 Yu Liu1
†Ying Yang1
†
Yu Liu1
†Ying Yang1
†
 Liming Chu
2
Liming Chu
2
 Shuai Ren
2Ying Li2Aimin Gao1Jing Wen1Wanling Deng1Yan Lu1Lingyin Kong2Bo Liang3*
Shuai Ren
2Ying Li2Aimin Gao1Jing Wen1Wanling Deng1Yan Lu1Lingyin Kong2Bo Liang3*
 Xiaoshan Shao
4*
Xiaoshan Shao
4*- 1 Department of Pediatric Endocrinology, Genetics and Metabolism, Guiyang Maternal and Child Health Care Hospital, Guiyang Children’s Hospital, Guiyang, China
- 2 Basecare Medical Device Co., Ltd., Suzhou, China
- 3 State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic and Developmental Sciences, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
- 4 Department of Renal Rheumatology and Immunology, Guiyang Maternal and Child Health Care Hospital, Guiyang Children’s Hospital, Guiyang, China
Interstitial chromosome 20q deletions, containing GNAS imprinted locus, are rarely reported in the past. Hereby, we presented a Chinese boy with a novel 4.36 Mb deletion at paternal 20q13.2-13.32, showing feeding difficulty, malnutrition, short stature, lower limb asymmetry, sightly abnormal facial appearance and mild intellectual abnormality. With 3 years’ growth hormone treatment, his height was increased from 90 to 113.5 cm. This report is the first time to describe the outcome of clinical treatment on a patient with this rare chromosomal 20 long arm interstitial deletion, containing GNAS locus, which may facilitate the diagnosis and treatment of this type of patient in the future.
Introduction
The most commonly observed chromosome 20 abnormality is a ring-shaped chromosome 20, r (20). Those patients with r (20) are characterized by growth retardation, intellectual problems, facial dysmorphism, seizures and specific electroencephalogram pattern (Porfirio et al., 1987). However, interstitial chromosomal deletions around 20q13.2-13.32, containing GNAS locus, are very rare. Till now, only 7 patients from 5 reports were shown to possess this type of deletions. They all shared similar phenotypes of feeding difficulty, growth retardation, abnormal facial characteristics, abnormal subcutaneous fat distribution and intellectual disability (Aldred et al., 2002; Geneviève et al., 2005; Butler et al., 132013; Balasubramanian et al., 2015; Solomon et al., 2011). Here, we detected a unique 4.36 Mb paternal deletion on chromosome 20q13.2-q13.32 in a Chinese boy via next generation sequencing (NGS)-based CNV analysis and SNP array-based haplotyping. He showed common phenotypes compared with the other patients. Remarkably, he had good response to growth hormone (GH) treatment, and his height was increased by 23.5 cm after about 3 years’ treatment.
Materials and Methods
Patient’s Informed Consent and Sample Collection
All genetic tests were based on peripheral blood samples collected from the patient and his parents. Informed consent was obtained from the patient and his parents for this study in accordance with the Helsinki principles for enrollment in research protocols approved by the Ethics Committee of Guiyang Maternal and Child Health Care Hospital, Guiyang Children’s Hospital.
Whole Genome Copy Number Variation Analysis
Genomic DNA (gDNA) was extracted from blood leukocytes of the patient and his parents using TIANamp Blood DNA kit (TIANGEN). CNVs analysis was first performed via an NGS-based low depth whole genome sequencing. The data was then interpreted by TMAP (Version 4.6), Picard (Version 2.18.17), and the method of LOWESS regression and circular binary segmentation were applied to analyze CNV (Xia et al., 2019).
Haplotyping via SNP Array
Illumina Infinium Asian Screening Array-24 v1.0 was used to identify the SNPs in the whole genome and the SNPs were adopted to haplotype linkage analysis according to the instructions in a previous paper (Zhang et al., 2017).
