
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 19 April 2022
Sec. Genomics of Plants and the Phytoecosystem
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.874648
This article is part of the Research Topic Genetics and Epigenetics: Plausible Role in Development of Climate Resilient Crops View all 17 articles
 Faisal Saeed1
Faisal Saeed1 Usman Khalid Chaudhry1
Usman Khalid Chaudhry1 Allah Bakhsh2
Allah Bakhsh2 Ali Raza3
Ali Raza3 Yasir Saeed4
Yasir Saeed4 Abhishek Bohra5
Abhishek Bohra5 Rajeev K. Varshney3,5,6*
Rajeev K. Varshney3,5,6*Plants offer a habitat for a range of interactions to occur among different stress factors. Epigenetics has become the most promising functional genomics tool, with huge potential for improving plant adaptation to biotic and abiotic stresses. Advances in plant molecular biology have dramatically changed our understanding of the molecular mechanisms that control these interactions, and plant epigenetics has attracted great interest in this context. Accumulating literature substantiates the crucial role of epigenetics in the diversity of plant responses that can be harnessed to accelerate the progress of crop improvement. However, harnessing epigenetics to its full potential will require a thorough understanding of the epigenetic modifications and assessing the functional relevance of these variants. The modern technologies of profiling and engineering plants at genome-wide scale provide new horizons to elucidate how epigenetic modifications occur in plants in response to stress conditions. This review summarizes recent progress on understanding the epigenetic regulation of plant stress responses, methods to detect genome-wide epigenetic modifications, and disentangling their contributions to plant phenotypes from other sources of variations. Key epigenetic mechanisms underlying stress memory are highlighted. Linking plant response with the patterns of epigenetic variations would help devise breeding strategies for improving crop performance under stressed scenarios.
Agriculture plays a vital role in feeding the rapidly growing world population. For fibre, fuel, and food, we usually depend on major crops such as cotton, maize, sugarcane, rice, barley, wheat, and soybean. An increase in world population day by day puts tremendous pressure on current food crop production systems (Chaudhry et al., 2021a; Junaid et al., 2021). Additionally, climate change results in several weather adversaries, and frequent disease and pest attacks threaten crop production worldwide (Raza et al., 2020). These stresses interfere with plant’s physiological, biochemical, molecular, and cellular mechanisms, ultimately reducing overall growth, and production (Raza et al., 2020; Raza et al., 2021). With time, for better growth and cultivation, human beings carried out an artificial selection of thousands of plants to get desired traits in plants (Herron et al., 2020). In recent years, new ways and tools have been discovered for the betterment of crops. For instance, affordable genetic systems for profiling of the plant genomes have led to the development of robust molecular diagnostics for rapid and precise selection of desirable crop plants (Gökçe and Chaudhry, 2020; Gökçe et al., 2021). In parallel, targeted genetic modification has been greatly benefitted by the availability of the whole genome sequence information in different crop species. For instance, genome editing techniques such as clustered regularly interspaced short palindromic repeats (CRISPR) technology using RNA-guided nucleases have facilitated the alteration of a variety of important plant phenotypes (Ma et al., 2016; Saeed et al., 2020; Dangol et al., 2021).
Feeding the growing world population would require harnessing the latest discoveries in plant epigenetics. A growing body of literature suggests that epigenetics contributes to many vital traits in different plant species. The term epigenetics refers to heritable changes in the phenotype, which are not due to a change in DNA sequence (Heard and Martienssen, 2014). In other words, epigenetics involves alterations in gene expressions that are stably transmitted from generation to generation. Plant epigenetics combines different research fields that help us understand how plants adjust their phenotypes other than modifying their DNA sequence under extreme stress conditions. The molecular processes encompassing epigenetics are DNA methylation and histone modification (Henikoff and Greally, 2016). The structure of chromatin is regulated by methylation of DNA and modification of histones, and these modifications remain crucial to the repression or activation of a gene (Ganai, 2020). The review article presents the recent advances in plant epigenetics, emphasizing plant stress response. The underlying mechanisms are discussed in the following sections.
Rollin Hotchkiss in 1948 identified DNA methylation. After 30 years, Holliday and Pugh proposed that DNA methylation is an essential epigenetic hallmark (Holliday and Pugh, 1975). Methylation of DNA is among the key epigenetic mechanisms regulating various bioprocesses. In plants, DNA methylation is initiated by the RNA-directed DNA methylation (RdDM) pathway. In all sequence contexts, DNA methyltransferases DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) catalyzes the process of methylation (Zhang et al., 2018a). RdDM is further divided into canonical and non-canonical pathways. In the canonical pathway, RNA polymerase IV synthesizes single-stranded RNA (ssRNA) that are changed into double-stranded RNAs (dsRNAs) by involving RNA Dependent RNA polymerase 2 (RDR2). DICER LIKE 3 (DCL3) involves in the cutting off this dsRNA. This dsRNA is than converted into 24 bases of small interfering RNA (siRNAs) (Zhang et al., 2018a). The second part of this pathway depends on the transcription of non-coding RNAs (ncRNAs) by the involvement of Pol V (Liu et al., 2018). Canonical pathway initiated by Pol IV- dependent 24 nt siRNAs whereas non-canonical pathway initiated by pol II and small RNAs (sRNAs) are involved. These sRNAs are produced from dsRNAs. sRNAs consist of 21–24 nt but are cut by different DCL proteins. These sRNAs are involved in triggereing post transcriptional gene silencing (PTGS) (Martínez de Alba et al., 2013; Cuerda-Gil and Slotkin, 2016). Methylation of cytosine involves a change in cytosine to 5- methylcytosine (5-mC), and it takes place when a methyl group is transferred at 5′ positions of S- adenosyl methionine. Cytosine methylation, though dependent on plant species, ranges from 6 to 25% therefore, plants have high levels of methylcytosine (Steward et al., 2000). Methylation of DNA cytosine occurs in three sequence contexts in plants, i.e., CpG, CpHpG, and CpHpH (H known as for T, A, and C). After replication, DNA methylation of CpG and CpHpG can easily be copied because the methylation at the symmetrical CG and CHG sites can be maintained during DNA replication. The methylation at the non-symmetrical CHH sites is not maintained during replication and occurs de novo (Karlsson et al., 2011). Plants store this epigenetic memory at the vegetative phase under different stresses and transfer it to the next generation, which gets established during the development of germline cells. DNA is methylated at both the gene body and the promoter regions, and due to this methylation, it allows the gene to remain suppressed. Thus, lower methylation helps increase the expression of a gene (Finnegan et al., 1998). Several processes are involved in the epigenetic mechanism. These mechanisms are cytosine methylation, chromatin proteins, and post-translational modifications (Abdolhamid Angaji et al., 2010).
Different developmental, physiological, and stress stimuli are involved in regulating DNA methylation in plants. Histone and DNA methylation are inter-reliant procedures. In Arabidopsis mutant met1, CpG causes the loss of methylation of H3K9 (Soppe et al., 2002; Tariq et al., 2003). But the loss of methylation of H3K9 in kryptonite (KYP) did not affect the methylation of CpG site (Jasencakova et al., 2003). Consequently, it shows that methylation of CpHpG site is partly dependent on the activity of KYP due to loss of H3K9 methylation (Jackson et al., 2002). The process of demethylation and methyltransferase both control the methylation of DNA. Demethylation follows two routes, i.e., the passive and the active. During the process of cell division, methylated DNA can vanish from the genome. If maintenance machinery present in dividing cells can be blocked. During the duplication of DNA inhibition of enzymatic activity, expression loss or elimination of DNA methyltransferase repair machinery leads toward extinction of 5-mC marks. This loss of 5-mC sites is known as passive DNA methylation (Feng et al., 2010).
Active DNA demethylation occurs by glycosylase activity by taking out the methylcytosines (Zhu et al., 2000). A single nucleotide gap is filled by demethylated cytosine with the help of the base excision repairing process (Agius et al., 2006). Many RNA molecules in the eukaryotic genome do not participate in protein production and are called non-coding RNAs (ncRNA) (Mishra and Bohra, 2018). On a size basis, these ncRNAs are divided into two types. i.e., small ncRNA and long non-coding RNA (lncRNA). These groups were divided based on the size of the transcripts. Small ncRNAs possess less than 200 nucleotides (Bohra et al., 2021), whereas lncRNAs contain more than 200 nucleotides (Ghildiyal and Zamore, 2009; Quinn and Chang, 2016). Small and lncRNAs are important epigenetic players in regulating the plant stress response, growth, and development (El-Shami et al., 2007; Heo et al., 2013). Research has shown that lncRNAs serve as epigenetic regulators of gene expression at different stages (Karlik et al., 2019). LncRNAs work as cis-acting elements near the RNA synthesis sites (Zhao et al., 2020b). Further, trans-acting factors, they can also work away from synthesis sites (Suksamran et al., 2020). LncRNAs transcribed by polymerase II, III, IV, and V. They are further divided into five categories depending on their positions in genome near or away from protein-coding genes. The five categories are sense, antisense, bidirectional, intronic, and large intergenic lncRNA. Different lncRNAs are differentially expressed under various stresses and were suggested to play an important role (Urquiaga et al., 2021). These ncRNAs are involved in different epigenetic regulation mechanisms such as histone modification and DNA methylation (Ariel et al., 2014). Double-stranded RNAs synthesized by RNA Dependent RNA polymerases (RDRs) during this process, small interfering RNA (siRNA) arises (Song et al., 2019). siRNA are produced during transcription or transpositional reactivation of transposable elements (TE) during stress conditions (Hou et al., 2019b).
Nucleosome architectures are altered in response to epigenetic changes; however, this alteration does not involve the DNA sequence (Zhao et al., 2021). Gene expression lowers or increases due to histone modification or DNA methylation (Singh and Prasad, 2021). During developmental processes or under any stresses, epigenetic changes in chromatin structure are regular and extremely active (Bhadouriya et al., 2021). Acetylation/deacetylation, SUMOylation, ubiquitin of histone proteins, phosphorylation/dephosphorylation are key processes involved in histone modification. These histone proteins are also altered chemically; they change their different physical or chemical properties (Boulanger et al., 2021). In the regulation of gene expression, the histones also release their subunits from the octamer core. These known modifications help increase DNA accessibility and speed up the selection process of binding proteins, which participate in DNA replication, transcription, or DNA repair (Pang et al., 2020). In the DNA methylation process, which occurs in the eukaryotic genome, at 50 positions of nitrogenous cytosine base, a methyl group attaches (-CH3) and forms a 5-mC (Pandey et al., 2017). These methylation processes can be asymmetrical and symmetrical; commonly CHH methylation process is known as asymmetrical, and on the other side, CHG and CG represent symmetrical methylation (Parent et al., 2021). As we discussed earlier RdDM is also common in plants (Singroha and Sharma, 2019). Cytosine methylation regulates gene expression by controlling the interaction of nucleic acid with transcription factors and chromatin proteins (Casati and Gomez, 2021). Patterns of DNA methylation are constant and particular to the exact cell type. These patterns are heritable and remain the same throughout life (Singh and Prasad, 2021).
Plant memorizes the epigenetic changes, and it helps them to adapt under biotic and abiotic stresses (Kinoshita and Seki, 2014; Crisp et al., 2016; He and Li, 2018). For instance, in Arabidopsis thaliana, two important factors that memorize during stresses are modification of histone and HSFA2 (Heat shock factors). When a plant faces heat shock, the level of H3K4 (H3 lysine K4 methylation) methylation remains high at least for 2 days. This process is also linked with transcriptional memory. The expression of heat stress response and transcriptional heat shock memory is dependent on the accumulation of H3K4 methylation and HSFA2. REF6, known as RELATIVE OF EARLY FLOWERING 6, exhibits a positive response and transfers as long-term memory of epigenetic changes in A. thaliana (Liu et al., 2019a). LSD-1 (Lysine-specific histone demethylase-1) in wheat recorded upregulation during heat stress as compared to normal plants. It is linked with modification of histone in the generation of transgenerational thermotolerance by heat priming. These changes induced by heat shock, transgenerational epigenetic memory, or changes in phenotype can be carried out at least two to three generations (Suter and Widmer, 2013; Zhonga et al., 2013). Priming of organismal stress response explains the events by which transient stimulus alters plant for future exposure to stress (Conrath et al., 2015). The term priming basically referred to immunity against pathogens but was later applied to abiotic stress. Priming is a reversible event because it only changed the phenotypical appearance of plant and does not change genetic makeup (Hilker et al., 2016). There are still many questions related to epigenetic memory. The specificity of stability of DNA and choramtin and their existence during mitosis and upkeep of memory. The mechanisms directly linked to chromatin changes which is further linked to transcriptional responses when plant faces stress are still not clear (Asensi-Fabado et al., 2017). A plant that once faces any harmful or stress conditions can recover from that stress, and epigenetic memory helps its future survival under stress conditions. As illustrated in Figure 1, the plant activates the epigenetic stress memory against future stresses, and the plant will remain protected.
