 Jun Fu1,2†
Jun Fu1,2† Jiewen Zhang
Jiewen Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 08 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.859688
Background: Mutations in the NIPA1 gene cause hereditary spastic paraplegia (HSP) type 6 (SPG6), which is a rare type of HSP with a frequency of less than 1% in Europe. To date, less than 30 SPG6 families and limited NIPA1 mutations have been reported in different ethnic regions. The clinical features are variable.
Methods: We screened for NIPA1 mutations by whole exome sequencing or next generation sequencing in 35 unrelated Chinese families with HSP. The clinical manifestations were evaluated.
Results: Two variants of NIPA1 were identified in three index patients (3/35, 8.6%), two of whom carried a previously reported common variant c.316G > A (p.G106R), and the third patient harbored a novel likely pathogenic variant c.126C > G (p.N42K). Both variants were de novo in the three index patients. The phenotype was pure HSP in two patients and complicated HSP with epilepsy in the third one.
Conclusion: NIPA1-related HSP is more common in China than it in Europe. Both pure and complicated form of HSP can be found. The variant c.316G > A is a hotspot mutation, and the novel variant c.126C > G expands the mutational spectrum. The phenomenon of de novo mutations in NIPA1 emphasizes the need to consider autosomal dominant HSP-related genes in sporadic patients.
Hereditary spastic paraplegia (HSP) comprises a group of clinically and genetically heterogeneous neurodegenerative disorders (Erfanian et al., 2021). Clinically, HSP is classified as pure form characterized by progressive lower limb weakness and spasticity, or complicated form with additional features (Harding, 1983). Thus far, more than 80 genes for HSPs have been identified (Erfanian et al., 2021). Mutations in the non-imprinted in Prader-Willi/Angelman syndrom 1 (NIPA1) gene have been identified as the cause of hereditary spastic paraplegia type 6 (SPG6) with an autosomal dominant (AD) mode of inheritance (Rainier et al., 2003). SPG6 is a very rare type of HSP, accounting for less than 1% of all ADHSP cases in Europe (Klebe et al., 2007). To date, less than 30 SPG6 families have been reported in different ethnic populations (Chen et al., 2005; Bien-Willner et al., 2006; Kaneko et al., 2006; Munhoz et al., 2006; Klebe et al., 2007; Kim et al., 2019). The phenotype was often pure form; however, complicated forms have also been described with polyneuropathy (Du et al., 2011), idiopathic generalized epilepsy (Svenstrup et al., 2011), cognitive impairment (Martinez-Lage et al., 2012), ataxia (Kim et al., 2019), or amyotrophic lateral sclerosis (ALS) (Tanti et al., 2020). The mutational spectrum of NIPA1 is quite limited with only seven mutations reported previously, and most of the SPG6 patients harbored a hotspot mutation c.316G > A (p.G106R) (Hedera, 2013).
In this study, we screened for NIPA1 mutations by whole exome sequencing or next generation sequencing in 35 Chinese HSP families. Finally, we identified a known variant c.316G > A (p.G106R) in two unrelated patients and a novel variant c.126C > G (p.N42K) in the third patient. Both variants were de novo in the three index patients. Detailed manifestations were described and a general review of NIPA1-related HSP was performed to elucidate the clinical and genetic features of this disease.
From 2018 to 2022, we performed genetic testing for 35 unrelated Chinese patients clinically diagnosed with HSP according to the Harding’s criteria (Harding, 1983) from Henan province. All index patients and some of their relatives underwent detailed clinical evaluation. The mode of inheritance was autosomal dominant in 12 families, autosomal recessive in two families, and apparently sporadic in 21 cases with no evidence of family history. Among the 35 index patients, 13 cases presented with a complicated phenotype. Three families were finally identified to be NIPA1-related SPG6. This study was approved by the Ethics Committee of Henan Provincial People’s Hospital. All participants gave their written informed consent.
