 Weigang Ji1
Weigang Ji1 Xueqian Wang
Xueqian Wang- 1Department of Pediatrics, Affiliated Matern & Child Care Hospital of Nantong University, Nantong, China
- 2Department of Prenatal Screening and Diagnosis Center, Affiliated Matern & Child Care Hospital of Nantong University, Nantong, China
- 3Department of Radiology, Affiliated Matern & Child Care Hospital of Nantong University, Nantong, China
- 4Department of Ultrasound, Affiliated Matern & Child Care Hospital of Nantong University, Nantong, China
- 5Nantong Institute of Genetics and Reproductive Medicine, Affiliated Matern & Child Care Hospital of Nantong University, Nantong, China
The SMPD4 gene encodes sphingomyelin phosphodiesterase 4, which preferentially hydrolyzes sphingomyelin over other phospholipids. The biallelic loss-of-function variants of SMPD4 have been identified in a group of children with neurodevelopmental disorder with microcephaly, arthrogryposis, and structural brain anomalies (NEDMABA). Here, we report a girl of Chinese ancestry with intrauterine growth restriction, microcephaly, postnatal developmental delay, arthrogryposis, hypertonicity, seizure, and hypomyelination on brain magnetic resonance imaging; biallelic null variants (c.1347C > G [p.Tyr449*]; Chr2 [GRCh37]: g.130877574_131221737del [whole-gene deletion]) were detected by whole-exome sequencing. Our case is the first report of NEDMABA of Chinese ancestry, confirming the involvement of SMPD4 in NEDMABA and expanding the mutation spectrum of this syndrome.
Introduction
Neurodevelopmental disorders are a group of highly heterogenous conditions characterized by an inability to reach cognitive, emotional, and motor developmental milestones. Neurodevelopmental disorders have a complex pathophysiology and an etiology that may involve genetic and environmental factors such as genetic syndromes, metabolic abnormalities, immunologic disorders, infection, physical trauma, and exposure to toxic agents. Many neurodevelopmental disorders accompanied by structural abnormalities have a chromosomal or monogenic etiology. Recently, neurodevelopmental disorder with microcephaly, arthrogryposis, and structural brain anomalies (NEDMABA) was described as an autosomal recessive disorder (Mendelian Inheritance in Man [MIM]: 618622) caused by homozygous or compound heterozygous mutations in the sphingomyelin phosphodiesterase 4 gene (SMPD4; MIM: 610457) on chromosome 2 (Magini et al., 2019). The majority of individuals with NEDMABA present with intrauterine growth restriction (IUGR), congenital microcephaly, neonatal respiratory distress, and arthrogryposis of the hands and feet. Magnetic resonance imaging (MRI) has revealed a simplified gyral pattern of the cerebral cortex, delayed myelination, thin corpus callosum, and hypoplasia of the brainstem and cerebellum (Magini et al., 2019; Monies et al., 2019; Ravenscroft et al., 2020).
Here, we report two compound heterozygous null variants of SPMD4 in a child of a Chinese family presenting with IUGR, microcephaly, postnatal developmental delay, hypomyelination, arthrogryposis, hypertonicity, and seizures. As the clinical manifestations were identical to NEDMABA, our findings provide additional evidence for the critical role of SMPD4 in this syndrome.
Case Presentation
A girl was the first child of a nonconsanguineous couple of Chinese ancestry. Her family history was unremarkable. She was naturally conceived when her mother was 22 years old. At the first trimester screening, nuchal translucency was 1.49 mm. Ultrasound examination revealed IUGR and microcephaly at 34 + 3 weeks with an estimated fetal weight of 1,695 g (−2.7 SD) (Figure 1A) and a head circumference of 274 mm (−3.6 SD) (Figure 1B). The child was born at 39 + 2 weeks gestation by vaginal delivery. The birth weight was 2,460 g (−2.2 SD), and the Apgar score was 9 at 0 min and 9 at 5 min (the reference population was Asian and Pacific Islander from the National Institute of Child Health and Human Development Fetal Growth Study (Buck Louis et al., 2015)). At 2 months, she was transferred to the pediatric intensive care unit (ICU) with acute wheezing bronchitis, developmental delay, and seizure; her body weight was 2,440 g (−3 SD) and head circumference was 32 cm (−3 SD) (the reference population was Chinese children (Zong and Li, 2013)). The physical examination found craniosynostosis with an anterior fontanelle (0.5 × 0.5 cm) and hypertonia. Arthrogryposis of the index fingers and thumbs was observed. The results of routine tests such as liver function, blood glucose, renal function, and newborn screening by tandem mass spectrometry were normal, and she passed the auditory brainstem response test. An MRI revealed a thin corpus callosum (Figure 1C), hypomyelination (Figure 1D), and decreased craniofacial ratio. No other abnormalities were found. She passed away 1 week after ICU admission from respiratory failure.