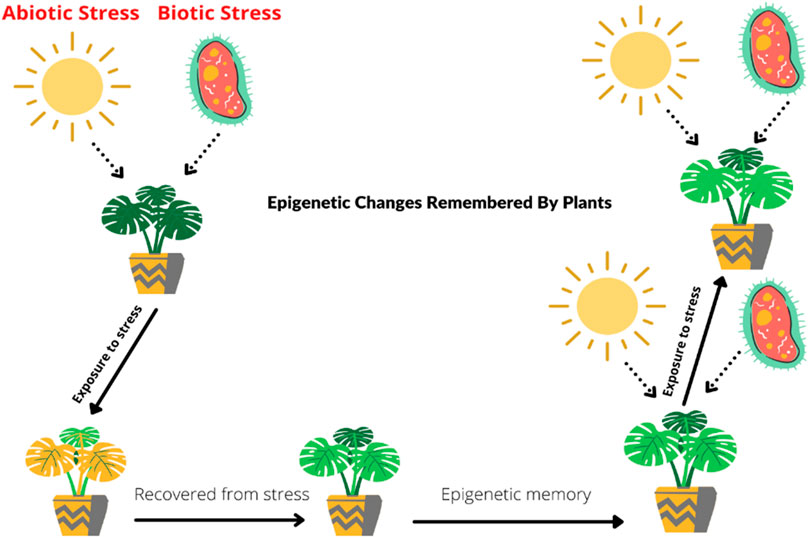
FIGURE 1. Mechanisms underlying epigenetic memory in plants during stress. Plants’ epigenetic memory helps protect them from different stresses. Whenever a plant faces stress regardless of its biotic or abiotic nature, it starts recovery against stress, and the plant epigenetic stress memory stores that information. Due to this stored memory, stress does not affect the plant on subsequent exposures.
On exposure to biotic stress, the defense machinery evokes the immune system, such as basal defense machinery and pathogen-associated molecular patterns (Muthamilarasan and Prasad, 2013). DNA methylation changes in plants as a defense response against biotic stress. The role of DNA methylation has been reported in A. thaliana (Dowen et al., 2012). In A. thaliana, the met1 and ddc mutants could not produce infectious symptoms by Pseudomonas syringae pv. tomato DC3000 due to the elimination of methylation capability of cytosine. Another gene named ELP2 initiates DNA methylation in Arabidopsis, and pathogen-altered methylation of DNA takes place (Dowen et al., 2012; Yu et al., 2013). Akimoto and colleagues artificially reduced the DNA methylation at the promoter region of R gene (Xa 21G) of rice (Akimoto et al., 2007). During pathogen infection, the influence on the expression of defense-related genes was shown, caused by hypomethylation of DNA. When plants are infected with Xanthomonas oryzae pv. oryzae, rice mutant lines with constitutive silencing of Xa21G gene showed more resistance as compared to wild type (Akimoto et al., 2007). With advancements in molecular biology, scientists found new pathways and events in virus-infected plants. Plants generally employ siRNA-mediated DNA methylation against viruses or biotic stresses; in this mechanism, the plant methylated its genomic parts (Emran et al., 2012).
Elucidation of the epigenetic alterations under biotic stress helps understand the plant-pathogen interaction (Zogli and Libault, 2017). In the analysis of plant-microbe relation, harmful pst introduced in Arabidopsis and DNA methylation changes occurred in all sequences of context. On the other side, when a non-harmful strain (i.e., bacterial strain) was introduced, only changes in CHG and CG methylation were detected (Dowen et al., 2012). Changes in methylation were common proximal to genes related to defense, and their activation correlated with transcription, so they have a role in reaction to pathogens (Yu et al., 2013).
Limited information is available on plant epigenetic effects created by fungal pathogens or oomycetes (Crespo-Salvador et al., 2018). Arabidopsis roots infected by cyst showed a huge change in DNA methylation and small RNA. In general, at the stage of infection, dynamic shifts take place (Joseph et al., 2021). However, DNA methylation change can be associated with several regions; transcriptional and epigenetic changes are related to each other and affect genes responsible for defense (Hewezi et al., 2017). In non-model crop species, understanding of epigenetic modifications is often limited, including patterns of DNA methylation (Herrera et al., 2016). In Brassica rapa, changes in DNA methylation were related to differences in floral morphology and less attraction of pollinators (Kellenberger et al., 2016). Detailed knowledge and high-resolution analysis will facilitate understanding plant-microbe interaction and underlying epigenetics changes (Richards et al., 2017). Plant-to-plant interactions facilitated by microbiota related to roots or allelochemicals obtained from plants may have some effect on chromatin arrangement (Venturelli et al., 2015).
Plants can be affected by neighbouring plants, microbes, and herbivores. Due to this, epigenetic changes affect plant phenotypes and plant’s interactions with other ogranisms (Latzel et al., 2012). Advances in plant epigenetics have deepened our understanding of the plant response against biotic stresses (Marfil et al., 2009). AGO4 mutants of A. thaliana lacking methylation are susceptible to pst (Dowen et al., 2012). Similarly, overexpression of histone lysine demethylase enhanced resistance against blight in rice (Li et al., 2013). Relation between epigenetics and plant interaction with fungal pathogens is evident from enhanced susceptibility A. thaliana mutants (dml1 dml2 ros1) (Le et al., 2014). Post-translation histone modifications are also included in defense against pathogens (Xia et al., 2013). Evidence of inclusion of epialleles in biotic relations was discovered from mutants of Arabidopsis (Johannes et al., 2009; Reinders et al., 2009). Furthermore, these lines were also used to identify epigenetic QTLs that established a connection between epigenetic modifications and phenotypic variability (Latzel et al., 2012).
Climate change is crucial concerning, the adaptation of the crops to the changing climate scenarios as well as the growth of future crops to ensure food security. Environmental changes trigger drought, salt, heat, and cold stresses (Raza, 2020; Raza et al., 2020). Any fluctuation in temperature negatively affects plants growth and development, resulting in poor yield (Raza, 2021; Raza et al., 2021). Over the past few decades, several studies have explained the mechanisms of abiotic stresses, but reports on epigenetic regulation are still limited (Raza et al., 2021). Cold/chilling stress influences plant metabolic enzyme activities, responsible for gene expression (Raza et al., 2020; He et al., 2021; Raza et al., 2022). Cold stress is another important abiotic stress that retards plant growth and yield. To mitigate the risks associated with cold stress, plants have evolved signaling system that stimulates the expression of cold-stress-related genes. Plant response to cold stress is well-characterized, and research highlights the profound role of C- REPEAT BINDING FACTOR (CBF)-COLD RESPONSIVE (COR) pathways. Cold stress is reported to stimulate transcription factors (TFs) expression, which includes CBF family proteins. The TFs bind to the promoter region of downstream COR genes that activate its gene expression (Zhu, 2016). A recent study in A. thaliana described that chromatin remodeler PICKLE (PKL) is involved in CBF-dependent cold stress response (Yang et al., 2019). Histone methylation and histone modifications play a significant role against cold stress. For example, acetylation of histone is found enriched in several cold-responsive genes (Park et al., 2018). It is regulated dynamically by histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Li et al., 2021). In recent research, A. thaliana plants were exposed to cold stress, and overexpression of histone deacetylase 2D (HD2D) exhibited lower lipid peroxidation with decreased accumulation of malondialdehyde contents that eliminated the oxidative burst (Han et al., 2016). Furthermore, plants under the influence of cold stress showed induction of histone acetylation in the promoter region of COR47 and COR15A (Pavangadkar et al., 2010). The H3K4me3 and H3K27me3 have been reported to be involved in response to vernalization. The study elucidated their regional regulation and contributions to epigenetic memory. The vernalized Brachypodium distachyon induced epigenetic changes that regulate multiple genes to coordinate biological processes (Huan et al., 2018).
A global rise in temperature has attracted the attention of plant scientists to improve crop’s adaptation to future scenarios (Jha et al., 2014; Haider et al., 2022). In response to heat stress, Heat Shock Transcription Factor A1s (HSFA1s) are the main TFs controlled by phosphorylation or dephosphorylation and protein-protein interactions. A temperature higher than required for the normal growth functioning of a plant instantly disrupts its photosynthetic machinery with the absorption of increased light. It damages the photosystem II, thylakoid protein phosphorylation. Heat stress stimulates hyper-phosphorylation likewise activates heat shock TFs. Acetylation is the epigenetic modification that alters the H2A and H3 histones, two important players associated with heat stress response in plants. For instance, actin-related protein 6 (ARP6) in Arabidopsis is reported to regulate gene expression. It encodes the SWR1 complex that is necessary for the insertion of H2A.Z histone in nucleosomes as a replacement for H2A histone (Nie and Wang, 2021). It is also reported as an indispensable event for temperature sensing. Moreover, acetylation of H3K56 is related to accumulating RNA polymerase II and activates TFs with exposure to heat stress (Haider et al., 2021). Research has demonstrated that the RdDM pathway and histone dynamics are involved in the responses against heat stress (Lämke et al., 2016; Yang et al., 2018). The heat shock proteins (HSPs) are primarily involved in conferring tolerance against heat stress in plants, regulating folding and unfolding of proteins (Singh et al., 2016). The heat stress stimulates the persistent expression of H3K4me3 and H3K9Ac on HSP18, APX2, and HSP70 genes (Lämke et al., 2016). In A. thaliana, the member of the F-Box family protein such as suppressor of DRM1 DRM2 CMT3 (SDC) proteins facilitates the degradation of the protein. Heat stress for a prolonged period induces transcriptional expression of a subgroup of genes, which ultimately assist the plant in recovering from heat stress (Popova et al., 2013; Sanchez and Paszkowski, 2014). The SDC gene targets the RdDM pathway that can be silenced epigenetically under normal growth conditions. However, its activation under heat stress suggests a transcriptional response, which overcomes the silencing effect of RdDM at some loci.
Like heat, salt stress is another key challenge to global agriculture (Jha et al., 2014). The plant faces salt stress with the higher accumulation of salt contents, mainly increased sodium ions (Na+) content that cause ionic toxicity. The growth and development of plants are impaired following secondary oxidative stress (Chaudhry et al., 2021b; Hafeez et al., 2021). The participation of Histone acetyltransferase (HAT) in regulating salt tolerance has been elucidated in A. thaliana (Zheng et al., 2019). Higher uptake and accumulation of Na+ in the GENERAL CONTROL NONDEREPRESSIBLE 5 (GCN5) mutant as compared to wild-type plants impaired the growth of the mutant in response to salt stress. Additionally, GCN5 can bind with cell wall synthesis genes such as CHITINASE-LIKE 1 (CTL1), and MYB54. Notably, lower H3K9ac and H3K14ac concentrations in mutant due to salt stress suggested that the GCN5 is a conserved epigenetic regulator (Zheng et al., 2019). A recent study on GCN5 in wheat has shown that target genes responsible for producing ROS species such as H2O2 (Zheng et al., 2021). The calcium ions (Ca2+) CALCINEURIN B-LIKE PROTEIN (CBL) CBL INTERACTING PROTEIN KINASE (CIPK) component performs an essential role in regulating cellular ionic homeostasis (Zhu, 2016). Higher Na+, lower K+, excessiveness of Mg2+, and higher pH levels stimulate cytosolic Ca2+ signaling for the activation of SALT OVERLY SENSITIVE3 (SOS3)-SOS2, CBL2/3-CIPK3/9/23/26, CBL1/9-CIPK23, and SCaBP1-CIPK11/14 that causes phosphorylation and regulation of the activity of H+ ATPase, Mg2+ transporter, Arabidopsis K+ TRANSPORTER (AKT1, K+ channel), and SOS1 (Na+/H+ antiporter) (Zhu, 2016). HIGH-AFFINITY K+ CHANNEL 1 (HKT1), which facilitates Na+ influx in plants, is vital transporter for coordinating with the SOS pathway to confer salt tolerance (Rus et al., 2001). In A. thaliana wild-type plants, a small RNA target region was identified at approximately 2.6 kb upstream of HKT1 that was reported to be highly methylated (Baek et al., 2011). It was reported that a lower DNA methylation level in RdDM mutant rdr2 led to an enhanced expression of HKT1, thus highlighting the role of RdDM-mediated regulation of gene expression (Miryeganeh, 2021). Another study in wheat revealed that salt stress-induced cytosine methylation caused suppression of TaHKT2 expression in roots and shoots of both tolerant and sensitive genotypes (Kumar et al., 2017). The TFs induced by salt stress include MYB74 of the R2R3-MYB family. The MYB74 promoter is extremely methylated due to the RdDM pathway, and in salt stress, 24-nt siRNA levels and DNA methylation were almost imperceptible at MYB74 is accompanied by the upregulated expression of MYB74 (Xu et al., 2015).