Genomic DNA was extracted from peripheral blood samples from all participants following standard procedures. Whole exome sequencing was performed on some probands using Agilent SureSelect Human All Exon 50-Mb kit (Agilent, Santa Clara, CA, United States) for exome enrichment and the Illumina HiSeq2500 platform (Illumina, San Diego, CA, United States). Next generation sequencing was also conducted on the other probands using a panel targeting more than 3,000 genes related to neurological diseases, including HSP. All identified variants were validated by Sanger sequencing. The variants with minor allele frequency (MAF) of >1% in the Single Nucleotide Polymorphism Database (dbSNP), the Genome Aggregation Database (gnomAD), Exome Aggregation Consortium (ExAC), and the 1,000 Genomes Project database (1000G) were excluded. In silico predictions of the functional effect of variants were performed with MutationTaster (https://www.mutationtaster.org), PolyPhen-2 (https://genetics.bwh.harvard.edu/pph2) and SIFT (https://sift.jcvi.org). Co-segregation analysis was further performed by Sanger sequencing in the family members. For de novo variants, paternity was confirmed by analysis of highly polymorphic unlinked microsatellite makers. The novel variants were assigned in accordance with the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015).
Genetic diagnosis of HSP was established for 25 families (25/35, 71.4%). The most frequently affected gene was SPAST (SPG4) (n = 9), followed by SPG7 (SPG7) (n = 4), SPG11 (SPG11) (n = 3), NIPA1 (SPG6) (n = 3, 3/35, 8.6%). Additional mutations were detected in ATL1 (SPG3A), CYP7B1 (SPG5A), KIAA0196 (SPG8), ALDH18A1 (SPG9B), KIF5A (SPG10), and REEP1 (SPG31) in each one patient. The clinical features and mutations were briefly summarized in the Supplementary Table S1.
Two variants of NIPA1 (NM_144599) were identified in three families (Figure 1). A previously reported heterozygous variant, c.316G > A (p.G106R) (Figure 1C) (Chen et al., 2015), was detected in two index patients (family 1 Ⅱ-1 and family 2 Ⅱ-1) (Figures 1A,B). This variant was only found in one daughter (family 1 Ⅲ-1) of the first index patient. Both parents of the two index patients did not harbor this variant, indicating that it was a de novo variant.
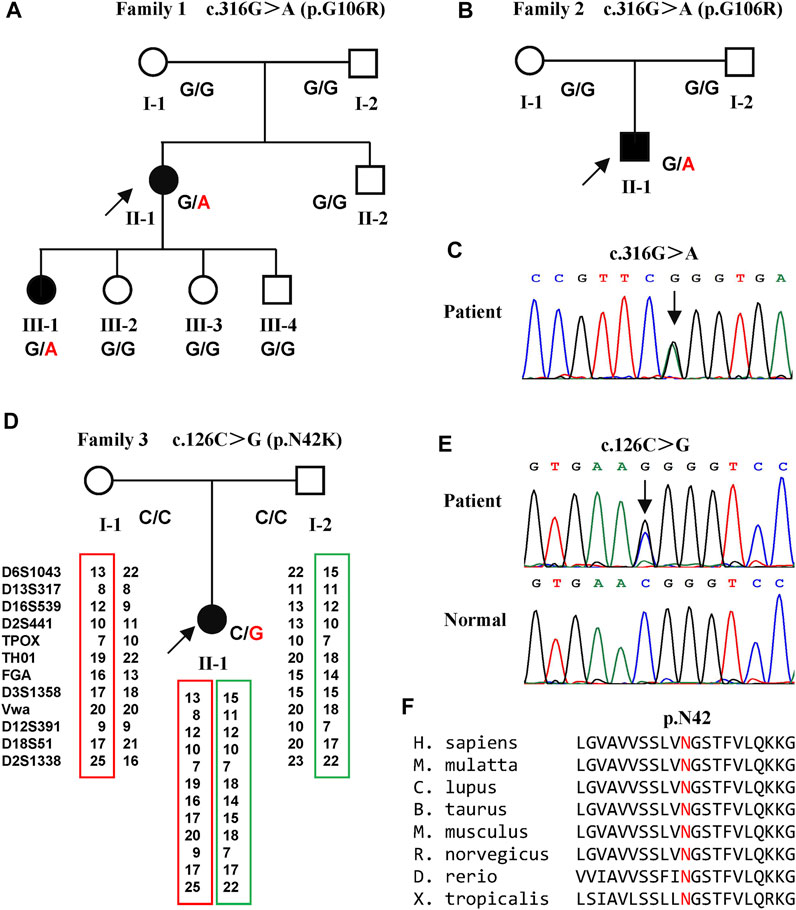
FIGURE 1. Pedigrees of the three families with SPG6 and genetic results. (A) Pedigree of family 1. The proband (arrow) and her elder daughter (Ⅲ-1) were heterozygous for the c.316G > A variant of NIPA1. (B) Pedigree of family 2. The proband (arrow) also carried the c.316G > A variant. (C) Sanger sequencing of the c.316G > A variant in the probands of family one and family 2. (D) Pedigree of family 3. The proband (arrow) carried the c.126C > G variant and her parents were normal. Paternity was confirmed by 12 highly polymorphic unlinked microsatellite makers. (E) Sanger sequencing of the c.126C > G variant in the proband of family three and her patients. (F) The asparagine at amino acid 42 was conserved in different species.