Results
Patient Characteristics and Clinical Observation
The patient in this case was the second child born from healthy Chinese parents, and his elder brother showed normal phenotype. The affected boy experienced intrauterine asphyxia during gestation and was born via caesarean section after full term of gestation. Birth measurements showed he was small for gestational age (SGA) (length = 47 cm; weight = 1.92 kg) and exhibited talipes equinus, so he accepted orthosis at one-year-old, lasting more than 6 months. According to his parents’ description, this patient experienced severe feeding difficulty after he was born. He also suffered from malnutrition, shortened left lower limb and small bilateral testes. Moreover, he had mild learning difficulties and achieved very poor grades in a mainstream class. In terms of his facial features, he had a triangular face, small and pointed chin and sparse-hair eyebrows (Figure 1) He first came to the clinic at 5 years of age due to severe postnatal growth retardation (height = 90 cm, <-3SD; weight = 10 kg). The patient’s pituitary hormone test exhibited increased level of thyroid-stimulating hormone (TSH: 5.85 μIU/ml), reduced but normal level of insulin-like growth factor-1 (IGF1: 3.25 nmol/L) and normal growth hormone (GH) release (peak GH level in L-dopa test = 24.5 ng/ml; peak GH level in Arginine test = 5.12 ng/ml).
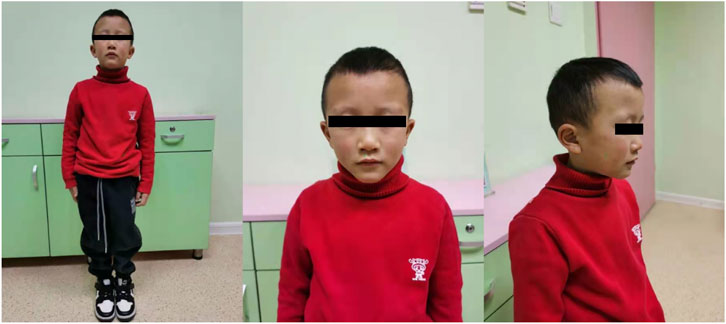
FIGURE 1. Front and side view of the patient. The patient had a triangular face, small and pointed chin and sparse-hair eyebrows, and shortened left lower limb.
Clinical Treatment and Follow up
After the initial attendance to the clinic, GH treatment was given to him for 2 years. Levothyroxine was added as an adjuvant therapy when TSH reached 7.77 μIU/ml after 3 months of GH treatment. His height was increased to 108.5 cm at 7 years old (about 9.25 cm/year, from 5–7 years old), and the TSH level was decreased to 4.98 μIU/ml. However, the patient’s family refused to continue using GH due to financial concerns at that time, so the boy only gained 1.5 cm in the following 6 months. After restoring the GH treatment, his growth rate increased again (3.5 cm/6 months), which means GH indeed promoted the growth of the patient, although he was still much shorter than the average (-3SD) (Figure 2). Thus, the patient were 90 cm (-4.93SD, < 3rd percentile, 5 years old) before GH treatment, 99.7 cm (-3.87SD, < 3rd percentile, 6 years old), 108.5 cm (-3.06SD, < 3rd percentile, 7 years old) and 113.5 cm (-3.03SD, < 3rd percentile, 8 years old) after GH treatment. The IGF1 levels were within normal range during GH treatment (Figure 2B). The patient was malnourished to some degree and we just advised him to increase his nutrition in the diet without special treatment.
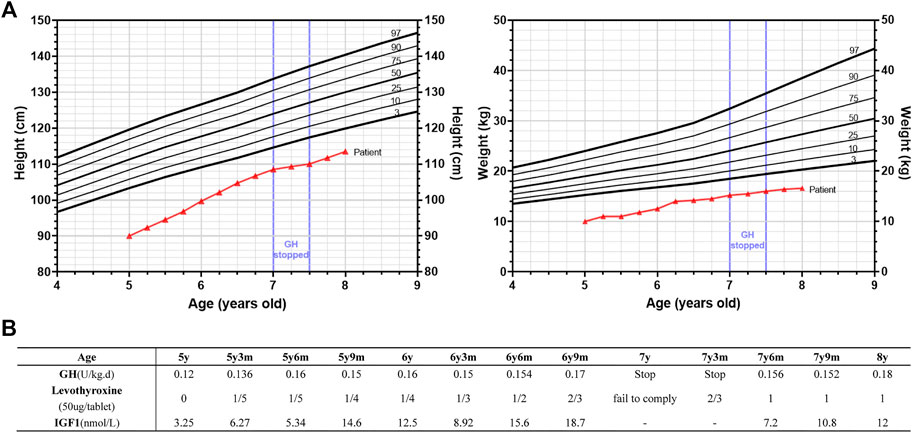
FIGURE 2. Growth of the patient’s height and weight during growth hormone (GH) and adjuvant levothyroxine treatment. (A) Height and weight standard deviation curve of Chinese 4–9 years old boys and the growth trend of the height (left panel) and weight (right panel) of the patient from 5y (years old) to 8y. The growth curve of the patient was indicated in red lines. GH treatment was suspended from 7y to 7y6m (months) as indicated. Different percentiles (3rd, 10th, 25th, 50th, 75th, 90th, and 97th) were as indicated. (B) The dosage of GH and levothyroxine used, and the corresponding IGF1 levels during this period. GH treatment was suspended from 7y to 7y6m (months) and levothyroxine was added as an adjuvant therapy when TSH reached 7.77 μIU/ml after 3 months of GH treatment.