Histone dynamics have been associated with drought stress response in plants (To and Kim, 2014). ABA-mediated signaling, playing an important role in drought stress in plants, is influenced by epigenetic modulation caused by either DNA methylation or histone acetylation. For example, analysis of ABA-deficient mutant maize (vp10) revealed differential methylation of several stress-responsive genes and TE. The key enzyme involved in the synthesis of ABA is NINE CIS-EPOXYCAROTENOID DIOXYGENASE 3 (NCED3) (Nambara and Marion-poll, 2003). Plant acclimatization to drought stress improved following deposition of H3K4me in the NCED3 gene that caused higher gene expression (Ding et al., 2011). Moreover, the elevated expression level has been noted in the genes RAP2.4, RD29A, RD29B, and RD22 in response to drought stress (Takahashi et al., 2000). The increased levels of H3K4me3 and H3K9Ac in the promoter regions of RAP2.4, RD22, RD29A, and RD29B also contributed to the activation of genes expression. It was suggested that histone marks in response to drought stress also varied with the intensity of stress. As H3K4me3 and H3K9Ac levels were higher with the exposure to severe drought in contrast to mild stress conditions (Kim et al., 2012). In A. thaliana, lower deposition of H3K27me3 in the gene body region of drought-associated TFs resulted in resistance to drought (Sebastian Ramirez-prado et al., 2019). The H3K27me3 reader protein is LIKE HETEROCHROMATIN PROTEIN 1 in the PRC1 complex (Mozgova and Hennig, 2015). Moreover, drought-stressed plants had modified DNA methylation levels that ultimately altered expression levels of several drought-responsive genes (Liang et al., 2014). The miniature inverted-repeat transposable element (MITE) inserts in the promoter region of the NAC gene suppress the expression with the deposition of RdDM and H3K9me3 (Mao et al., 2015). Likewise, zinc finger gene ZmMYB087 is related to the metabolism group MYB transcription factor to regulate the biosynthesis of the secondary cell wall. CW-type zinc finger protein participates in the methylation of histone H3 that is essential for epigenetic memory (Sallam and Moussa, 2021).
Optimal nutrient supply is essential for plant growth and development, but the excessive nutrients in the soil impair it by causing nutrient stress (Salim and Raza, 2020). Higher nitrogen in the soil down-regulated the expression of root nitrogen transporter, NRT2.1. The gene repression necessitates the involvement of the HIGH NITROGEN INSENSITIVE 9 (HNI9) in depositing H3K27me3 on the NRT2.1 gene (Widiez et al., 2011). Iron homeostasis in Arabidopsis was negatively regulated by PRMT5-mediated H4R3 symmetric dimethylation (H4R3sme2) (Fan et al., 2013). The PRMT5 linked with the bHLH genes, i.e., AtbHLH38 and AtbHLH100, for the symmetrical demethylation of H4R3 with no change in its gene expression (PRMT5) (Fan et al., 2013). Histone acetyltransferase GCN5 is involved in regulating iron homeostasis by FERRIC REDUCTASE DEFECTIVE 3 (FRD3) (Xing et al., 2015). The GCN5 can directly bind to the promoter region of iron-associated genes, which includes FRD3, to modulate the acetylation levels of H3K6 and H3K14 (Xing et al., 2015). The H3K4me3 acetylation and histone variant H2A.Z have a key role in response to phosphorus-deficient soil conditions. The protein PHD ALFIN-LIKE 6 (AL6) binds to the H3K4me3 mark that influences the maturation of transcript and stability of vital genes necessary for elongation of root hairs (Chandrika et al., 2013). Additionally, histone modifications in response to deficient phosphorus showed vast remodeling of DNA methylation (Secco et al., 2015). Gene expression levels of DNA methylase were induced on exposure to limiting phosphorus conditions (Yong-Villalobos et al., 2015). Table 1 enlists various crops where epigenetic mechanisms controlling response to various stresses have been elucidated.
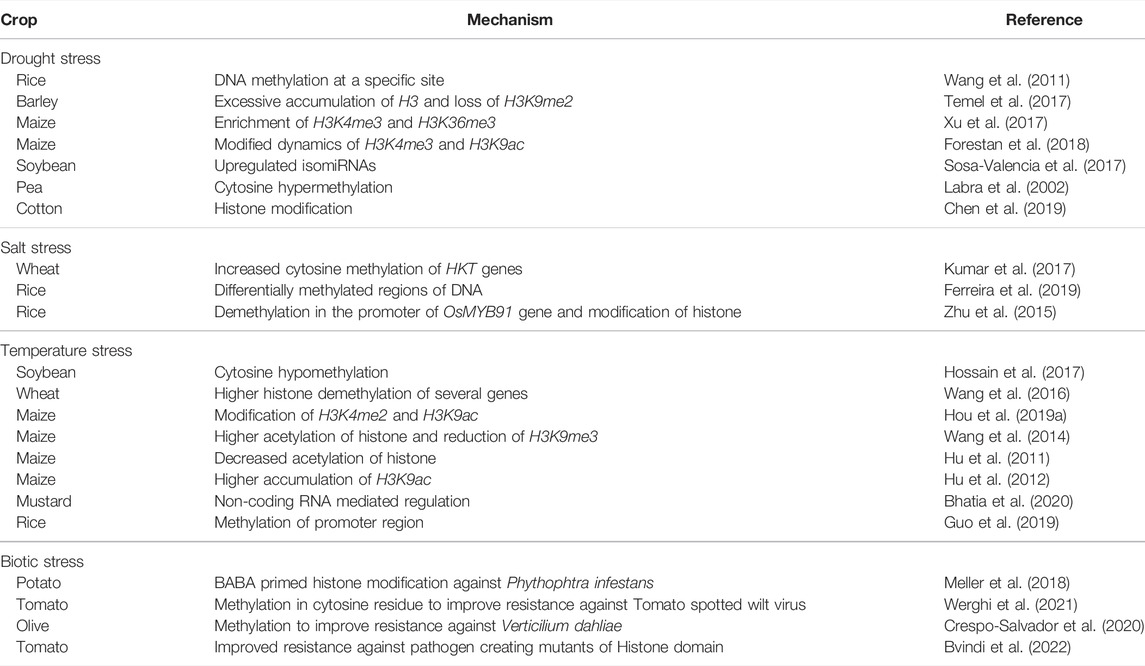
TABLE 1. Stress-related epigenetic mechanisms for improved crop development under stress conditions.
Development of experimental populations based on the two genotypes that have little DNA sequence polymorphism but show extensive variation in their methylation patterns could help greatly to overcome the confounding effect of DNA methylation and DNA sequence variation, exemplified by epigenetic recombinant inbred lines (epiRILs) in Arabidopsis based on Columbia (with wild type DDM1 allele) and Col-ddm1 mutant (Johannes et al., 2009). Similarly, Arabidopsis epiRILs showed distinct phenotypic variations with altered resistance stress (Lloyd and Lister, 2022). Such experimental populations laid a foundation to study epigenetic contributions to novel phenotypic variations. The combined studies of epiRILs and natural accessions suggested that epigenetic diversity is an important component of functional biodiversity. Additionally, comprehensive analysis of genes responsible for epigenetic machinery in epiRILs led to the mechanisms engaged in heritable DNA methylation and the resulting impact on plant phenotype (Johannes et al., 2009). DNA methylation is inherited in a stable Mendelian fashion in some genomic regions (Colomé-Tatche et al., 2012). The epiRIL population of ddm1, showed stable inheritance of differentially methylated regions for several generations without disrupting the DNA sequence and established a role for epigenetic quantitative trait loci (epiQTL) in phenotypic manifestation (Zhang et al., 2013).
The epiRILs of Arabidopsis differ for DNA methylation but show very minute changes in their DNA sequences. The development of epiRILs is similar to the creation of classic RILs, which includes the crossing of two genetically divergent parents and subsequently, inbred lines are established. DNA methylation exhibited developmental phenotypic changes that suggested the suppression of phenotypic plasticity, and it can be assessed with the construction of epiRILs (Bossdorf et al., 2010). The F2 progenies can be screened for the homozygosity of alleles to confirm the function of DNA methylation machinery and subsequently maintain the epigenetic chimeric chromosome developed by recombination at the F1 meiosis stage. The epiRILs harbor phenotypic variations for plant morphological characteristics, growth rate, and abiotic stress responses. Studies on epiRILs showed that epigenetic variations contributed to the functional diversity that had a similar impact on populations as noticed in genetic diversity. Similarly, higher epigenetic changes resulted in improved plant phenotypes translating to higher productivity and resilience in populations (Latzel et al., 2012). The heritable phenotypic variations among epiRILs in response to drought stress revealed phenotypic plasticity with the significant variation in root: shoot ratio that has the potential for developing stress-resilient plants (Zhang et al., 2013). The crop improvement is based on the stable transmission of epialleles in inheritance. With the introduction of methods for creating epiRILs in crop plants, the generation of epimutagenesis and engineered epigenetic changes would contribute to bridging the gaps for harnessing epigenetic variations for trait improvement to confer stress tolerance.
In recent years, several methods have been discovered to profile large-scale epigenetic modifications. With rapid progress in the field of biological sciences now, these methods are accurate, precise and affordable. Sodium bisulphite seqeucning and methylated immunoprecipitation sequencing (MeDIP-seq) in combination with latest sequencing technologies like single-molecule real-time sequencing (SMRT-seq) and Nanopore sequencing (Liang et al., 2019) are used genome-wide methylation profiling.
DNA methylation can be detected by sodium bisulfite, which converts demethylated cytosines into uracil (Kushwaha et al., 2016). However, it did not affect the 5-mC (Papanicolau-Sengos and Aldape, 2022). The sodium bisulfite method of detection of epigenetic changes is a standard technology for the detection of 5-mC due to its capacity to provide huge information about methylated DNA segments (Zhao et al., 2020a). Different methods after conversion are also used for further analysis, such as PCR analysis, sequencing of the methylated genome is widely used. It can provide information about even a single methylated nucleotide (Matzke and Mosher, 2014). Frommer and colleagues pioneered DNA methylation analysis based on bisulfite genomic sequencing. In this method, firstly, DNA is denatured by using alkali and treated with bisulfite. After that, in the second part, the region of interest was amplified by PCR using bisulfite-specific primers (Frommer et al., 1992). On the other sides, this sequencing method has several disadvantages, and we cannot differentiate between 5-mC and 5-hydroxymethyl cytosine. Unsuccessful reaction of bisulfite may cause a failure to covert entire demethylated cytosines to uracil, thus leading to false-positive results (Fraga and Esteller, 2002). When template DNA is treated with bisulfite, it is difficult to design primers for multiplex PCR reactions (Callinan and Feinberg, 2006). To overcome these problems, a modified method of bisulfite sequencing, reduced representation bisulfite sequencing (RRBS), was proposed (Meissner et al., 2005). This method gained traction among researchers due to its low cost and coverage of all regions. This method only required specific genomic sequences (Hahn et al., 2014). This can help detect different levels of methylation and can analyze the specific genes under stress and control conditions. RRBS technique provides useful data for further methylation measurements. Examination of wild tobacco plants under salt and low temperature analyzed by bisulfite sequencing revealed demethylation at GC sites in coding regions, and demethylation was found in promoter regions (Rehman and Tanti, 2020). In 2019, Liu and colleagues demonstrated two methods for the detection of 5-mC and 5-hydroxymethyl cytosine. One is ten-eleven translocation (TET)-assisted pyridine borane sequencing (TAPS) (Liu et al., 2019b) and its modified version long-read TET-assisted pyridine borane sequencing (lrTAPS) (Liu et al., 2020). With the help of pyridine borane, TAPS uses TET oxidation of 5-methyl cytosine and 5-hydroxymethyl cytosine to produce 5-carboxylcytosine that ultimately reduces to dihydrouracil (DHU). After that, the PCR reaction converts DHU to thymine and at last recognizes 5-mC and 5-hydroxymethyl cytosine (Liu et al., 2019b).
Methylated DNA immunoprecipitation (MeDIP) is one of the sensitive and widely used methods for methylome studies of plants. It is a purification method of gDNA fragments that contain methylated sites (Zhang et al., 2006). In this method, the fluorescent dye uses chemical derivatives of normal and modified deoxyribonucleotides to covalently bind to 6-methylated adenine (6 mA). Further, this 6 mA was quantified by using Capillary Electrophoresis with Laser-induced fluorescence (CE-LIF). The quest for alternatives like light sources instead of expensive lasers would impart cost-effectiveness to this method (Nguyen and Kang, 2019).