A previously unreported variant, c.126C > G (p.N42K) (Figure 1E) in exon one of NIPA1, was found in the third index patient (family 3 Ⅱ-1) (Figure 1D). This variant was neither found in ExAC nor 1000G, and predicted to be damaging by in silico analysis. The amino acid asparagine at position of 42 was conserved in different species (Figure 1F). Both parents of the third index did not harbor this variant, and true parenthood was confirmed by 12 highly informative unlinked microsatellite markers (Figure 1D). Thus, the variant c.126C > G was also de novo. According to the standards of ACMG, the novel variant c.126C > G was classified as likely pathogenic (evidence PS2+PM2+PP3).
The index patient of family 1 (Ⅱ-1) presented with gradually progressive lower limb weakness and stiffness since the age of 23 years (Table 1). She deteriorated and was assisted by a walker in the past 2 years. A history of generalized epilepsy was reported since 10 years old. She was treated with oral valproic acid irregularly, and no seizures occurred in the past 5 years. Neurological examination at the age of 35 years revealed marked spasticity, moderate weakness, and hyperactive deep tendon reflexes that were more prominent in the lower extremities. Bilateral ankle clonus, extensor plantar responses and pes cavus were also observed. There was no impairment of cognition, sensation, sphincter or cerebellar function. Brain and spine MRI were normal. Her elder daughter (family 1 Ⅲ-1), now 15 years old, had no symptoms of spasticity, but revealed hyperactive deep tendon reflexes in the lower limbs and extensor plantar responses (Table 1). Examination of both parents was normal.
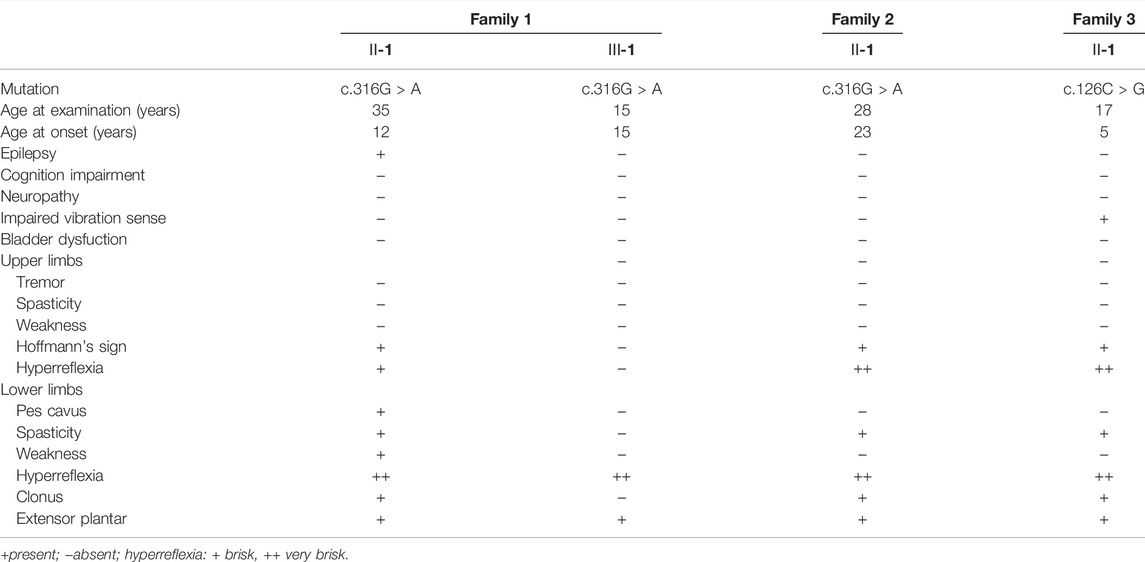
TABLE 1. Clinical features of affected family members carrying NIPA1 mutations.