Genetic Analysis in the Patient and his Parents
In contrast to the parents, who were healthy and had normal height (father’s height = 175 cm; mother’s height = 160 cm), the patient himself showed short stature, intellectual abnormality and subclinical hypothyroidism (Figure 3A). There was also no consanguinity between the parents. We hypothesized the patient may suffer from an autosomal recessive disease that was inherited from his parents, or an autosomal dominant disease that resulted from a de novo mutation. Thus, whole exome sequencing (WES) and whole genome copy number variation (CNV) analysis were carried out in this patient and his parents. WES did not reveal any relevant pathogenic variants in the family, while CNV analysis showed the presence of 46,XY,del (20) (q13.2-q13.32) in the patient’s genome (Supplementary Figure S1A). This deletion was about 4.36 Mb in size. However, the parents’ genomes were shown to be intact. Further comparison between the proband and his parents’ haplotypes confirmed his paternal copy of chromosome 20q13.2-q13.32 was lost (Figures 3B,C). Since the patient’s brother and both parents showed normal phenotype, and the parents both showed no such deletion in chromosome 20 (Supplementary Figure S1B), so this rare deletion was implied to be a de novo mutation.
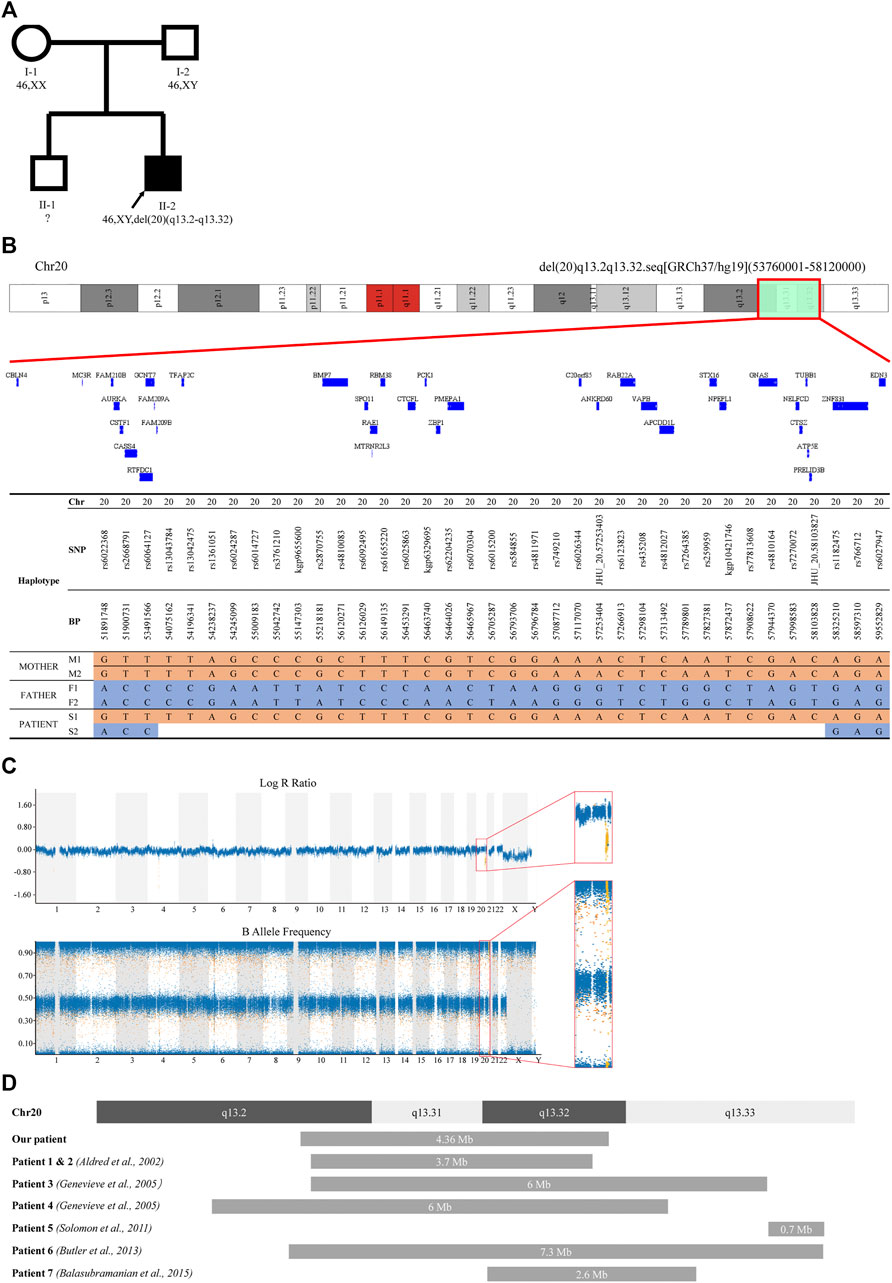
FIGURE 3. Pedigree and CNV analysis of the family. (A) Familial pedigree showing the phenotype and genotype of the family. Proband (II-2) had a heterozygous deletion of chromosome 20q13.2-q13.32. The genotype of proband’s elder brother (II-1) was not assessed. (B) A schematic structure of the deleted regions detected in the patient (II-2) and comparison with his parents (I-1 and I-2). Only SNP positions at which both parents were homozygous for opposite alleles were showed. (C) B allele frequency plot and log R ratio plot of the patient’s SNP array result. (D) A schematic diagram of reported deletions around 20q13.32 compared with the deletion identified in this study.
Discussion
In this case report, we presented a Chinese boy showing learning difficulties, growth retardation and endocrine abnormalities, with a rare de novo 4.36 Mb deletion on paternal 20q13.2-q13.32. Via exploring the online database (http://grch37.ensembl.org/index.html), this region was found consisting of 35 protein coding genes and 27 non-coding genes. Among them, there are 9 OMIM genes (MC3R, AURKA, PCK1, VAPB, TUBB1, STX16, ATP5E, GNAS and EDN3). Noteworthy, GNAS is a haploinsufficient gene locus highly associated with his phenotype (Turan and Bastepe, 2013).