Bisulfite sequencing can only detect changes at C, but it cannot be distinguished between 5mC and 5hmC, and also it needs a reference genome (Flusberg et al., 2010). There are several methods of methylation detection, such as HPLC-MS/MS that can detect even the low to the lowest amount of 6-methyl adenine in plant genome (Huang et al., 2015). Another method, dot blot assay, is used with specific antibodies applied, but this approach has a limit of detection (Liang et al., 2019). These two methods are widely used for the detection of 6-methyl adenine, but these approaches are unable to reveal the location of methylated sites.
The nucleotide sequencing technologies have evolved rapidly over the past 20 years (Varshney et al., 2007; Bohra et al., 2020; Varshney et al., 2021). First-generation sequencing technologies invented by Sanger and Maxam Gilbert revolutionized the molecular biology field (Sanger et al., 1977). In the mid-90s first Sanger sequencing platform (ABI 370) was made commercially available (Watts and MacBeath, 2003). The Sanger sequencing is the preferred technique for plant molecular biologists. It deals with the DNA genome of plants to the sequence. The ABI 370 xl DNA sequencer has maximum accuracy of up to 99.99%. It can generate reads as small as 1.9 kb to as long as 84 kb, and in 3-h run, it can create up to 300–400 bp reads (Liu et al., 2014). The major drawback associated with this sequencing technology is its time and resource-intensive nature. These bottlencks were overcome by the introduction of the second-generation of sequencing or next generation seqeucning (NGS) in 2005. This NGS technologies not only cut down the cost of sequencing but also produce million of reads in less time (Kchouk et al., 2017). Further development of the third-generation sequencing methods overcame many problems related to the second-generation sequencing methods, including sample preparation, product amplification, and time. The third-generation sequencing technique SMRT-seq (Single-molecule real-time sequencing) offers exact sequences and measures the nucleotide energy rate during sequencing. This sequencing method detects DNA changes at a single base. The NGS-based methologies have helped greatly to study genome-wide methylation patterns, as exemplified in Arabidopsis thaliana (Liang et al., 2018), rice (Zhang et al., 2018b), and fig (Ficus carcia) (Usai et al., 2021). The recently developed PacBio sequencing method requires only 5 h from sample to produce reads, and also it reduces costs. The only disadvantage of this system is a higher rate of error (14%) (Chin et al., 2016; Kchouk et al., 2017). The in situ sequencing (ISS) represents a fourth-generation platform that directly sequences the nucleic acids with a higher accuracy (Mignardi and Nilsson, 2014; Ke et al., 2016).
Biotic and abiotic stresses deteriorate the yield and quality of crop plants. Conventional breeding approaches are increasingly constrained to rapidly develop plant varieties having adaptation to changing climatic conditions. It is the need of the hour to utilize different approachesfor accelerated crop improvement (Watson et al., 2018). The development of stress-resilient crops requires plant breeding approaches to exploit variations beyond DNA sequence (Fiaz et al., 2019). Plant stress memoryrelated genes or factors can be used by genome editing techniques for a better understanding of the regulatory mechanisms underlying stress response. Plant response to various stresses (biotic and abiotic) is known to involve several genes (Raza et al., 2021). Epigenetic changes can be induced in plants, and these can be used as a helpful strategy for the improvement of crops and can accelerate the breeding process, as shown in Figure 2. Nevertheless, these epigenetic changes can be induced artificially by editing of epigenome or chemically treated to create mutation (Figure 2). The CRISPR/Cas9 protein is successfully utilized as a dCas9 to modify epigenetic changes. The dCas9 protein is attached with the epigenetic modifier to targeted modifications that result in altered gene expression (Adli, 2018).
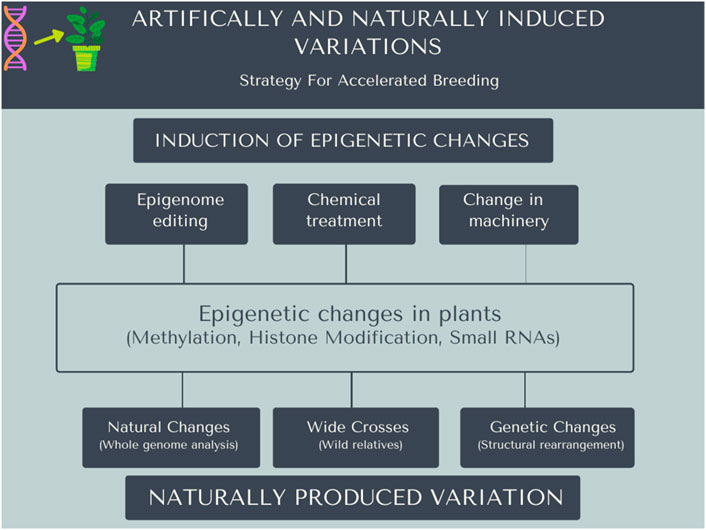
FIGURE 2. Artificially or naturally induced mutations in the plant genome can be helpful for accelerated breeding. Epigenetic changes can be induced in the plant genome using different methods, i.e., naturally or artificially. Artificially it can be induced using editing techniques, chemically treated plants to create mutations, and alter the machinery using different molecular approaches. Wide crosses of plants and naturally occurring changes in the genome in nature can be useful for accelerated breeding.
After the modification in Cas9 protein and the emergence of CRISPR/dCas9 technology, scientists start regulatory and reporter genes to investigate the ability of dCas9 for epigenome editing (Zezulin and Musunuru, 2018). It was reported that the plant genome was modified by methylation and demethylation at target DNA and resultantly developed late-flowering phenotypes (Adli, 2018). It has been reported that the fusion of repressor domains (such as KRAB/SID) with dCas9 led a significant improvement of transcriptional repression (Gilbert et al., 2013). Furthermore, the fusion of active transcriptional domain VP16/VP64 activates the expressional level of the gene of interest that permits the screening of stress-tolerant genotypes (Miglani et al., 2020). Nowadays, synthetic regulation of transcriptional modifications has been successfully considered for traits improvement by the CRISPR technique (Piatek et al., 2015). This technique has been used for precise and spatial modification by avoiding undesirable pleiotropic effects. For instance, the promoter of OsRAV2 gene, a TF responsible for salt stress, was genetically manipulated by CRISPR/Cas technique. The impaired growth of mutant lines by salt stress confirmed the important role of GT1 during normal plant growth and development (Duan et al., 2016). Conventional plant breeding has served humanity and this planet for a century and has always helped produce high-yield crops and fulfill the requirement of food for humans (Hickey et al., 2019). To this end, emerging plant breeding technologies such as epigenome editing can help to achieve food security targets by protecting plants from biotic or abiotic stresses.
Understanding the interactions between epigenetics and plant stress response has huge potential to develop modern crops adapted to future climatic conditions. Unlike abiotic stress, epigenetic regulation of biotic stress tolerance of plants is more complex, and it remains more challenging to carry out experiments for different species in vitro. In the future, biotechnologists, ecologists, and molecular biologists may collaborate to find out the mechanism involved between epigenetics and stress response. Methylome profiling has remained a challenge owing to the cost and technological considerations. However, recent advances in high-throughput assays have helped relieve this bottleneck. Interdisciplinary research efforts may help to sequence the genomes of diverse accessions and also play a vital role in establishing genomic tools to find out the epigenetic changes to stresses (Schrey et al., 2013). The growing information on whole genomes and gene content would inspire future researchers to decipher the epigenetics-mediated response of plants to a variety of biotic stresses. The foremost objective will be to survey epigenetic variation and quantify its effects on phenotypes in different crops (Kawakatsu et al., 2016). Furthermore, the candidate loci for the target traits can be identified, and epigenetic profiling may be precisely targeted to the candidate regions (Aller et al., 2018). A key challenge to decipher the role of epigenetic regulation is the application of stress treatments in controlled conditions. In the natural environment, plants often deal with multiple stresses at one time. With the rapid advances in genome-wide methylation profiling and gene editing techniques, we envisage a better understanding of the epigenetic changes controlling plant stress response, which will be crucial to precisely manipulate the epigenetic regulatory mechanism for improved crop performance.
FS and ABa conceived the idea. FS, UC, AR, ABa, and YS contributed to writing and literature search. FS, AR, ABo, and RKV proofread and edited the manuscript. All authors have proofread and approved the final version of the manuscript.
RKV is thankful to Bill & Melinda Gates Foundation, United States (The Tropical Legumes III project, OPP1114827), and Australia-India Strategic Research Fund from the Department of Biotechnology, Government of India, for supporting this work in part.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdolhamid Angaji, S., Hedayati, S. S., Poor, R. H., Poor, S. S., Shiravi, S., and Madani, S. (2010). Application of RNA Interference in Plants. Plant Omics 3, 77–84.
Adli, M. (2018). The CRISPR Tool Kit for Genome Editing and beyond. Nat. Commun. 9. doi:10.1038/s41467-018-04252-2
Agius, F., Kapoor, A., and Zhu, J.-K. (2006). Role of the Arabidopsis DNA Glycosylase/lyase ROS1 in Active DNA Demethylation. Proc. Natl. Acad. Sci. U.S.A. 103, 11796–11801. doi:10.1073/pnas.0603563103
Akimoto, K., Katakami, H., Kim, H.-J., Ogawa, E., Sano, C. M., Wada, Y., et al. (2007). Epigenetic Inheritance in rice Plants. Ann. Bot. 100, 205–217. doi:10.1093/aob/mcm110
Aller, E. S. T., Jagd, L. M., Kliebenstein, D. J., and Burow, M. (2018). Comparison of the Relative Potential for Epigenetic and Genetic Variation to Contribute to Trait Stability. G3 Genes, Genomes, Genet. 8, 1733–1746. doi:10.1534/g3.118.200127
Ariel, F., Jegu, T., Latrasse, D., Romero-Barrios, N., Christ, A., Benhamed, M., et al. (2014). Noncoding Transcription by Alternative Rna Polymerases Dynamically Regulates an Auxin-Driven Chromatin Loop. Mol. Cel 55, 383–396. doi:10.1016/j.molcel.2014.06.011
Asensi-Fabado, M.-A., Amtmann, A., and Perrella, G. (2017). Plant Responses to Abiotic Stress: The Chromatin Context of Transcriptional Regulation. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 1860, 106–122. doi:10.1016/j.bbagrm.2016.07.015
Baek, D., Jiang, J., Chung, J.-S., Wang, B., Chen, J., Xin, Z., et al. (2011). Regulated AtHKT1 Gene Expression by a Distal Enhancer Element and DNA Methylation in the Promoter Plays an Important Role in Salt Tolerance. m 52, 149–161. doi:10.1093/pcp/pcq182
Bhadouriya, S. L., Mehrotra, S., Basantani, M. K., Loake, G. J., and Mehrotra, R. (2020). Role of Chromatin Architecture in Plant Stress Responses: An Update. Front. Plant Sci. 11, 603380. doi:10.3389/FPLS.2020.603380/FULL
Bhatia, G., Singh, A., Verma, D., Sharma, S., and Singh, K. (2020). Genome-wide Investigation of Regulatory Roles of lncRNAs in Response to Heat and Drought Stress in Brassica Juncea (Indian Mustard). Environ. Exp. Bot. 171, 103922. doi:10.1016/j.envexpbot.2019.103922
Bohra, A., Gandham, P., Rathore, A., Thakur, V., Saxena, R. K., Naik, S. J. S., et al. (2021). Identification of microRNAs and Their Gene Targets in Cytoplasmic Male Sterile and fertile Maintainer Lines of Pigeonpea. Planta 253, 1–15. doi:10.1007/s00425-021-03568-6
Bohra, A., Jha, U. C., Godwin, I., and Varshney, R. K. (2020). Genomic Interventions for Sustainable Agriculture. Plant Biotechnol. J. 18, 2388–2405.