The index patient of family 2 (Ⅱ-1) was a 28 year-old man with the complaint of gradually progressive leg stiffness and shaking for 5 years (Table 1). He had no epilepsy or cognition impairment. Neurological examination revealed hyperactive deep tendon reflexes in both upper and lower limbs, bilateral ankle clonus, and extensor plantar responses. Both of his parents were normal on examination.
The index patient of family 3 (Ⅱ-1) was referred with the early onset gait disturbance since the age of 5 years (Table 1). She did not have any other medical problems and her parents were normal. Upon examination at age 17, a moderate spasticity especially in the lower limbs was found associated with mild diminished vibration sensation distally. MRI studies showed thoracic spinal cord atrophy. Electromyography and nerve conduction velocity studies were unremarkable.
In this study, we detected three patients with NIPA1 mutations amongst 35 Chinese HSP families. Thus, the mutation frequency was 8.6%. NIPA1 mutation was reported to be a rare cause of HSP (Klebe et al., 2007). Though identified in different ethnic populations (Chen et al., 2005; Kaneko et al., 2006; Bien-Willner et al., 2006; Munhoz et al., 2006; Klebe et al., 2007; Kim et al., 2019), less than 30 families with NIPA1-related SPG6 have been reported since the year of 2003 (Rainier et al., 2003) (Table 2). In the previous genetic screening studies of HSP, there was no NIPA1 mutation identified in German (Beetz et al., 2008), Italian (D'Amore et al., 2018), Korean (Yang et al., 2021) or Japanese (Ishiura et al., 2014) patients, and only one case carrying NIPA1 mutation found in France (Klebe et al., 2007), Hungarian (Balicza et al., 2016) and Danish (Svenstrup et al., 2011) patients, respectively (Table 3). However, previous studies in Chinese patients revealed a high NIPA1 mutation frequency of 3.6% (Lu et al., 2018), and it was the third most common cause of ADHSP (Dong et al., 2018). The higher mutation rate of NIPA1 in our study may be due to small sample size. Together with our study, NIPA1-related SPG6 was more common in China than it in Europe or other Asian countries.
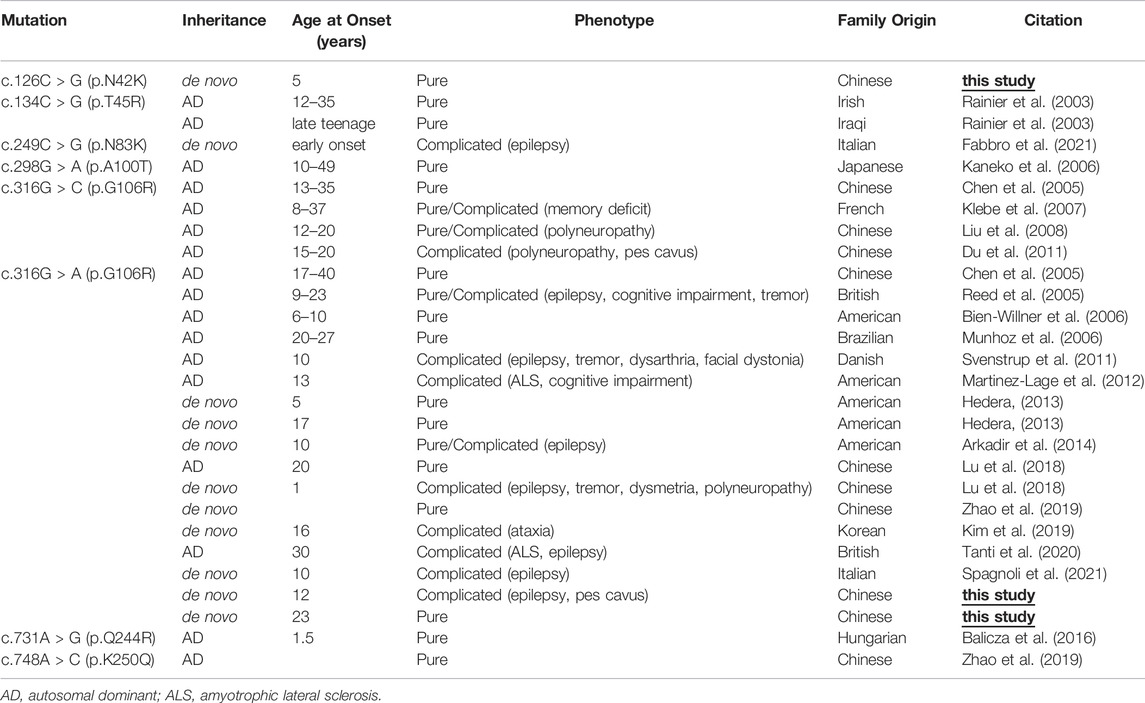
TABLE 2. Clinical features and NIPA1 mutations of SPG6 families reported in the literature and in the present study.