GNAS is an important imprinted locus that is responsible for the production of multiple transcripts: paternally expressed GNAS-A/B, GNAS-AS1, GNAS-XLαs and maternally expressed NESP55, while Gαs is biallelically expressed in most tissues, with maternal expression in some specific tissues (eg. neonatal brown tissue, gonads and thyroids) (Bastepe, 2007; Turan and Bastepe, 2015). Gsα is the most well-studied transcript expressed from GNAS locus, encoding the alpha subunit of G-protein involving in guanine nucleotide-binding. G-protein can trigger complex signaling networks via GDP-GTP exchange. The activated form of GTP-bound Gsα can stimulate different effectors, such as adenylyl cyclase, and thereby initiate various cellular responses, including in endocrine glands (Cabrera-Vera et al., 2003; Bastepe and Jüppner, 2005; Turan and Bastepe, 2015). For example, adenylyl cyclase facilitates the production of cAMP, which can bind to the protein kinase A (PKA) and further lead to the activation of its catalytic subunit. Catalytic-active PKA is involved in the iodine uptake and the expression of genes associated with thyroid hormone production (Pereira et al., 2020). As a result, mutation of GNAS can cause different types of endocrine-related diseases, such as McCune-Albright syndrome (OMIM#174800), progressive osseous heteroplasia (OMIM#166350) and pseudohypoparathyroidism (PHP).
It is worth to note that different forms of PHP can be triggered by heterozygous mutation of GNAS, such as PHP-Ia, PHP-1b, and pseudopseudohypoparathyroidism (PPHP) (Mantovani et al., 2020). PHP-Ia and PPHP are caused by inactivating mutations in Gs-a, while PHP-1b is due to an abnormal methylation on GNAS imprinted regions (Turan and Bastepe, 2015; Mantovani et al., 2020). PHP-Ia patients are characterized by short stature, subcutaneous ossification, brachydactyly and variable degrees of intellectual disability, so called Albright’s Hereditary Osteodystrophy (AHO), as well as parathyroid hormone (PTH) resistance. It may also show resistance to other hormones, such as growth hormone releasing hormone (GHRH) and TSH (Aldred et al., 2000; Weinstein et al., 2001; Levine, 2002). PPHP patients show most AHO phenotypes, but not presenting hormone resistance (Aldred et al., 2000; Mantovani et al., 2020). PPHP often develops when the paternal Gs-a is deficient, while PHP-Ia develops when the maternal one is inactivated (Davies and Hughes, 1993). The less severe phenotype of PPHP may be due to the tissue specific paternal imprint of Gs-a. In our case, the phenotype and genotype of our patient is more consistent with the PPHP. However, deletion of other genes may also play roles in the resulting phenotypes.