Bossdorf, O., Arcuri, D., Richards, C. L., and Pigliucci, M. (2010). Experimental Alteration of DNA Methylation Affects the Phenotypic Plasticity of Ecologically Relevant Traits in Arabidopsis thaliana. Evol. Ecol. 24, 541–553. doi:10.1007/s10682-010-9372-7
Boulanger, M., Chakraborty, M., Tempé, D., Piechaczyk, M., and Bossis, G. (2021). SUMO and Transcriptional Regulation: The Lessons of Large-Scale Proteomic, Modifomic and Genomic Studies. Moleculesmdpi.com 26, 828. doi:10.3390/molecules26040828
Bvindi, C., Lee, S., Tang, L., Mickelbart, M. V., Li, Y., and Mengiste, T. (2022). Main Manuscript for Improved Pathogen and Stress Tolerance in Tomato Mutants of SET Domain Histone 3 Lysine Methyltransferases. bioRxiv 2022. 02.19.481141. doi:10.1101/2022.02.19.481141
Callinan, P. A., and Feinberg, A. P. (2006). The Emerging Science of Epigenomics. Hum. Mol. Genet. 15, R95, R101 Spec No. doi:10.1093/hmg/ddl095
Casati, P., and Gomez, M. S. (2021). Chromatin Dynamics during DNA Damage and Repair in Plants: New Roles for Old Players. J. Exp. Bot. 72, 4119–4131. doi:10.1093/jxb/eraa551
Chandrika, N. N. P., Sundaravelpandian, K., Yu, S-M., and Schmidt, W. (2013). ALFIN‐LIKE 6 Is Involved in Root Hair Elongation during Phosphate Deficiency in Arabidopsis, Wiley Online Libr. New Phytol. 198, 709–720. 2013, undefined. doi:10.1111/nph.12194
Chaudhry, U. K., Gökçe, Z. N. Ö., and Gökçe, A. F. (2021a). Drought and Salt Stress Effects on Biochemical Changes and Gene Expression of Photosystem II and Catalase Genes in Selected Onion Cultivars. Biologia (Bratisl). 76, 3107–3121. doi:10.1007/S11756-021-00827-5
Chaudhry, U. K., Junaid, M. D., and Gökçe, A. F. (2021b). “Influence of Environmental Adversities on Physiological Changes in Plants,” in Developing Climate-Resilient Crops (Boca Raton, FL: CRC Press), 85–110. doi:10.1201/9781003109037-5-5
Chen, H., Feng, H., Zhang, X., Zhang, C., Wang, T., and Dong, J. (2019). An Arabidopsis E3 Ligase HUB2 Increases Histone H2B Monoubiquitination and Enhances Drought Tolerance in Transgenic Cotton. Plant Biotechnol. J. 17, 556–568. doi:10.1111/pbi.12998
Chin, C. S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing. Nat. Methods 13, 1050–1054. doi:10.1038/nmeth.4035
Colomé-Tatché, M., Cortijo, S., Wardenaar, R., Morgado, L., Lahouz, B., Sarazin, A., et al. (2012). Features of the Arabidopsis Recombination Landscape Resulting from the Combined Loss of Sequence Variation and DNA Methylation. Proc. Natl. Acad. Sci. U. S. A. 109, 16240–16245. doi:10.1073/pnas.1212955109
Conrath, U., Beckers, G. J. M., Langenbach, C. J. G., and Jaskiewicz, M. R. (2015). Priming for Enhanced Defense. Annu. Rev. Phytopathol. 53, 97–119. doi:10.1146/annurev-phyto-080614-120132
Crespo-Salvador, Ó., Escamilla-Aguilar, M., López-Cruz, J., López-Rodas, G., and González-Bosch, C. (2018). Determination of Histone Epigenetic marks in Arabidopsis and Tomato Genes in the Early Response to Botrytis Cinerea. Plant Cel Rep 37, 153–166. doi:10.1007/S00299-017-2218-9
Crespo-Salvador, Ó., Sánchez-Giménez, L., López-Galiano, M. J., Fernández-Crespo, E., Schalschi, L., García-Robles, I., et al. (2020). The Histone marks Signature in Exonic and Intronic Regions Is Relevant in Early Response of Tomato Genes to Botrytis Cinerea and in miRNA Regulation. Plants 9. doi:10.3390/plants9030300
Crisp, P. A., Ganguly, D., Eichten, S. R., Borevitz, J. O., and Pogson, B. J. (2016). Reconsidering Plant Memory: Intersections between Stress Recovery, RNA Turnover, and Epigenetics. Sci. Adv. 2. doi:10.1126/sciadv.1501340
Cuerda-Gil, D., and Slotkin, R. (2016). Non-canonical RNA-Directed DNA Methylation. nature.com. Available at: https://idp.nature.com/authorize/casa?redirect_uri=https://www.nature.com/articles/nplants2016163&casa_token=Odyo1slr4hcAAAAA:Z0_pz-xDixXs-Ia1AlsPnEBxI-LNNuf5I9_DjNoj3WMfEJNXESQh4yrZGMPpTmSMpHJAw9JZ2TKVeIEtXg.doi:10.1038/nplants.2016.163
Dangol, S. Das., Hashmi, M. H., Saeed, F., Yel, I., Öztürk, A., and Bakhsh, A. (2021). Utilizing RNA-Based Approaches to Understand Plant-Insect Interactions. Available at: https://books.google.com/books?hl=en&lr=&id=V8IoEAAAQBAJ&oi=fnd&pg=PA392&dq=SD+dangol+et+al+2021&ots=wu5CIsvrvO&sig=g4vB7Sq3mGe4fg-Lfnu3Fgdj9ks (Accessed October 12, 2021).doi:10.1007/978-3-030-64994-4_17
Ding, Y., Avramova, Z., and Journal, M. F.-T. P. (2011). The Arabidopsis Trithorax‐like Factor ATX1 Functions in Dehydration Stress Responses via ABA‐dependent and ABA‐independent Pathways, Wiley Online Libr. Plant J. 66, 735–744. 2011, undefined. doi:10.1111/j.1365-313X.2011.04534.x
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread Dynamic DNA Methylation in Response to Biotic Stress. Proc. Natl. Acad. Sci. U. S. A. 109. doi:10.1073/pnas.1209329109
Duan, Y., Li, J., Qin, R., Xu, R., Li, H., Yang, Y.-C., et al. (2016). Identification of a Regulatory Element Responsible for Salt Induction of rice OsRAV2 through Ex Situ and In Situ Promoter Analysis. Plant J. 90, 49–62. Springer. doi:10.1007/s11103-015-0393-z
El-Shami, M., Pontier, D., Lahmy, S., Braun, L., Picart, C., Vega, D., et al. (2007). Reiterated WG/GW Motifs Form Functionally and Evolutionarily Conserved ARGONAUTE-Binding Platforms in RNAi-Related Components. Genes Dev. 21, 2539–2544. doi:10.1101/gad.451207
Emran, A. M., Tabei, Y., Kobayashi, K., Yamaoka, N., and Nishiguchi, M. (2012). Molecular Analysis of Transgenic Melon Plants Showing Virus Resistance Conferred by Direct Repeat of Movement Gene of Cucumber green Mottle Mosaic Virus. Plant Cel Rep 31, 1371–1377. doi:10.1007/S00299-012-1237-9
Fan, H., Zhang, Z., Wang, N., Cui, Y., Sun, H., Liu, Y., et al. (2013). SKB1/PRMT5‐mediated Histone H4R3 Dimethylation of Ib Subgroup bHLH Genes Negatively Regulates Iron Homeostasis in Arabidopsis thaliana, Wiley Online Libr. Plant J. 77, 209–221. doi:10.1111/tpj.12380
Feng, S., Jacobsen, S. E., and Reik, W. (2010). Epigenetic Reprogramming in Plant and Animal Development. Science 330 (80), 622–627. doi:10.1126/science.1190614
Ferreira, L. J., Donoghue, M. T. A., Barros, P., Saibo, N. J., Santos, A. P., and Oliveira, M. M. (2019). Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes 3, 4. doi:10.3390/epigenomes3010004
Fiaz, S., Ahmad, S., Noor, M., Wang, X., Younas, A., Riaz, A., et al. (2019). Applications of the CRISPR/Cas9 System for rice Grain Quality Improvement: Perspectives and Opportunities. Int. J. Mol. Sci. 20 (4), 888. 2019, undefined. doi:10.3390/ijms20040888
Finnegan, E. J., Genger, R. K., Kovac, K., Peacock, W. J., and Dennis, E. S. (1998). DNA Methylation and the Promotion of Flowering by Vernalization. Natl. Acad. Sci. 95, 5824–5829. Available at: https://www.pnas.org/content/95/10/5824.short (Accessed October 6, 2021). doi:10.1073/pnas.95.10.5824
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct Detection of DNA Methylation during Single-Molecule, Real-Time Sequencing. Nat. Methods 7, 461–465. doi:10.1038/nmeth.1459
Forestan, C., Farinati, S., Rouster, J., and Genetics, H. L. (2018). Control of Maize Vegetative and Reproductive Development, Fertility, and rRNAs Silencing by HISTONE DEACETYLASE 108. Genetics 208 (4), 1443–1466. 2018, undefined. doi:10.1534/genetics.117.300625
Fraga, M. F., and Esteller, M. (2002). DNA Methylation: A Profile of Methods and Applications. Biotechniques 33, 632–649. doi:10.2144/02333rv01
Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., et al. (1992). A Genomic Sequencing Protocol that Yields a Positive Display of 5- Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. U. S. A. 89, 1827–1831. doi:10.1073/pnas.89.5.1827
Ganai, S. A. (2020). “Summa of Erasers of Histone Acetylation with Special Emphasis on Classical Histone Deacetylases (HDACs),” in Histone Deacetylase Inhibitors in Combinatorial Anticancer Therapy (Springer Singapore), 67–74. doi:10.1007/978-981-15-8179-3_3
Ghildiyal, M., and Zamore, P. D. (2009). Small Silencing RNAs: An Expanding Universe. Nat. Rev. Genet. 10, 94–108. doi:10.1038/nrg2504
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., et al. (2013). XCRISPR-mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 154, 442. doi:10.1016/j.cell.2013.06.044
Gökçe, A. F., Chaudhry, U. K., and Junaid, M. D. (2021). “Mapping QTLs for Abiotic Stress,” in Developing Climate-Resilient Crops (Boca Raton, FL: CRC Press), 175–201. doi:10.1201/9781003109037-9-9
Gökçe, A. F., and Chaudhry, U. K. (2020). “Use of QTL in Developing Stress Tolerance in Agronomic Crops,” in Agronomic Crops (Springer), 527–556. doi:10.1007/978-981-15-0025-1_25
Guo, H., Wu, T., Li, S., He, Q., Yang, Z., Zhang, W., et al. (2019). The Methylation Patterns and Transcriptional Responses to Chilling Stress at the Seedling Stage in rice. Int. J. Mol. Sci. 20, 5089. doi:10.3390/ijms20205089
Hafeez, M. B., Raza, A., Zahra, N., Shaukat, K., Akram, M. Z., Iqbal, S., et al. (2021). Gene Regulation in Halophytes in Conferring Salt Tolerance. Handbook of Bioremediation, 341–370. doi:10.1016/b978-0-12-819382-2.00022-3
Hahn, M. A., Li, A. X., Wu, X., and Pfeifer, G. P. (2014). “Single Base Resolution Analysis of 5-methylcytosine and 5-hydroxymethylcytosine by RRBS and TAB-RRBS,” in Cancer Epigenetics: Risk Assessment, Diagnosis, Treatment, and Prognosis (New York: Springer), 273–287. doi:10.1007/978-1-4939-1804-1_14
Haider, S., Iqbal, J., Naseer, S., Yaseen, T., Shaukat, M., Bibi, H., et al. (2021). Molecular Mechanisms of Plant Tolerance to Heat Stress: Current Landscape and Future Perspectives. Plant Cel Rep 40, 2247–2271. doi:10.1007/s00299-021-02696-3
Haider, S., Raza, A., Iqbal, J., Shaukat, M., and Mahmood, T. (2022). Analyzing the Regulatory Role of Heat Shock Transcription Factors in Plant Heat Stress Tolerance: a Brief Appraisal. Mol. Biol. Rep. 1–15. doi:10.1007/s11033-022-07190-x
Han, Z., Yu, H., Zhao, Z., Hunter, D., Luo, X., Duan, J., et al. (2016). AtHD2D Gene Plays a Role in Plant Growth, Development, and Response to Abiotic Stresses in Arabidopsis Thaliana. Front. Plant Sci. 7. doi:10.3389/fpls.2016.00310
He, H., Lei, Y., Yi, Z., Raza, A., Zeng, L., Yan, L., et al. (2021). Study on the Mechanism of Exogenous Serotonin Improving Cold Tolerance of Rapeseed (Brassica Napus L.) Seedlings. Plant Growth Regul. 94, 161–170. doi:10.1007/s10725-021-00700-0