TABLE 3. NIPA1 mutation rate in different regions.
The phenotype of our patients with SPG6 was similar to the other reports. The age of disease onset was usually in the second and third decades, although variability could also be observed (Table 2). The index patient of family three in our study had an early onset age of 5 years, and it could be as early as 1 year (Lu et al., 2018). The disease often progressed slowly, while some patients deteriorated and required walking aids in their twenties or thirties as the first index patient in our study (Bien-Willner et al., 2006; Svenstrup et al., 2011; Hedera, 2013). SPG6 is known as a generally pure form of HSP; however, more cases with a complicated phenotype have also been reported. The co-morbidities included idiopathic generalized epilepsy (Svenstrup et al., 2011), polyneuropathy (Liu et al., 2008; Du et al., 2011), cognitive impairment (Martinez-Lage et al., 2012), ataxia (Kim et al., 2019), postural tremor (Svenstrup et al., 2011; Lu et al., 2018) and amyotrophic lateral sclerosis (ALS) (Tanti et al., 2020). Until now, epilepsy has been described in eight families with SPG6 (Reed et al., 2005; Svenstrup et al., 2011; Arkadir et al., 2014; Lu et al., 2018; Tanti et al., 2020; Fabbro et al., 2021; Spagnoli et al., 2021), including the first index patient in our study, who presented with a complicated form of HSP. Why NIPA1 mutation might cause epilepsy is unclear. The other patients in the present study showed a pure form of HSP.
The NIPA1 gene has five coding exons located at 15q11.2, and encodes a nine transmembrane protein as an intracellular magnesium transporter (Goytain et al., 2007). To date, only seven missense variants of NIPA1 have been previously reported (Table 2; Figure 2). In our study, we identified the most common variant c.316G > A (p.G106R) in two unrelated patients. It has been discovered in more than 10 HSP families from China (Chen et al., 2005; Lu et al., 2018; Zhao et al., 2019), Britain (Reed et al., 2005), Brazil (Munhoz et al., 2006), Danmark (Svenstrup et al., 2011), Italy (Spagnoli et al., 2021), Korea (Kim et al., 2019) and America (Hedera 2013). Thus, it was considered to be a hotspot mutation of NIPA1 with the mechanism of DNA methylation in the coding regions (Beetz et al., 2008). Patients carrying the variant c.316G > A could present with pure or complicated HSP (Tanti et al., 2020). We further identified a novel variant c.126C > G (p.N42K) in the third index patient. It occurred in the first transmembrane domain and was near to the reported pathogenic variant c.134C > G (p.T45R) (Rainier et al., 2003). By analysis of in silico predictions and family segregation, c.126C > G was classified as likely pathogenic; therefore, it expanded the mutational spectrum of NIPA1. The patient carrying this novel variant presented with a pure HSP. The disease mechanism of NIPA1-related SPG6 is likely to be toxic gain of function (Zhao et al., 2008).
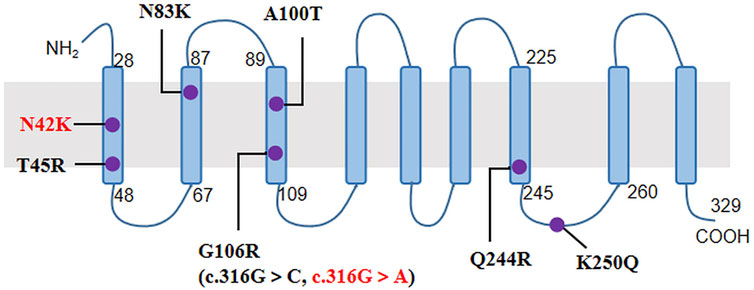
FIGURE 2. The nine transmembrane domains of NIPA1 protein and localization of NIPA1 mutations. The two mutations identified in our patients were indicated by red colour.