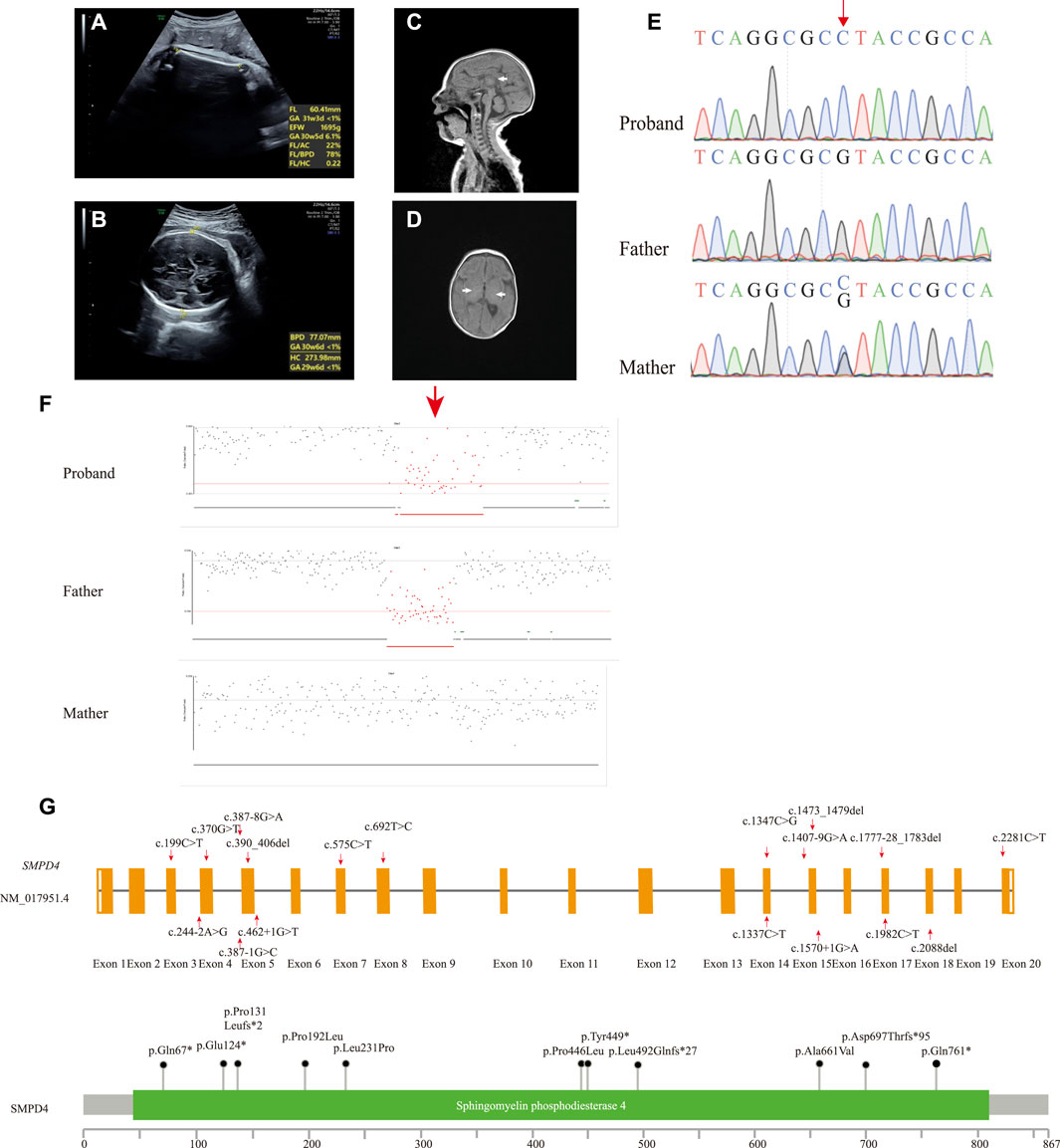
FIGURE 1. (A,B) Prenatal ultrasound image of fetus at 34 + 3 weeks showing IUGR (A) and microcephaly (B). MR image of the 2-month-old patient. (C) T1-weighted image (T1WI) of median sagittal section showing thin corpus callosum, splenium, and unclear body (white arrow). (D) T1WI axial section absent of high signal of bilateral posterior limbs of the internal capsule showing hypomyelination (white arrow). (E) Sanger sequencing results of the proband and her parents. The single-nucleotide substitution is indicated by the red arrow. (F) Deletion in the proband was inherited from the father (red arrow). (G) Schematic presentation of linear SMPD4 transcript (NM_017951.4) (up) and protein (down) with all variants reported.