He, Y., and Li, Z. (2018). Epigenetic Environmental Memories in Plants: Establishment, Maintenance, and Reprogramming. Trends Genet. 34, 856–866. doi:10.1016/j.tig.2018.07.006
Heard, E., and Martienssen, R. A. (2014). Transgenerational Epigenetic Inheritance: Myths and Mechanisms. Cell 157, 95–109. doi:10.1016/j.cell.2014.02.045
Henikoff, S., and Greally, J. M. (2016). Epigenetics, Cellular Memory and Gene Regulation. Curr. Biol. 26, R644–R648. doi:10.1016/j.cub.2016.06.011
Heo, J. B., Lee, Y. S., and Sung, S. (2013). Epigenetic Regulation by Long Noncoding RNAs in Plants. Chromosom. Res. 21, 685–693. doi:10.1007/s10577-013-9392-6
Herrera, C. M., Medrano, M., and Bazaga, P. (2016). Comparative Spatial Genetics and Epigenetics of Plant Populations: Heuristic Value and a Proof of Concept. Mol. Ecol. 25, 1653–1664. doi:10.1111/mec.13576
Herron, S. A., Rubin, M. J., Ciotir, C., Crews, T. E., Van Tassel, D. L., and Miller, A. J. (2020). Comparative Analysis of Early Life Stage Traits in Annual and Perennial Phaseolus Crops and Their Wild Relatives. Front. Plant Sci. 11. doi:10.3389/FPLS.2020.00034/FULL
Hewezi, T., Lane, T., Piya, S., Rambani, A., Rice, J. H., and Staton, M. (2017). Cyst Nematode Parasitism Induces Dynamic Changes in the Root Epigenome. Plant Physiol. 174, 405–420. doi:10.1104/pp.16.01948
Hickey, L. T., Hafeez, N. A, Robinson, H., Jackson, S. A., Leal-Bertioli, S. C. M., et al. (2019). Breeding Crops to Feed 10 Billion. Nat. Biotechnol. 37, 744–754. doi:10.1038/s41587-019-0152-9
Hilker, M., Schwachtje, J., Baier, M., Balazadeh, S., Bäurle, I., Geiselhardt, S., et al. (2016). Priming and Memory of Stress Responses in Organisms Lacking a Nervous System. Biol. Rev. 91, 1118–1133. doi:10.1111/brv.12215
Holliday, R., and Pugh, J. E. (1975). DNA Modification Mechanisms and Gene Activity during Development. Sci.187 (80), 226–232. doi:10.1126/science.111109810.1126/science.187.4173.226
Hossain, M., Kawakatsu, T., Kim, K., Zhang, N., Nguyen, C. T., Khan, S. M., et al. 2017, undefined (2017). Divergent Cytosine DNA Methylation Patterns in Single‐cell, Soybean Root Hairs. Wiley Online Libr. 214, 808–819. doi:10.1111/nph.14421
Hou, H., Zhao, L., Zheng, X., Gautam, M., Yue, M., Hou, J., et al. (2019a). Dynamic Changes in Histone Modification Are Associated with Upregulation of Hsf and rRNA Genes during Heat Stress in maize Seedlings. Protoplasma 256, 1245–1256. doi:10.1007/S00709-019-01364-4
Hou, J., Lu, D., Mason, A. S., Li, B., Xiao, M., An, S., et al. (2019b). Non-coding RNAs and Transposable Elements in Plant Genomes: Emergence, Regulatory Mechanisms and Roles in Plant Development and Stress Responses. Planta 250, 23–40. doi:10.1007/s00425-019-03166-7
Hu, X., Lu, M., Li, C., Liu, T., Wang, W., Wu, J., et al. (2011). Differential Expression of Proteins in maize Roots in Response to Abscisic Acid and Drought. Acta Physiol. Plant 33, 2437–2446. doi:10.1007/S11738-011-0784-Y
Hu, X., Wu, X., Li, C., Lu, M., Liu, T., Wang, Y., et al. (2012). Abscisic Acid Refines the Synthesis of Chloroplast Proteins in Maize (Zea mays) in Response to Drought and Light. PLoS One 7. doi:10.1371/JOURNAL.PONE.0049500
Huan, Q., Mao, Z., Chong, K., and Phytologist, J. Z.-N. (2018). Global Analysis of H3K4me3/H3K27me3 in Brachypodium Distachyon Reveals VRN3 as Critical Epigenetic Regulation point in Vernalization and Provides Insights into Epigenetic Memory, Wiley Online Libr. New Phytol. 219, 1373–1387. 2018, undefined. doi:10.1111/nph.15288
Huang, W., Xiong, J., Yang, Y., Liu, S. M., Yuan, B. F., and Feng, Y. Q. (2015). Determination of DNA Adenine Methylation in Genomes of Mammals and Plants by Liquid Chromatography/mass Spectrometry. RSC Adv. 5, 64046–64054. doi:10.1039/c5ra05307b
Jackson, J. P., Lindroth, A. M., Cao, X., and Jacobsen, S. E. (2002). Control of CpNpG DNA Methylation by the KRYPTONITE Histone H3 Methyltransferase. Nature 416, 556–560. doi:10.1038/nature731
Jasencakova, Z., Soppe, W. J. J., Meister, A., Gernand, D., Turner, B. M., and Schubert, I. (2003). Histone Modifications in Arabidopsis - High Methylation of H3 Lysine 9 Is Dispensable for Constitutive Heterochromatin. Plant J. 33, 471–480. doi:10.1046/J.1365-313X.2003.01638.X
Jha, U. C., Bohra, A., and Singh, N. P. (2014). Heat Stress in Crop Plants: Its Nature, Impacts and Integrated Breeding Strategies to Improve Heat Tolerance. Plant Breed 133, 679–701. doi:10.1111/pbr.12217
Johannes, F., Porcher, E., Teixeira, F. K., Saliba-Colombani, V., Simon, M., Agier, N., et al. (2009). Assessing the Impact of Transgenerational Epigenetic Variation on Complex Traits. Plos Genet. 5. doi:10.1371/journal.pgen.1000530
Joseph, T. J., Chandhini, S., Das, S., Mysore, S. K., and Shah, M. J. (2021). Methylation Status of Arabidopsis DNA Repair Gene Promoters during Agrobacterium Infection Reveals Epigenetic Changes in Three Generations. Plant Mol. Biol. Rep. doi:10.1007/S11105-021-01287-6
Junaid, M. D., Chaudhry, U. K., and Gökçe, A. F. (2021). Climate Change and Plant Growth – South Asian Perspective. Clim. Change Plants, 37–53. doi:10.1201/9781003108931-3-3
Karlik, E., Ari, S., and Gozukirmizi, N. (2019). LncRNAs: Genetic and Epigenetic Effects in Plants. Biotechnol. Biotechnol. Equip. 33, 429–439. doi:10.1080/13102818.2019.1581085
Karlsson, M., Weber, W., and Fussenegger, M. (2011). De Novo design and Construction of an Inducible Gene Expression System in Mammalian Cells. Methods Enzymol. 497, 239–253. doi:10.1016/B978-0-12-385075-100011-110.1016/b978-0-12-385075-1.00011-1
Kawakatsu, T., Huang, S., Jupe, F., Sasaki, E., and Cell, R. S. (2016). Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions Elsevier Enhanced Reader, 2016, U. Elsevier, 492–505. Available at: https://www.sciencedirect.com/science/article/pii/S0092867416308522 (Accessed October 6, 2021).
Kchouk, M., Gibrat, J. F., and Elloumi, M. (2017). Generations of Sequencing Technologies: From First to Next Generation. Biol. Med. 09. doi:10.4172/0974-8369.1000395
Ke, R., Mignardi, M., Hauling, T., and Nilsson, M. (2016). Fourth Generation of Next-Generation Sequencing Technologies: Promise and Consequences. Hum. Mutat. 37, 1363–1367. doi:10.1002/humu.23051
Kellenberger, R. T., Schlüter, P. M., and Schiestl, F. P. (2016). Herbivore-Induced DNA Demethylation Changes floral Signalling and Attractiveness to Pollinators in brassica Rapa. PLoS One 11. doi:10.1371/JOURNAL.PONE.0166646
Kim, J. M., To, T. K., and Seki, M. (2012). An Epigenetic Integrator: New Insights into Genome Regulation, Environmental Stress Responses and Developmental Controls by HISTONE DEACETYLASE 6. m 53, 794–800. doi:10.1093/pcp/pcs004
Kinoshita, T., and Seki, M. (2014). Epigenetic Memory for Stress Response and Adaptation in Plants. Plant Cel Physiol 55, 1859–1863. doi:10.1093/pcp/pcu125
Kumar, S., Beena, A. S., Awana, M., and Singh, A. (2017). Physiological, Biochemical, Epigenetic and Molecular Analyses of Wheat (Triticum aestivum) Genotypes with Contrasting Salt Tolerance. Front. Plant Sci. 8. doi:10.3389/FPLS.2017.01151/FULL
Kushwaha, M., Rostain, W., Prakash, S., Duncan, J. N., and Jaramillo, A. (2016). Using RNA as Molecular Code for Programming Cellular Function. ACS Synth. Biol. 5, 795–809. doi:10.1021/acssynbio.5b00297
Labra, M., Ghiani, A., Citterio, S., Sgorbati, S., Sala, F., Vannini, C., et al. (2002). Analysis of Cytosine Methylation Pattern in Response to Water Deficit in Pea Root Tips. Plant Biol. 4, 694–699. doi:10.1055/S-2002-37398
Lämke, J., Brzezinka, K., Altmann, S., and Bäurle, I. (2016). A Hit‐and‐run Heat Shock Factor Governs Sustained Histone Methylation and Transcriptional Stress Memory. EMBO J. 35, 162–175. doi:10.15252/EMBJ.201592593
Latzel, V., Zhang, Y., Karlsson Moritz, K., Fischer, M., and Bossdorf, O. (2012). Epigenetic Variation in Plant Responses to Defence Hormones. Ann. Bot. 110, 1423–1428. doi:10.1093/aob/mcs088
Le, T. N., Schumann, U., Smith, N. A., Tiwari, S., Khang Au, P. C., Zhu, Q. H., et al. (2014). DNA Demethylases Target Promoter Transposable Elements to Positively Regulate Stress Responsive Genes in Arabidopsis. Genome Biol. 15. doi:10.1186/s13059-014-0458-3
Li, S., He, X., Gao, Y., Zhou, C., Chiang, V. L., and Li, W. (2021). Histone Acetylation Changes in Plant Response to Drought Stress. Genes (Basel). 12, 1409. doi:10.3390/genes12091409
Li, T., Chen, X., Zhong, X., Zhao, Y., Liu, X., Zhou, S., et al. (2013). Jumonji C Domain Protein JMJ705-Mediated Removal of Histone H3 Lysine 27 Trimethylation Is Involved in Defense-Related Gene Activation in rice. Plant Cell 25, 4725–4736. doi:10.1105/tpc.113.118802
Liang, D., Zhang, Z., Wu, H., Huang, C., Shuai, P., Ye, C. Y., et al. (2014). Single-base-resolution Methylomes of Populus Trichocarpa Reveal the Association between DNA Methylation and Drought Stress. BMC Genet. 15. doi:10.1186/1471-2156-15-S1-S9
Liang, Z., Riaz, A., Chachar, S., Ding, Y., Du, H., and Gu, X. (2019). Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant. doi:10.1016/j.molp.2019.12.007
Liang, Z., Shen, L., Cui, X., Bao, S., Geng, Y., Yu, G., et al. (2018). DNA N6-Adenine Methylation in Arabidopsis thaliana. Dev. Cel 45, 406–416. e3. doi:10.1016/j.devcel.2018.03.012
Liu, J., Feng, L., Gu, X., Deng, X., Qiu, Q., Li, Q., et al. (2019a). An H3K27me3 Demethylase-HSFA2 Regulatory Loop Orchestrates Transgenerational Thermomemory in Arabidopsis. Cell Res 29, 379–390. doi:10.1038/s41422-019-0145-8
Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., et al. (2014). Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 251364. doi:10.1155/2012/251364
Liu, W., Duttke, S. H., Hetzel, J., Groth, M., Feng, S., Gallego-Bartolome, J., et al. (2018). RNA-directed DNA Methylation Involves Co-transcriptional Small-RNA-Guided Slicing of Polymerase V Transcripts in Arabidopsis. Nat. Plants 4, 181–188. doi:10.1038/s41477-017-0100-y
Liu, Y., Cheng, J., Siejka-Zielińska, P., Weldon, C., Roberts, H., Lopopolo, M., et al. (2020). Accurate Targeted Long-Read DNA Methylation and Hydroxymethylation Sequencing with TAPS. Genome Biol. 21. doi:10.1186/s13059-020-01969-6
Liu, Y., Siejka-Zielińska, P., Velikova, G., Bi, Y., Yuan, F., Tomkova, M., et al. (2019b). Bisulfite-free Direct Detection of 5-methylcytosine and 5-hydroxymethylcytosine at Base Resolution. Nat. Biotechnol. 37, 424–429. doi:10.1038/s41587-019-0041-2