Interestingly, all of the three index patients in our study harbored a de novo mutation, which was not common in HSP (Lu et al., 2018). Most of the previously reported SPG6 patients were familial cases with autosomal dominant inheritance (Rainier et al., 2003). The de novo mutations of NIPA1 were only documented in several cases since the first report by Hedera (Hedera et a., 2013). In such sense, the ADHSP-related genes need to be considered in the screening of HSP patients without family history.
Recently, a meta-analysis provided evidence for the association of NIPA1 repeat expansions with ALS, which showed an overall increased risk of ALS in those with expanded (>8) GCG repeat length (Tazelaar et al., 2019). In addition to NIPA1, repeat expansions in C9orf72 and ATXN2 have also been reported in ALS (Elden et al., 2010; DeJesus-Hernandez et al., 2011). However, NIPA1 repeat length was not confirmed to be a modifier of the C9orf72 ALS disease risk (Corrado et al., 2019).
In summary, we reported three SPG6 families, which indicated that NIPA1 mutations were more common in China. The phenotype of SPG6 included both pure and complicated HSP. The variant c.316G > A of NIPA1 was a hotspot mutation, and the novel variant c.126C > G expanded the mutational spectrum. The phenomenon of de novo mutations in NIPA1 emphasized the need to consider ADHSP-related genes in sporadic patients.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of Henan Provincial People’s Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
JZ contributed to conception and design of the study. JF, MM, and GL contributed to patient material and clinical data. JF wrote the first draft of the manuscript. All authors contributed to article revision, read, and approved the submitted version.
This study was funded by the National Natural Science Foundation of China (No. 81873727).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the patients and their family members for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.859688/full#supplementary-material
Arkadir, D., Noreau, A., Goldman, J. S., Rouleau, G. A., and Alcalay, R. N. (2014). Pure Hereditary Spastic Paraplegia Due to Ade Novomutation in theNIPA1gene. Eur. J. Neurol. 21 (1), e2. doi:10.1111/ene.12284
Balicza, P., Grosz, Z., Gonzalez, M. A., Bencsik, R., Pentelenyi, K., Gal, A., et al. (2016). Genetic Background of the Hereditary Spastic Paraplegia Phenotypes in Hungary - an Analysis of 58 Probands. J. Neurol. Sci. 364, 116–121. doi:10.1016/j.jns.2016.03.018
Beetz, C., Schüle, R., Klebe, S., Klimpe, S., Klopstock, T., Lacour, A., et al. (2008). Screening of Hereditary Spastic Paraplegia Patients for Alterations at NIPA1 Mutational Hotspots. J. Neurol. Sci. 268 (1-2), 131–135. doi:10.1016/j.jns.2007.11.015
Bien-Willner, R., Sambuughin, N., Holley, H., Bodensteiner, J., and Sivakumar, K. (2006). Childhood-Onset Spastic Paraplegia with NIPA1 Gene Mutation. J. Child. Neurol. 21 (11), 974–977. doi:10.1177/08830738060210111501
Chen, S., Song, C., Guo, H., Xu, P., Huang, W., Zhou, Y., et al. (2005). Distinct Novel Mutations Affecting the Same Base in the NIPA1 Gene Cause Autosomal Dominant Hereditary Spastic Paraplegia in Two Chinese Families. Hum. Mutat. 25 (2), 135–141. doi:10.1002/humu.20126
Corrado, L., Brunetti, M., Di Pierro, A., Barberis, M., Croce, R., Bersano, E., et al. (2019). Analysis of the GCG Repeat Length in NIPA1 Gene in C9orf72-Mediated ALS in a Large Italian ALS Cohort. Neurol. Sci. 40 (12), 2537–2540. doi:10.1007/s10072-019-04001-3