Molecular Findings
Peripheral blood samples were collected from the proband and her parents for trio-whole exome sequencing (WES). Genomic DNA was extracted from the blood samples using the SolPure Blood DNA Kit (Guangzhou Magen Biotechnology Co., Guangzhou, China). Exome capture was performed using xGen Exome Research panel v1 (Integrated DNA Technologies, Coralville, IA, United States), and sequencing was performed using a NovaSeq 6,000 system (Illumina, San Diego, CA, United States). The sequences were aligned to a human reference sequence (NCBI Genome build GRCh37) with the Burrows–Wheeler Aligner (0.7.10-r789) (Li and Durbin, 2010), and coverage above 20× was >98%. The Genome Analysis Toolkit (4.1.8) pipeline was used to detect single-nucleotide and insertion/deletion (INDEL) polymorphisms (McKenna et al., 2010), with 39,816 variants identified. The variants were annotated with ANNOVAR (2019–10-24) according to GRCh37 (Wang et al., 2010), and variant interpretation was performed according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines and Clinical Genome Resource specifications (Richards et al., 2015; Zhang et al., 2020). We prioritized variants that were previously reported, considered loss-of-function (nonsense, frameshift, or splice sites mutations) or absent in gnomAD. IUGR (Human Phenotype Ontology [HP]: 0001511), central nervous system hypomyelination (HP: 0003429), seizure (HP: 0001250), and microcephaly (HP: 0000252) were used to narrow down the candidate gene list. We identified a novel homozygous nonsense variant in SMPD4 (NM_017951.4: c.1347C > G [p.Tyr449*]). This variant had a CADD_PHRED score of 34 and was absent in gnomAD (PM2_Supporting) and was predicted to cause a premature termination in exon 14 of 20 that likely results in nonsense-mediated mRNA decay (PVS1). The variant was, therefore, classified as likely pathogenic. The 3-dimensional structure of the SMPD4 protein was obtained using the AlphaFold Protein Structure database (Varadi et al., 2022), and mutations were predicted using ChimeraX1.3 (Pettersen et al., 2021) (Supplementary Figure S1A). The mutation was heterozygous in the mother and absent in the father, suggesting that it was hemizygous in the setting of a paternal deletion. Sanger sequencing was performed to confirm the result (Figure 1E). NextGENe software (SoftGenetics, State College, PA, United States) was used to analyze WES copy number variations (CNVs), which revealed a paternally inherited 344-kb contiguous gross deletion (Chr2 [GRCh37]: g.130877574_131221737del) that encompassed the whole SMPD4 gene (Figure 1F). This region contained part or all of 29 genes, two of which had Online MIM phenotypes (SMPD4 and coiled-coil domain–containing 115 [CCDC115]), with no haploinsufficient genes identified. CCDC115 is responsible for the congenital disorder of glycosylation, type IIo (MIM: 616828), an autosomal recessive metabolic disorder characterized by infantile onset of progressive liver failure, hypotonia, and delayed psychomotor development. There were no rare variants of the CCDC115 gene. Consequently, the c.1347C > G mutation was determined to be hemizygous, and SMPD4 was identified as the causative gene in our patient. The steps and methodology for molecular diagnosis are summarized in Supplementary Figure S1B.
Discussion and Conclusion
SMPD4 encodes sphingomyelin phosphodiesterase 4 (SMPD4), which preferentially hydrolyzes sphingomyelin over other phospholipids (Krut et al., 2006). Sphingomyelin is required for the proper functioning of the nervous system; an imbalance between sphingomyelin synthesis and degradation has been linked to a variety of neurologic pathologies including Niemann–Pick disease and Alzheimer’s disease (Bienias et al., 2016). In fibroblasts derived from affected individuals, SMPD4 deletion results in aberrant mitosis and increased susceptibility to apoptotic cell death (Magini et al., 2019), which are mechanisms that have been shown to underlie human microcephaly and a simplified gyral pattern (Adachi et al., 2011).