Lloyd, J. P. B., and Lister, R. (2022). Epigenome Plasticity in Plants. Nat. Rev. Genet. 23, 55–68. doi:10.1038/s41576-021-00407-y
Ma, X., Zhu, Q., Chen, Y., and Liu, Y. G. (2016). CRISPR/Cas9 Platforms for Genome Editing in Plants: Developments and Applications. Mol. Plant 9, 961–974. doi:10.1016/j.molp.2016.04.009
Mao, H., Wang, H., Liu, S., Li, Z., Yang, X., Yan, J., et al. (2015). A Transposable Element in a NAC Gene Is Associated with Drought Tolerance in maize Seedlings. Nat. Commun. 6. doi:10.1038/ncomms9326
Marfil, C. F., Camadro, E. L., and Masuelli, R. W. (2009). Phenotypic Instability and Epigenetic Variability in a Diploid Potato of Hybrid Origin, Solanum Ruiz-Lealii. BMC Plant Biol. 9. doi:10.1186/1471-2229-9-21
Martínez de Alba, A. E., Elvira-Matelot, E., and Vaucheret, H. (2013). Gene Silencing in Plants: A Diversity of Pathways. Biochim. Biophys. Acta - Gene Regul. Mech. 1829, 1300–1308. doi:10.1016/j.bbagrm.2013.10.005
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA Methylation: An Epigenetic Pathway of Increasing Complexity. Nat. Rev. Genet. 15, 570. doi:10.1038/nrg3794
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced Representation Bisulfite Sequencing for Comparative High-Resolution DNA Methylation Analysis. Nucleic Acids Res. 33, 5868–5877. doi:10.1093/nar/gki901
Meller, B., Kuźnicki, D., Arasimowicz-Jelonek, M., Deckert, J., and Floryszak-Wieczorek, J. (2018). Baba-primed Histone Modifications in Potato for Intergenerational Resistance to Phytophthora Infestans. Front. Plant Sci. 9. doi:10.3389/fpls.2018.01228
Miglani, G., Kaur, A., and Kaur, L. (2019). Plant Gene Expression Control Using Genome-And Epigenome-Editing Technologies. Taylor Fr 34, 1–63. 2020, undefined. doi:10.1080/15427528.2019.1678541
Mignardi, M., and Nilsson, M. (2014). Fourth-generation Sequencing in the Cell and the Clinic. Genome Med. 6. doi:10.1186/gm548
Miryeganeh, M. (2021). Plants’ Epigenetic Mechanisms and Abiotic Stress. Genes (Basel) 12. doi:10.3390/genes12081106
Mishra, A., and Bohra, A. (2018). Non-coding RNAs and Plant Male Sterility: Current Knowledge and Future Prospects. Plant Cel Rep 37, 177–191. doi:10.1007/s00299-018-2248-y
Mozgova, I., and Hennig, L. (2015). The Polycomb Group Protein Regulatory Network. Annu. Rev. Plant Biol. 66, 269–296. doi:10.1146/ANNUREV-ARPLANT-043014-115627
Muthamilarasan, M., and Prasad, M. (2013). Plant Innate Immunity: An Updated Insight into Defense Mechanism. J. Biosci. 38, 433–449. doi:10.1007/s12038-013-9302-2
Nambara, E., and Marion-Poll, A. (2003). ABA Action and Interactions in Seeds. Trends Plant Sci. 8, 213–217. doi:10.1016/S1360-1385(03)00060-8
Nguyen, B. T., and Kang, M. J. (2019). Application of Capillary Electrophoresis with Laser-Induced Fluorescence to Immunoassays and Enzyme Assays. Molecules 24. doi:10.3390/molecules24101977
Nie, W.-F., and Wang, J. (20212021). Actin-Related Protein 4 Interacts with PIE1 and Regulates Gene Expression in Arabidopsis. Genes 12 (520 12), 520. doi:10.3390/GENES12040520
Pandey, G., Yadav, C. B., Sahu, P. P., Muthamilarasan, M., and Prasad, M. (2017). Salinity Induced Differential Methylation Patterns in Contrasting Cultivars of Foxtail Millet (Setaria Italica L.). Plant Cel Rep 36, 759–772. doi:10.1007/s00299-016-2093-9
Pang, M., Sun, X., Ausió, J., and Ausió, J. (2020). Histone H4 Variant, H4G, Drives Ribosomal RNA Transcription and Breast Cancer Cell Proliferation by Loosening Nucleolar Chromatin Structure, Wiley Online Libr. J. Cel Physiol 235, 235. 2020, undefined. doi:10.1002/jcp.29770
Papanicolau-Sengos, A., and Aldape, K. (2022). DNA Methylation Profiling: An Emerging Paradigm for Cancer Diagnosis. Annu. Rev. Pathol. Mech. Dis. 17. doi:10.1146/annurev-pathol-042220-022304
Parent, J., Cahn, J., Herridge, R. P., Grimanelli, D., and Robert, A. M. (2021). Small RNAs Guide Histone Methylation in Arabidopsis Embryos. genesdev.cshlp.org 35 (11-12), 841–846. 2021, undefined. doi:10.1101/gad.343871.120
Park, J., Lim, C. J., Shen, M., Park, H. J., Cha, J. Y., Iniesto, E., et al. (2018). Epigenetic Switch from Repressive to Permissive Chromatin in Response to Cold Stress. Proc. Natl. Acad. Sci. U. S. A. 115. doi:10.1073/pnas.1721241115
Pavangadkar, K., Thomashow, M. F., and Triezenberg, S. J. (2010). Histone Dynamics and Roles of Histone Acetyltransferases during Cold-Induced Gene Regulation in Arabidopsis. Plant Mol. Biol. 74, 183–200. doi:10.1007/S11103-010-9665-9
Piatek, A., Ali, Z., Baazim, H., Al-Shareef, S., and Aouida, M. (2015). RNA‐guided Transcriptional Regulation in Planta via Synthetic dCas9‐based Transcription Factors. Wiley Online Libr. 13, 578–589. biotechnology, and 2015, undefined. doi:10.1111/pbi.12284
Popova, O. V., Dinh, H. Q., Aufsatz, W., and Jonak, C. (2013). The RdDM Pathway Is Required for Basal Heat Tolerance in Arabidopsis. Mol. Plant 6, 396–410. doi:10.1093/mp/sst023
Quinn, J. J., and Chang, H. Y. (2016). Unique Features of Long Non-coding RNA Biogenesis and Function. Nat. Rev. Genet. 17, 47–62. doi:10.1038/nrg.2015.10
Raza, A., Ashraf, F., Zou, X., Zhang, X., and Tosif, H. (2020). “Plant Adaptation and Tolerance to Environmental Stresses: Mechanisms and Perspectives,” in Plant Ecophysiology and Adaptation under Climate Change: Mechanisms and Perspectives I: General Consequences and Plant Responses (Springer Singapore), 117–145. doi:10.1007/978-981-15-2156-0_5
Raza, A. (2021). Eco-physiological and Biochemical Responses of Rapeseed (Brassica Napus L.) to Abiotic Stresses: Consequences and Mitigation Strategies. J. Plant Growth Regul. 40, 1368–1388. doi:10.1007/s00344-020-10231-z
Raza, A. (2020). Metabolomics: a Systems Biology Approach for Enhancing Heat Stress Tolerance in Plants. Plant Cel Rep. doi:10.1007/s00299-020-02635-8
Raza, A., Su, W., Jia, Z., Luo, D., Zhang, Y., Gao, A., et al. (2022). Mechanistic Insights into Trehalose-Mediated Cold Stress Tolerance in Rapeseed (Brassica Napus L.) Seedlings. Front. Plant Sci. 13, 857980. doi:10.3389/fpls.2022.857980
Raza, A., Tabassum, J., Kudapa, H., and Varshney, R. K. (2021). Can Omics Deliver Temperature Resilient Ready-To-Grow Crops? Crit. Rev. Biotechnol. 41, 1209–1232. doi:10.1080/07388551.2021.1898332
Rehman, M., and Tanti, B. (2020). Understanding Epigenetic Modifications in Response to Abiotic Stresses in Plants. Biocatal. Agric. Biotechnol. 27. doi:10.1016/j.bcab.2020.101673
Reinders, J., Wulff, B. B. H., Mirouze, M., Marí-Ordóñez, A., Dapp, M., Rozhon, W., et al. (2009). Compromised Stability of DNA Methylation and Transposon Immobilization in Mosaic Arabidopsis Epigenomes. Genes Dev. 23, 939–950. doi:10.1101/gad.524609
Richards, C., Alonso, C., Becker, C., Bossdorf, O., Bucher, E., Colomé-Tatché, M., et al. (2017). Ecological Plant Epigenetics: Evidence from Model and Non‐model Species, and the Way Forward, Wiley Online Libr. Ecol. Lett. 20, 1576–1590. 2017, undefined. doi:10.1111/ele.12858
Rus, A., Yokoi, S., Sharkhuu, A., Reddy, M., Lee, B. H., Matsumoto, T. K., et al. (2001). AtHKT1 Is a Salt Tolerance Determinant that Controls Na+ Entry into Plant Roots. Proc. Natl. Acad. Sci. U. S. A. 98, 14150–14155. doi:10.1073/pnas.241501798
Saeed, F., Hashmi, M. H., Hossain, M. J., Ali, M. A., and Bakhsh, A. (2020). Transgenic Technology Based Value Addition in Plant Biotechnology, 123–156. doi:10.1016/b978-0-12-818632-9.00006-xTransgenic Technologies for Efficient Insect Pest Management in Crop Plants
Salim, N., and Raza, A. (2020). Nutrient Use Efficiency (NUE) for Sustainable Wheat Production: a Review. Taylor Fr 43, 297–315. doi:10.1080/01904167.2019.1676907
Sallam, N., and Moussa, M. (2021). DNA Methylation Changes Stimulated by Drought Stress in ABA-Deficient maize Mutant Vp10. Plant Physiol. Biochem. 160, 218–224. doi:10.1016/j.plaphy.2021.01.024
Sanchez, D. H., and Paszkowski, J. (2014). Heat-Induced Release of Epigenetic Silencing Reveals the Concealed Role of an Imprinted Plant Gene. Plos Genet. 10. doi:10.1371/JOURNAL.PGEN.1004806
Sanger, F., Nicklen, S., and Coulson, A. (1977). DNA Sequencing with Chain-Terminating. Proc. Natl. Acad. Sci. USA 74, 5463–5467. Available at: https://www.pnas.org/content/74/12/5463.short.doi:10.1073/pnas.74.12.5463
Schrey, A. W., Alvarez, M., Foust, C. M., Kilvitis, H. J., Lee, J. D., Liebl, A. L., et al. (2013). Ecological Epigenetics: Beyond MS-AFLP. Integr. Comp. Biol., 340–350. doi:10.1093/icb/ict012
Sebastian Ramirez-Prado, J., Latrasse, D., Yaneth Rodriguez-Granados, N., Huang, Y., Manza-Mianza, D., Brik-Chaouche, R., et al. (2019). The Polycomb Protein LHP1 Regulates Arabidopsis thaliana Stress Responses through the Repression of the MYC2‐dependent branch of Immunity. Wiley Online Libr. 100, 1118–1131. doi:10.1111/tpj.14502
Secco, D., Wang, C., Shou, H., Schultz, M. D., Chiarenza, S., Nussaume, L., et al. (2015). Stress Induced Gene Expression Drives Transient DNA Methylation Changes at Adjacent Repetitive Elements. Elife 4. doi:10.7554/eLife.09343.001
Singh, R. K., Jaishankar, J., Muthamilarasan, M., Shweta, S., Dangi, A., and Prasad, M. (20162016). Genome-wide Analysis of Heat Shock Proteins in C4 Model, Foxtail Millet Identifies Potential Candidates for Crop Improvement under Abiotic Stress. Sci. Rep. 61 (6), 1–14. doi:10.1038/srep32641
Singh, R. K., and Prasad, M. (2021). Delineating the Epigenetic Regulation of Heat and Drought Response in Plants. Crit. Rev. Biotechnol. doi:10.1080/07388551.2021.1946004
Singroha, G., and Sharma, P. (2019). Epigenetic Modifications in Plants under Abiotic Stress. Epigenetics. doi:10.5772/intechopen.84455
Song, X., Li, Y., Cao, X., and Qi, Y. (2019). MicroRNAs and Their Regulatory Roles in Plant-Environment Interactions. Annu. Rev. Plant Biol. 70, 489–525. doi:10.1146/annurev-arplant-050718-100334
Soppe, W. J. J., Jasencakova, Z., Houben, A., Kakutani, T., Meister, A., Huang, M. S., et al. (2002). DNA Methylation Controls Histone H3 Lysine 9 Methylation and Heterochromatin Assembly in Arabidopsis. EMBO J. 21, 6549–6559. doi:10.1093/emboj/cdf657
Sosa-Valencia, G., Romero-Pérez, P. S., Miguel Palomar, V., Covarrubias, A. A., and Reyes, J. L. (2017). Insights into the Function of the phasiRNA-Triggering miR1514 in Response to Stress in Legumes. Plant Signal. Behav. 12. doi:10.1080/15592324.2017.1284724