D'Amore, A., Tessa, A., Casali, C., Dotti, M. T., Filla, A., Silvestri, G., et al. (2018). Next Generation Molecular Diagnosis of Hereditary Spastic Paraplegias: An Italian Cross-Sectional Study. Front. Neurol. 9, 981. doi:10.3389/fneur.2018.00981
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72 (2), 245–256. doi:10.1016/j.neuron.2011.09.011
Dong, E.-L., Wang, C., Wu, S., Lu, Y.-Q., Lin, X.-H., Su, H.-Z., et al. (2018). Clinical Spectrum and Genetic Landscape for Hereditary Spastic Paraplegias in China. Mol. Neurodegeneration 13 (1), 36. doi:10.1186/s13024-018-0269-1
Du, J., Hu, Y.-c., Tang, B.-s., Chen, C., Luo, Y.-y., Zhan, Z.-x., et al. (2011). Expansion of the Phenotypic Spectrum of SPG6 Caused by Mutation in NIPA1. Clin. Neurol. Neurosurg. 113 (6), 480–482. doi:10.1016/j.clineuro.2011.02.011
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 466 (7310), 1069–1075. doi:10.1038/nature09320
Erfanian Omidvar, M., Torkamandi, S., Rezaei, S., Alipoor, B., Omrani, M. D., Darvish, H., et al. (2021). Genotype-phenotype Associations in Hereditary Spastic Paraplegia: a Systematic Review and Meta-Analysis on 13,570 Patients. J. Neurol. 268 (6), 2065–2082. doi:10.1007/s00415-019-09633-1
Fabbro, D., Mio, C., Fogolari, F., and Damante, G. (2021). A Novel De Novo NIPA1 Missense Mutation Associated to Hereditary Spastic Paraplegia. J. Hum. Genet. 66 (12), 1177–1180. doi:10.1038/s10038-021-00941-x
Goytain, A., Hines, R. M., El-Husseini, A., and Quamme, G. A. (2007). NIPA1(SPG6), the Basis for Autosomal Dominant Form of Hereditary Spastic Paraplegia, Encodes a Functional Mg2+ Transporter. J. Biol. Chem. 282 (11), 8060–8068. doi:10.1074/jbc.M610314200
Harding, A. E. (1983). Classification of the Hereditary Ataxias and Paraplegias. The Lancet 321 (8334), 1151–1155. doi:10.1016/s0140-6736(83)92879-9
Hedera, P. (2013). Recurrent De Novo c.316G>A Mutation in NIPA1 Hotspot. J. Neurol. Sci. 335 (1-2), 231–232. doi:10.1016/j.jns.2013.09.015
Ishiura, H., Takahashi, Y., Hayashi, T., Saito, K., Furuya, H., Watanabe, M., et al. (2014). Molecular Epidemiology and Clinical Spectrum of Hereditary Spastic Paraplegia in the Japanese Population Based on Comprehensive Mutational Analyses. J. Hum. Genet. 59 (3), 163–172. doi:10.1038/jhg.2013.139
Kaneko, S., Kawarai, T., Yip, E., Salehi-Rad, S., Sato, C., Orlacchio, A., et al. (2006). NovelSPG6 Mutation p.A100T in a Japanese Family with Autosomal Dominant Form of Hereditary Spastic Paraplegia. Mov Disord. 21 (9), 1531–1533. doi:10.1002/mds.21005
Kim, A., Kumar, K. R., Davis, R. L., Mallawaarachchi, A. C., Gayevskiy, V., Minoche, A. E., et al. (2019). Increased Diagnostic Yield of Spastic Paraplegia with or without Cerebellar Ataxia through Whole-Genome Sequencing. Cerebellum 18 (4), 781–790. doi:10.1007/s12311-019-01038-0
Klebe, S., Lacour, A., Durr, A., Stojkovic, T., Depienne, C., Forlani, S., et al. (2007). NIPA1 (SPG6) Mutations Are a Rare Cause of Autosomal Dominant Spastic Paraplegia in Europe. Neurogenetics 8 (2), 155–157. doi:10.1007/s10048-006-0074-9
Liu, S. G., Zhao, J. J., Zhuang, M. Y., Li, F. F., Zhang, Q. J., Huang, S. Z., et al. (2008). Clinical and Genetic Study of SPG6 Mutation in a Chinese Family with Hereditary Spastic Paraplegia. J. Neurol. Sci. 266 (1-2), 109–114. doi:10.1016/j.jns.2007.09.024
Lu, C., Li, L.-X., Dong, H.-L., Wei, Q., Liu, Z.-J., Ni, W., et al. (2018). Targeted Next-Generation Sequencing Improves Diagnosis of Hereditary Spastic Paraplegia in Chinese Patients. J. Mol. Med. 96 (7), 701–712. doi:10.1007/s00109-018-1655-4