To date, 20 SMPD4 variants have been identified in 29 genetically confirmed individuals from 16 unrelated families (including our case) (Table 1); 28 individuals presented part or all of the manifestations including IUGR, microcephaly, arthrogryposis, a thin corpus callosum, and a simplified gyral pattern. However, only one individual showed distinct symptoms of brain atrophy and skeletal dysplasia (UPN-1246) (Monies et al., 2019). Notably, this individual shared the same homozygous null variant with another case of Arab descent (Family 5) who presented typical symptoms. Thus, skeletal dysplasia and brain atrophy are not variant-specific features. No animal models are currently available to confirm the phenotypes of SPMD4 knockout; therefore, additional cases and functional analyses are needed to determine why the same null mutation resulted in two distinct phenotypes.
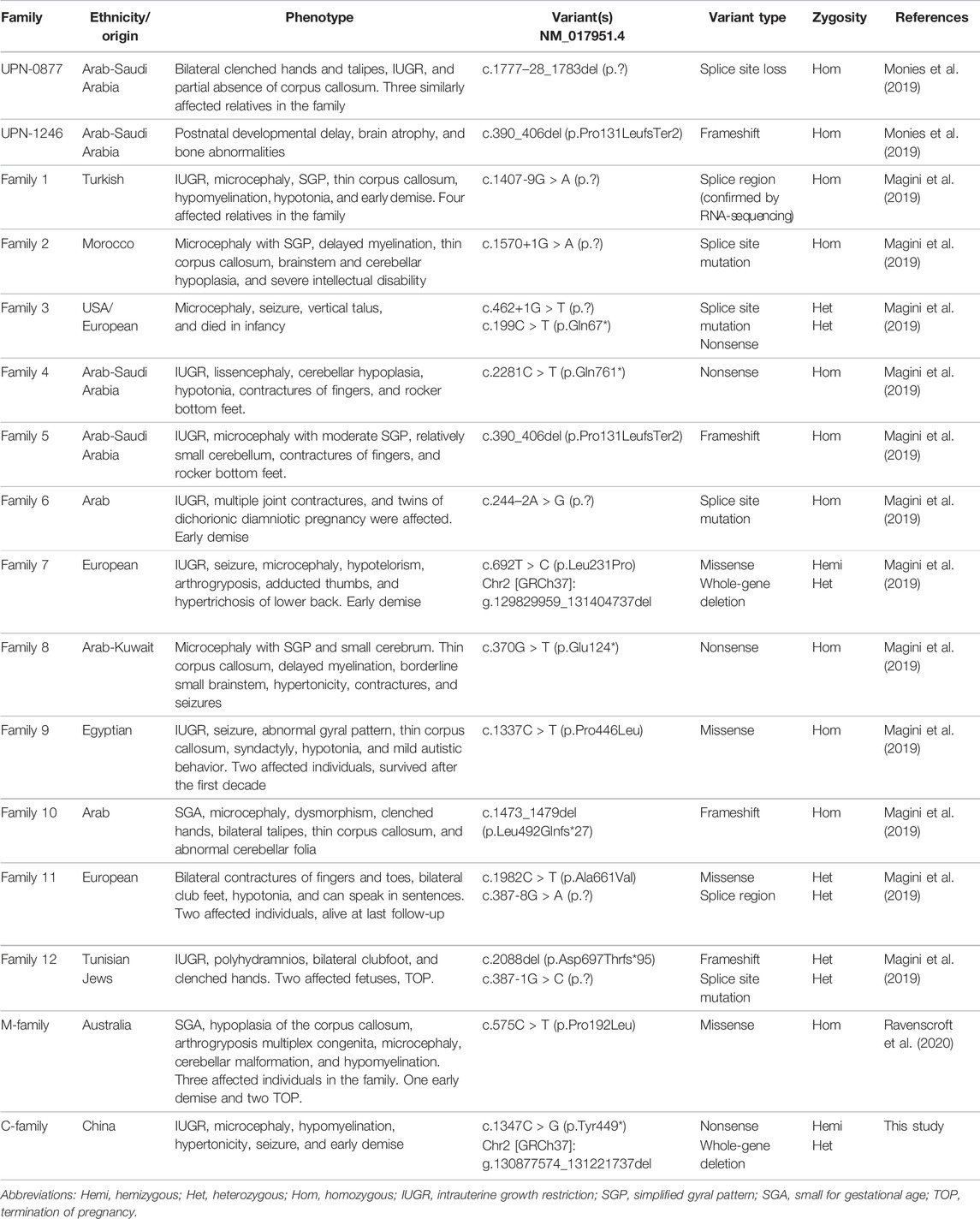
TABLE 1. SMPD4 variants and affected families.
