12864-021-07501-9
Wang, Y., Song, F., Zhu, J., Zhang, S., Yang, Y., Chen, T., et al. (2017). GSA: Genome Sequence Archive. Genom. Proteom. Bionf. 15, 14–18. doi:10.1016/j.gpb.2017.01.001
Wang, Z., Chen, J., and Shi, J. (2011). Progress on WUS/CLV Feedback Regulatory Mechanisms in Plant Stem Cells. Sci. Silvae Sin. 47 (004), 159–165. doi:10.3724/SP.J.1011.2011.00353
Wang, Z., Wang, G., Cai, Q., Jiang, Y., Wang, C., Xia, H., et al. (2021). Genomewide Comparative Analysis of Codon Usage Bias in Three Sequenced Jatropha Curcas. J. Genet. 100, 20. doi:10.1007/s12041-021-01271-9
Webster, G. L. (1994). Classification of the Euphorbiaceae. Ann. Mo. Botanical Gard. 81, 3–32. doi:10.2307/2399908
Wu, P., Zhou, C., Cheng, S., Wu, Z., Lu, W., Han, J., et al. (2015). Integrated Genome Sequence and Linkage Map of Physic Nut (Jatropha Curcas L.), a Biodiesel Plant. Plant J. 81, 810–821. doi:10.1111/tpj.12761
Wu, X., Chory, J., and Weigel, D. (2007). Combinations of WOX Activities Regulate Tissue Proliferation during Arabidopsis Embryonic Development. Dev. Biol. 309, 306–316. doi:10.1016/j.ydbio.2007.07.019
Yang, R., Wu, Z., Bai, C., Sun, Z., Wang, M., Huo, Y., et al. (2021). Overexpression of PvWOX3a in Switchgrass Promotes Stem Development and Increases Plant Height. Hortic. Res. 8, 252. doi:10.1038/s41438-021-00678-w
Yang, Z., Gong, Q., Qin, W., Yang, Z., Cheng, Y., Lu, L., et al. (2017). Genome-Wide Analysis of WOX Genes in Upland Cotton and Their Expression Pattern under Different Stresses. BMC Plant Biol. 17, 113. doi:10.1186/s12870-017-1065-8
Yoo, S. C., Cho, S. H., and Paek, N. C. (2013). Rice WUSCHEL-Related Homeobox 3A (OsWOX3A) Modulates Auxin-Transport Gene Expression in Lateral Root and Root Hair Development. Plant Signal. Behav. 8, e25929. doi:10.4161/psb.25929
Zhang, F., and Tadege, M. (2015). Repression of AS2 by WOX Family Transcription Factors is Required for Leaf Development in Medicago and Arabidopsis. Plant Signal. Behav. 10, e993291. doi:10.4161/15592324.2014.993291
Zhang, L., He, L., Fu, Q., and Xu, Z. (2013). Selection of Reliable Reference Genes for Gene Expression Studies in the Biofuel Plant Jatropha Curcas Using Real-Time Quantitative PCR. Int. J. Mol. Sci. 14, 24338–24354. doi:10.3390/ijms141224338
Zhang, N., Yu, H., Yu, H., Cai, Y., Huang, L., Xu, C., et al. (2018). A Core Regulatory Pathway Controlling Rice Tiller Angle Mediated by the LAZY1-Dependent Asymmetric Distribution of Auxin. Plant Cell 30, 1461–1475. doi:10.1105/tpc.18.00063
Zhang, X., Zong, J., Liu, J., Yin, J., and Zhang, D. (2010). Genome-Wide Analysis of WOX Gene Family in Rice, Sorghum, Maize, Arabidopsis and Poplar. J. Integr. Plant Biol. 52, 1016–1026. doi:10.1111/j.1744-7909.2010.00982.x
Zhao, S., Jiang, Q., Ma, J., Zhang, X., Zhao, Q., Wang, X., et al. (2014). Characterization and Expression Analysis of WOX5 Genes from Wheat and its Relatives. Gene 537, 63–69. doi:10.1016/j.gene.2013.12.022
Zhao, Y., Hu, Y., Dai, M., Huang, L., and Zhou, D. (2009). The WUSCHEL-Related Homeobox Gene WOX11 is Required to Activate Shoot-Borne Crown Root Development in Rice. Plant Cell 21, 736–748. doi:10.1105/tpc.108.061655
Zhou, M., Tong, C., and Shi, J. (2007a). Analysis of Codon Usage between Different Poplar Species. J. Genet. Genomics 34, 555–561. doi:10.1016/S1673-8527(07)60061-7
Zhou, M., Tong, C., and Shi, J. (2007b). A Preliminary Analysis of Synonymous Codon Usage in Poplar Species. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao 33 (4), 285–293. doi:10.2471/BLT.13.118778
Zhou, S., Jiang, W., Long, F., Cheng, S., Yang, W., Zhao, Y., et al. (2017). Rice Homeodomain Protein WOX11 Recruits a Histone Acetyltransferase Complex to Establish Programs of Cell Proliferation of Crown Root Meristem. Plant Cell 29, 1088–1104. doi:10.1105/tpc.16.00908
Zou, Z., Yang, L., Gong, J., Mo, Y., Wang, J., Cao, J., et al. (2016). Genome-Wide Identification of Jatropha Curcas Aquaporin Genes and the Comparative Analysis Provides Insights into the Gene Family Expansion and Evolution in Hevea Brasiliensis. Front. Plant Sci. 7, 395. doi:10.3389/fpls.2016.00395
Keywords: WOX genes, Euphorbiaceae, Jatropha curcas, bioinformatics analysis, gene expression
Citation: Wang Z, Cai Q, Xia H, Han B, Li M, Wang Y, Zhu M, Jiao C, Wang D, Zhu J, Yuan W, Zhu D, Xu C, Wang H, Zhou M, Zhang X, Shi J and Chen J (2022) Genome-Wide Identification and Comparative Analysis of WOX Genes in Four Euphorbiaceae Species and Their Expression Patterns in Jatropha curcas. Front. Genet. 13:878554. doi: 10.3389/fgene.2022.878554
Received: 18 February 2022; Accepted: 22 April 2022;
Published: 30 June 2022.
Edited by:
Xiaoming Song, North China University of Science and Technology, ChinaReviewed by:
Mingyong Tang, Key Laboratory of Tropical Plant Resource and Sustainable Use, Xishuangbanna Tropical Botanical Garden (CAS), ChinaFei Chen, Hainan University, China
Wen Xiaopeng, Guizhou University, China
Copyright © 2022 Wang, Cai, Xia, Han, Li, Wang, Zhu, Jiao, Wang, Zhu, Yuan, Zhu, Xu, Wang, Zhou, Zhang, Shi and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhui Chen, Y2hlbmpoQG5qZnUuZWR1LmNu