To date, there were only 7 patients reported to carry this rare deletion around 20q13.32 containing GNAS imprinted locus (Figure 3D), and 4 of them were confirmed to have paternal deletion (Table 1). Almost all of those affected by 20q13.32 deletion containing GNAS showed reduced birth weight, abnormal facial appearance, feeding difficulties, intellectual disabilities and growth retardation (Aldred et al., 2002; Geneviève et al., 2005; Butler et al., 132013; Balasubramanian et al., 2015; Solomon et al., 2011). The two cases reported by Aldred et al. showed obesity, and those reported by Genevieve et al. had uneven subcutaneous fat distribution, while the patient reported by Balasubramanian et al. had reduced adipose tissue (Aldred et al., 2002; Geneviève et al., 2005; Balasubramanian et al., 2015). Compared with those previous reports, the patient in our case showed similar triangular face, sparse-hair eyebrows, low birth weight, feeding problems, reduced adipose tissue, severe growth delay and intellectual abnormalities. However, when compared with UPD(20)mat and Silver Russell Syndrome (SRS)-like phenotypes, the clinical manifestations in our patient (paternal 20q13.2-q13.32 deletion) was a little bit different. As previously reported, patients with UPD(20)mat generally have phenotypic features such as growth retardation, including intrauterine growth retardation (IUGR), feeding difficulties, about 1/5 patients shared phenotypes like: hypotonia, dysmorphic features, developmental delay (DD) and Silver-Russel-syndrome like phenotypes [http://cs-tl.de/DB/CA/UPD/0-Start.html (accessed 02/25/2022)]. And the clinical diagnosis of SRS was based on six features: pre- and postnatal growth failure, relative macrocephaly, prominent forehead, body asymmetry, and feeding difficulties (Netchine–Harbison clinical scoring system (NH-CSS)) (Tannorella et al., 2021). So our patient shared growth retardation and feeding difficulties with UPD(20)mat patients, and shortened left lower limb fits the phenotype of Silver Russell Syndrome, 3/6 met the NH-CSS criteria. Accordingly, phenotypes of this 20q deletions were different from that of UPD(20)mat and SRS, mainly in the triangular face, sparse-hair eyebrows, reduced adipose tissue, and intellectual abnormalities. In mice and humans, loss of function of XLαs was associated with intrauterine and perinatal growth retardation, as well as poor adaption to feeding (He et al., 2015), which was similar to our patient. But XLαs deficiency also caused hypoglycemia and disrupted glucose counter-regulation which did not occur in our patient (He et al., 2015). Considering that the paternal GNAS allele was deleted, it was also predicted that the patient might have some clinical features of Albright hereditary osteodystrophy (AHO). In our case, The patient had part of the characteristics of AHO, such as: short stature, brachydactyly, he also had limb asymmetry, but he did not have other characteristics of AHO such as obesity, round face and subcutaneous ossification.