All reported SMPD4 variants are summarized in Table 1; Figure 1F including nonsense mutations (4/20), splice site/region mutations (7/20), frameshifts (3/20), missense mutations (4/20), and gross deletions (2/20); of these, 15/20 are null mutations. Individuals with biallelic null mutations always exhibit more severe phenotypes, such as brain structural abnormalities, arthrogryposis, and early death (Magini et al., 2019; Monies et al., 2019; Ravenscroft et al., 2020). Four individuals from two families (Families 9 and 11) harboring non-null mutations survived into childhood and showed some motor skill and mental development (Magini et al., 2019). Our proband carried the compound heterozygous nonsense mutation and a whole-gene deletion, both of which abolished the protein and appeared to cause severe neonatal developmental delay, microcephaly with craniosynostosis, and early demise, which are among the most severe manifestations of NEDMABA.
Although only 28 individuals with NEDMABA have been reported, this may be an underestimate; the prevalence estimated based on the gene carrier rate (Guo and Gregg, 2019) calculated from loss-of-function variants of SMPD4 in gnomAD is about one in 1,580,000. This may be attributable to the fact that the typical symptoms of NEDMABA are nonspecific, making clinical diagnosis difficult. In many countries, chromosomal microarray is the first-tier genetic test for individuals with developmental disabilities or congenital anomalies, with diagnosis rates of 10–20% (Miller et al., 2010). Since 2011, WES has been increasingly used to determine the etiology of genetic disorders; more than 20% of patients can be diagnosed using this method (Yang et al., 2013; Meng et al., 2017). WES can identify single-nucleotide variations (SNVs) and small INDELs. However, large CNVs are missed by the standard analysis pipeline. Recently, several algorithms for WES-based CNV detection have been developed based on comparisons of depth of coverage (Yang et al., 2013; Backenroth et al., 2014; Fromer and Purcell, 2014; Talevich et al., 2016; Meng et al., 2017) and could detect the CNVs of the exon level which are smaller than those detected by CMA. Thus, WES could replace chromosomal microarray as a more cost-effective genetic test for detecting CNVs and diagnosing highly heterogenous conditions such as NEDMABA. In our case, the standard pipeline only identified a maternally inherited homozygous variant. There are several possible explanations for this observation including uniparental disomy, de novo mutation, and deletion of the corresponding region on another allele. The risk to the siblings of the affected individual should be discussed in genetic counseling sessions, although that is likely to vary according to the situation. Based on the paternally inherited deletion identified by CNV WES, we predict a 25% risk.
In conclusion, our study reveals for the first time the NEDMABA phenotype of an individual of Chinese ancestry and provides further evidence for the role of SMPD4 in this syndrome.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Affiliated Maternity and Child Health Care Hospital of Nantong University (Y2019038). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
WJ and XW designed the study. XW and XK performed the genetic and bioinformatic analyses. HY, JX and XW prepared the figure and drafted the manuscript. WJ and XK conducted the clinical evaluations and collected clinical data. All authors read and approved the final article.