Martinez-Lage, M., Molina-Porcel, L., Falcone, D., McCluskey, L., Lee, V. M.-Y., Van Deerlin, V. M., et al. (2012). TDP-43 Pathology in a Case of Hereditary Spastic Paraplegia with a NIPA1/SPG6 Mutation. Acta Neuropathol. 124 (2), 285–291. doi:10.1007/s00401-012-0947-y
Munhoz, R. P., Kawarai, T., Teive, H. A., Raskin, S., Sato, C., Liang, Y., et al. (2006). Clinical and Genetic Study of a Brazilian Family with Spastic Paraplegia (SPG6 Locus). Mov Disord. 21 (2), 279–281. doi:10.1002/mds.20775
Rainier, S., Chai, J.-H., Tokarz, D., Nicholls, R. D., and Fink, J. K. (2003). NIPA1 Gene Mutations Cause Autosomal Dominant Hereditary Spastic Paraplegia (SPG6). Am. J. Hum. Genet. 73 (4), 967–971. doi:10.1086/378817
Reed, J. A., Wilkinson, P. A., Patel, H., Simpson, M. A., Chatonnet, A., Robay, D., et al. (2005). A Novel NIPA1 Mutation Associated with a Pure Form of Autosomal Dominant Hereditary Spastic Paraplegia. Neurogenetics 6 (2), 79–84. doi:10.1007/s10048-004-0209-9
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Spagnoli, C., Schiavoni, S., Rizzi, S., Salerno, G. G., Frattini, D., Koskenvuo, J., et al. (2021). SPG6 (NIPA1 Variant): A Report of a Case with Early-Onset Complex Hereditary Spastic Paraplegia and Brief Literature Review. J. Clin. Neurosci. 94, 281–285. doi:10.1016/j.jocn.2021.10.026
Svenstrup, K., Møller, R. S., Christensen, J., Budtz-Jørgensen, E., Gilling, M., and Nielsen, J. E. (2011). NIPA1mutation in Complex Hereditary Spastic Paraplegia with Epilepsy. Eur. J. Neurol. 18 (9), 1197–1199. doi:10.1111/j.1468-1331.2011.03359.x
Tanti, M., Cairns, D., Mirza, N., McCann, E., and Young, C. (2020). Is NIPA1-Associated Hereditary Spastic Paraplegia Always 'pure'? Further Evidence of Motor Neurone Disease and Epilepsy as Rare Manifestations. Neurogenetics 21 (4), 305–308. doi:10.1007/s10048-020-00619-0
Tazelaar, G. H. P., Dekker, A. M., van Vugt, J. J. F. A., van der Spek, R. A., Westeneng, H.-J., Kool, L. J. B. G., et al. (2019). Association of NIPA1 Repeat Expansions with Amyotrophic Lateral Sclerosis in a Large International Cohort. Neurobiol. Aging 74, e9. doi:10.1016/j.neurobiolaging.2018.09.012
Yang, J. O., Yoon, J.-Y., Sung, D. H., Yun, S., Lee, J.-J., Jun, S. Y., et al. (2021). The Emerging Genetic Diversity of Hereditary Spastic Paraplegia in Korean Patients. Genomics 113 (6), 4136–4148. doi:10.1016/j.ygeno.2021.10.014
Zhao, J., Matthies, D. S., Botzolakis, E. J., Macdonald, R. L., Blakely, R. D., and Hedera, P. (2008). Hereditary Spastic Paraplegia-Associated Mutations in the NIPA1 Gene and its Caenorhabditis elegans Homolog Trigger Neural Degeneration In Vitro and In Vivo through a Gain-Of-Function Mechanism. J. Neurosci. 28 (51), 13938–13951. doi:10.1523/JNEUROSCI.4668-08.2008
Keywords: NIPA1, hereditary spastic paraplegia, hotspot mutation, de novo, epilepsy, SPG6
Citation: Fu J, Ma M, Li G and Zhang J (2022) Clinical and Genetic Features of Chinese Patients With NIPA1-Related Hereditary Spastic Paraplegia Type 6. Front. Genet. 13:859688. doi: 10.3389/fgene.2022.859688
Received: 21 January 2022; Accepted: 22 March 2022;
Published: 08 April 2022.
Edited by:
Sadeq Vallian, University of Isfahan, IranReviewed by:
Alessio Di Fonzo, IRCCS Ca' Granda Foundation Maggiore Policlinico Hospital, ItalyCopyright © 2022 Fu, Ma, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiewen Zhang, emhhbmdqaWV3ZW45MEAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.