Steward, N., Kusano, T., and Sano, H. (2000). Expression of ZmMET1, a Gene Encoding a DNA Methyltransferase from maize, Is Associated Not Only with DNA Replication in Actively Proliferating Cells, but Also with Altered DNA Methylation Status in Cold-Stressed Quiescent Cells. Nucleic Acids Res. 28, 3250–3259. doi:10.1093/nar/28.17.3250
Suksamran, R., Saithong, T., Thammarongtham, C., and Kalapanulak, S. (2020). Genomic and Transcriptomic Analysis Identified Novel Putative Cassava lncRNAs Involved in Cold and Drought Stress. Genes (Basel) 11. doi:10.3390/genes11040366
Suter, L., and Widmer, A. (2013). Phenotypic Effects of Salt and Heat Stress over Three Generations in Arabidopsis thaliana. PLoS One 8. doi:10.1371/JOURNAL.PONE.0080819
Takahashi, S., Katagiri, T., Yamaguchi-Shinozaki, K., and Shinozaki, K. (2000). An Arabidopsis Gene Encoding a Ca2+ -binding Protein Is Induced by Abscisic Acid during Dehydration. m 41, 898–903. doi:10.1093/pcp/pcd010
Tariq, M., Saze, H., Probst, A. V., Lichota, J., Habu, Y., and Paszkowski, J. (2003). Erasure of CpG Methylation in Arabidopsis Alters Patterns of Histone H3 Methylation in Heterochromatin. Proc. Natl. Acad. Sci. 100, 8823–8827. doi:10.1073/PNAS.1432939100
Temel, A., Janack, B., and Agronomy, K. H. (2017). Drought Stress-Related Physiological Changes and Histone Modifications in Barley Primary Leaves at HSP17 Gene, mdpi.com. Agronomy 7 (2), 43. 2017, undefined. doi:10.3390/agronomy7020043
To, T. K., and Kim, J. M. (2014). Epigenetic Regulation of Gene Responsiveness in Arabidopsis. Front. Plant Sci. 4. doi:10.3389/FPLS.2013.00548/FULL
Urquiaga, M. C. de. O., Thiebaut, F., Hemerly, A. S., and Ferreira, P. C. G. (2021). From Trash to Luxury: The Potential Role of Plant LncRNA in DNA Methylation during Abiotic Stress. Front. Plant Sci. 11. doi:10.3389/fpls.2020.603246
Usai, G., Vangelisti, A., Simoni, S., Giordani, T., Natali, L., Cavallini, A., et al. (2021). DNA Modification Patterns within the Transposable Elements of the Fig (Ficus Carica l.) Genome. Plants 10, 1–13. doi:10.3390/plants10030451
Varshney, R. K., Bohra, A., Yu, J., Graner, A., Zhang, Q., and Sorrells, M. E. (2021). Designing Future Crops: Genomics-Assisted Breeding Comes of Age. Trends Plant Sci. 26, 631–649. doi:10.1016/j.tplants.2021.03.010
Varshney, R. K., Marcel, T. C., Ramsay, L., Russell, J., Röder, M. S., Stein, N., et al. (2007). A High Density Barley Microsatellite Consensus Map with 775 SSR Loci. Theor. Appl. Genet. 114, 1091–1103. doi:10.1007/s00122-007-0503-7
Venturelli, S., Belz, R. G., Kämper, A., Berger, A., von Horn, K., Wegner, A., et al. (2015). Plants Release Precursors of Histone Deacetylase Inhibitors to Suppress Growth of Competitors. Plant Cell 27, 3175–3189. doi:10.1105/tpc.15.00585
Wang, P., Zhao, L., Hou, H., Zhang, H., Huang, Y., Wang, Y., et al. (2014). Epigenetic Changes Are Associated with Programmed Cell Death Induced by Heat Stress in Seedling Leaves of Zea Mays. m 56, 965–976. doi:10.1093/pcp/pcv023
Wang, W. S., Pan, Y. J., Zhao, X. Q., Dwivedi, D., Zhu, L. H., Ali, J., et al. (2011). Drought-induced Site-specific DNA Methylation and its Association with Drought Tolerance in rice (Oryza Sativa L.). J. Exp. Bot. 62, 1951–1960. doi:10.1093/jxb/erq391
Wang, Y., Li, H., Sun, Q., and Yao, Y. (2016). Characterization of Small RNAs Derived from tRNAs, rRNAs and snoRNAs and Their Response to Heat Stress in Wheat Seedlings. PLoS One 11. doi:10.1371/JOURNAL.PONE.0150933
Watson, A., Ghosh, S., Williams, M., and plants, W. C.-N. (20182017). undefinedSpeed Breeding Is a Powerful Tool to Accelerate Crop Research and Breeding. nature.com. doi:10.1101/161182
Watts, D., and MacBeath, J. R. E. (2003). “Automated Fluorescent DNA Sequencing,” in DNA Sequencing Protocols (Totowa, NJ: Humana Press), 153–170. doi:10.1385/1-59259-113-2:153
Werghi, S., Herrero, F. A., Fakhfakh, H., and Gorsane, F. (2021). Auxin Drives Tomato Spotted Wilt Virus (TSWV) Resistance through Epigenetic Regulation of Auxin Response Factor ARF8 Expression in Tomato. Gene. doi:10.1016/j.gene.2021.145905
Widiez, T., El Kafafi, E. S., Girin, T., Berr, A., Ruffel, S., Krouk, G., et al. (2011). High Nitrogen Insensitive 9 (HNI9)-Mediated Systemic Repression of Root NO 3- Uptake Is Associated with Changes in Histone Methylation. Proc. Natl. Acad. Sci. U. S. A. 108, 13329–13334. doi:10.1073/pnas.1017863108
Xia, S., Cheng, Y., Huang, S., Win, J., Soards, A., and Jinn, T. (2013). Regulation of Transcription of Nucleotide-Binding Leucine-Rich Repeat-Encoding Genes SNC1 and RPP4 via. Artic. Plant Physiol. 162 (3), 1694–1705. doi:10.1104/pp.113.214551
Xing, J., Wang, T., Liu, Z., Xu, J., Yao, Y., Hu, Z., et al. (2015). General Control Nonrepressed Protein5-Mediated Histone Acetylation of Ferric Reductase Defective3 Contributes to Iron Homeostasis in Arabidopsis. Plant Physiol. 168, 1309–1320. doi:10.1104/pp.15.00397
Xu, J., Qi, W., Freeling, M., Xuecai, Z., Xu, Y., Mao, Y., et al. (2017). Natural Antisense Transcripts Are Significantly Involved in Regulation of Drought Stress in maize. Nucleic Acids Res. 45, 5126–5141. doi:10.1093/nar/gkx085
Xu, W., Dubos, C., and Lepiniec, L. (2015). Transcriptional Control of Flavonoid Biosynthesis by MYB-bHLH-WDR Complexes. Trends Plant Sci. 20, 176–185. doi:10.1016/j.tplants.2014.12.001
Yang, C., Shen, W., Chen, H., Chu, L., Xu, Y., Zhou, X., et al. (2018). Characterization and Subcellular Localization of Histone Deacetylases and Their Roles in Response to Abiotic Stresses in Soybean. BMC Plant Biol. 18. doi:10.1186/S12870-018-1454-7
Yang, R., Hong, Y., Ren, Z., Tang, K., Zhang, H., Zhu, J. K., et al. (2019). A Role for PICKLE in the Regulation of Cold and Salt Stress Tolerance in Arabidopsis. Front. Plant Sci. 10. doi:10.3389/fpls.2019.00900
Yong-Villalobos, L., González-Morales, S. I., Wrobel, K., Gutiérrez-Alanis, D., Cervantes-Peréz, S. A., Hayano-Kanashiro, C., et al. (2015). Methylome Analysis Reveals an Important Role for Epigenetic Changes in the Regulation of the Arabidopsis Response to Phosphate Starvation. Proc. Natl. Acad. Sci. U. S. A. 112, E7293–E7302. doi:10.1073/pnas.1522301112
Yu, A., Lepère, G., Jay, F., Wang, J., Bapaume, L., Wang, Y., et al. (2013). Dynamics and Biological Relevance of DNA Demethylation in Arabidopsis Antibacterial Defense. Proc. Natl. Acad. Sci. U. S. A. 110, 2389–2394. doi:10.1073/pnas.1211757110
Zezulin, A., and Musunuru, K. (2018). Turning up the Heat with Therapeutic Epigenome Editing. Cell Stem Cell 22, 10–11. doi:10.1016/j.stem.2017.12.013
Zhang, H., Lang, Z., and Zhu, J. K. (2018a). Dynamics and Function of DNA Methylation in Plants. Nat. Rev. Mol. Cel Biol. 19, 489–506. doi:10.1038/s41580-018-0016-z
Zhang, Q., Liang, Z., Cui, X., Ji, C., Li, Y., Zhang, P., et al. (2018b). N 6 -Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and its Association with Gene Expression, Plant Development, and Stress Responses. Mol. Plant 11, 1492–1508. doi:10.1016/j.molp.2018.11.005
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W. L., Chen, H., et al. (2006). Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 126, 1189–1201. doi:10.1016/j.cell.2006.08.003
Zhang, Y. Y., Fischer, M., Colot, V., and Bossdorf, O. (2013). Epigenetic Variation Creates Potential for Evolution of Plant Phenotypic Plasticity. New Phytol. 197, 314–322. doi:10.1111/nph.12010
Zhao, B., Xi, Y., Kim, J., and Advances, S. S.-S. (20212021). undefinedChromatin Architectural Proteins Regulate Flowering Time by Precluding Gene Looping. advances.sciencemag.org. Available at: https://advances.sciencemag.org/content/7/24/eabg3097.abstract (Accessed October 6, 2021).730973108doi:10.1126/sciadv.abg3097
Zhao, L. Y., Song, J., Liu, Y., Song, C. X., and Yi, C. (2020a). Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell 11, 792–808. doi:10.1007/s13238-020-00733-7
Zhao, M., Wang, T., Sun, T., Yu, X., Tian, R., and Zhang, W. H. (2020b). Identification of Tissue-specific and Cold-Responsive lncRNAs in Medicago Truncatula by High-Throughput RNA Sequencing. BMC Plant Biol. 20. doi:10.1186/s12870-020-2301-1
Zheng, M., Lin, J., Liu, X., Chu, W., Li, J., Gao, Y., et al. (2021). Histone Acetyltransferase TaHAG1 Acts as a Crucial Regulator to Strengthen Salt Tolerance of Hexaploid Wheat. Plant Physiol. 186, 1951–1969. doi:10.1093/plphys/kiab187
Zheng, M., Liu, X., Lin, J., Liu, X., Wang, Z., Xin, M., et al. (2019). Histone Acetyltransferase GCN5 Contributes to Cell wall Integrity and Salt Stress Tolerance by Altering the Expression of Cellulose Synthesis Genes. Plant J. 97, 587–602. doi:10.1111/tpj.14144
Zhonga, S. H., Liu, J. Z., Jin, H., Lin, L., Li, Q., Chen, Y., et al. (2013). Warm Temperatures Induce Transgenerational Epigenetic Release of RNA Silencing by Inhibiting siRNA Biogenesis in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 110, 9171–9176. doi:10.1073/pnas.1219655110
Zhu, B., Zheng, Y., Angliker, H., Schwarz, S., Thiry, S., Siegmann, M., et al. (2000). 5-Methylcytosine DNA Glycosylase Activity Is Also Present in the Human MBD4 (G/T Mismatch Glycosylase) and in a Related Avian Sequence. Nucleic Acids Res. 28, 4157–4165. doi:10.1093/nar/28.21.4157
Zhu, J. K. (2016). Abiotic Stress Signaling and Responses in Plants. Cell 167, 313–324. doi:10.1016/j.cell.2016.08.029
Zhu, N., Cheng, S., Liu, X., Du, H., Dai, M., Zhou, D. X., et al. (2015). The R2R3-type MYB Gene OsMYB91 Has a Function in Coordinating Plant Growth and Salt Stress Tolerance in rice. Plant Sci. 236, 146–156. doi:10.1016/j.plantsci.2015.03.023
Keywords: biotechnology, epigenetics, food security, abiotic stress, biotic stress, stress memory
Citation: Saeed F, Chaudhry UK, Bakhsh A, Raza A, Saeed Y, Bohra A and Varshney RK (2022) Moving Beyond DNA Sequence to Improve Plant Stress Responses. Front. Genet. 13:874648. doi: 10.3389/fgene.2022.874648
Received: 12 February 2022; Accepted: 31 March 2022;
Published: 19 April 2022.
Edited by:
Vinod K. K, Indian Agricultural Research Institute (ICAR), IndiaReviewed by:
Muthusamy Ramakrishnan, Nanjing Forestry University, ChinaCopyright © 2022 Saeed, Chaudhry, Bakhsh, Raza, Saeed, Bohra and Varshney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajeev K. Varshney, cmFqZWV2LnZhcnNobmV5QG11cmRvY2guZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.