Funding
This study was funded by the Clinical Medical Project of Nantong University (Grant No. 2019LZ014); Innovative and Entrepreneurial Ph.D. Program of Jiangsu Province; and Jiangsu Health Innovation Team Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all of the participation of the family members in this study. Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.872264/full#supplementary-material
References
Adachi, Y., Poduri, A., Kawaguch, A., Yoon, G., Salih, M. A., Yamashita, F., et al. (2011). Congenital Microcephaly with a Simplified Gyral Pattern: Associated Findings and Their Significance. AJNR Am. J. Neuroradiol. 32, 1123–1129. doi:10.3174/ajnr.A2440
Backenroth, D., Homsy, J., Murillo, L. R., Glessner, J., Lin, E., Brueckner, M., et al. (2014). CANOES: Detecting Rare Copy Number Variants from Whole Exome Sequencing Data. Nucleic Acids Res. 42, e97. doi:10.1093/nar/gku345
Bienias, K., Fiedorowicz, A., Sadowska, A., Prokopiuk, S., and Car, H. (2016). Regulation of Sphingomyelin Metabolism. Pharmacol. Rep. 68, 570–581. doi:10.1016/j.pharep.2015.12.008
Buck Louis, G. M., Grewal, J., Albert, P. S., Sciscione, A., Wing, D. A., Grobman, W. A., et al. (2015). Racial/Ethnic Standards for Fetal Growth: The NICHD Fetal Growth Studies. Am. J. Obstet. Gynecol. 213, 449.e1–449.e41. doi:10.1016/j.ajog.2015.08.032
Fromer, M., and Purcell, S. M. (2014). Using XHMM Software to Detect Copy Number Variation in Whole‐Exome Sequencing Data. Curr. Protoc. Hum. Genet. 81, 7.23.1–7.23.21. doi:10.1002/0471142905.hg0723s81
Guo, M. H., and Gregg, A. R. (2019). Estimating Yields of Prenatal Carrier Screening and Implications for Design of Expanded Carrier Screening Panels. Genet. Med. 21, 1940–1947. doi:10.1038/s41436-019-0472-7
Krut, O., Wiegmann, K., Kashkar, H., Yazdanpanah, B., and Krönke, M. (2006). Novel Tumor Necrosis Factor-Responsive Mammalian Neutral Sphingomyelinase-3 Is a C-Tail-Anchored Protein. J. Biol. Chem. 281, 13784–13793. doi:10.1074/jbc.M511306200
Li, H., and Durbin, R. (2010). Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Magini, P., Smits, D. J., Vandervore, L., Schot, R., Columbaro, M., Kasteleijn, E., et al. (2019). Loss of SMPD4 Causes a Developmental Disorder Characterized by Microcephaly and Congenital Arthrogryposis. Am. J. Hum. Genet. 105, 689–705. doi:10.1016/j.ajhg.2019.08.006
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Meng, L., Pammi, M., Saronwala, A., Magoulas, P., Ghazi, A. R., Vetrini, F., et al. (2017). Use of Exome Sequencing for Infants in Intensive Care Units. JAMA Pediatr. 171, e173438. doi:10.1001/jamapediatrics.2017.3438
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 86, 749–764. doi:10.1016/j.ajhg.2010.04.006
Monies, D., Abouelhoda, M., Assoum, M., Moghrabi, N., Rafiullah, R., Almontashiri, N., et al. (2019). Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 105, 879. doi:10.1016/j.ajhg.2019.04.01110.1016/j.ajhg.2019.09.019
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX : Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 30, 70–82. doi:10.1002/pro.3943
Ravenscroft, G., Clayton, J. S., Faiz, F., Sivadorai, P., Milnes, D., Cincotta, R., et al. (2020). Neurogenetic Fetal Akinesia and Arthrogryposis: Genetics, Expanding Genotype-Phenotypes and Functional Genomics. J. Med. Genet. 58, 609–618. doi:10.1136/jmedgenet-2020-106901
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. Plos Comput. Biol. 12, e1004873. doi:10.1371/journal.pcbi.1004873
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 50, D439–D444. doi:10.1093/nar/gkab1061
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 369, 1502–1511. doi:10.1056/NEJMoa1306555
Zhang, J., Yao, Y., He, H., and Shen, J. (2020). Clinical Interpretation of Sequence Variants. Curr. Protoc. Hum. Genet. 106, e98. doi:10.1002/cphg.98
Keywords: SMPD4, neurodevelopmental disorder (NDD), null variants, microcephaly, structural brain anomalies, arthrogryposis, early death
Citation: Ji W, Kong X, Yin H, Xu J and Wang X (2022) Case Report: Novel Biallelic Null Variants of SMPD4 Confirm Its Involvement in Neurodevelopmental Disorder With Microcephaly, Arthrogryposis, and Structural Brain Anomalies. Front. Genet. 13:872264. doi: 10.3389/fgene.2022.872264
Received: 09 February 2022; Accepted: 18 March 2022;
Published: 16 May 2022.
Edited by:
Dan Koboldt, Nationwide Children’s Hospital, United StatesReviewed by:
Louise Bicknell, University of Otago, New ZealandMuhammad Umair, King Abdullah International Medical Research Center (KAIMRC), Saudi Arabia
Copyright © 2022 Ji, Kong, Yin, Xu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueqian Wang, c3RldmVuLndvbmcuYmlvQGZveG1haWwuY29t