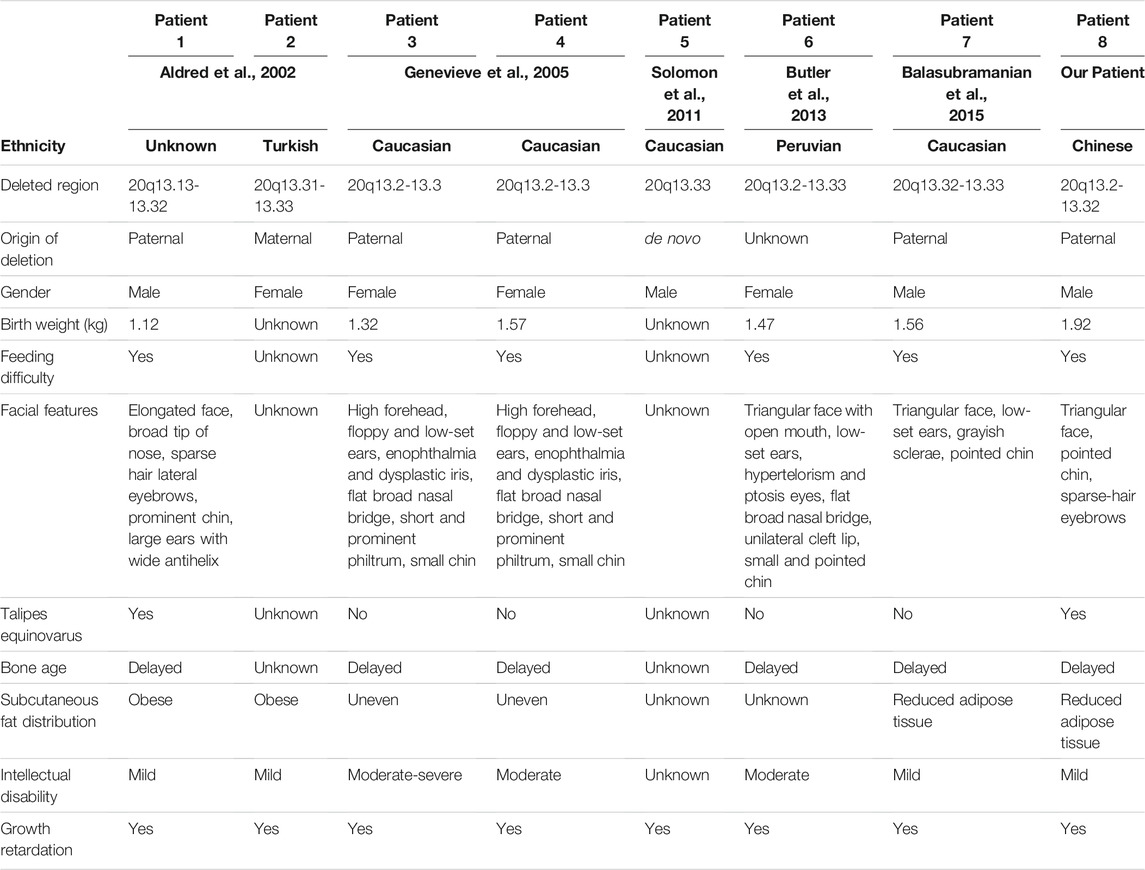
TABLE 1. Clinical features of patients with deletion around 20q13.32 containing GNAS locus.
The dosage of GH was based on the guideline recommendations for the clinical use of recombinant human growth hormone in Chinese children: childhood GHD: 0.075- 0.15 U/(kg d), ISS: 0.125-0.2 U/(kg d) (Subspecialty Group of End, 2013). The amount of GH used was in this range (Figure 2B). Considering the initial age (5 years) and bone age of the child, and the space for subsequent height gain is still relatively large, our initial GH dose is slightly smaller (0.12 U/(kg d) at 5 years old), and then gradually increased to 0.18 U/(kg d) (at 8 years old) because the growth rate is not very satisfactory. A recent study showed that, after 1 year of GH treatment, HtSDS (Height standard deviation score) increased in 13 ISS children with normal IGF1 level (start at -2.06, end at -1.76) and 17 GHD children with normal IGF1 level (start at -2.23, end at -1.87) (Soliman et al., 2021). GH therapy was relatively effective in SRS patients. Meropi Toumba et al. repoted that 26 SRS patients received GH treatment for at least 7 years [median duration of treatment was 9.8 years (range 7.0-15.7)], and the median height of the 26 patients at the commencement of treatment increased significantly by the end of treatment (height SDS: start at -2.74, end at -1.33) (Toumba et al., 2010). As for GH treatment in PPHP patient, R Manfredi et al. repoted a female PPHP patient, who received a 3-yr-6 months GH treatment. As a result, a significant improvement of growth velocity was obtained, while the height SDS failed to improve (-3.30 pretreatment (10 years old); -3.14(11 years old), -3.36(12 years old) and -3.59 (13 years old) after the first, the second and the third year of treatment, respectively) (Manfredi et al., 1993). As for our patient, although he failed to return to normal height with GH treatment, his height SD changed more in the first 2 years (5–7 years old) than that in those reports (Figure 2A).
Although GH treatment did not restore the child’s height to normal, the increase of about 9.25 cm/year in height during GH treatment (GH treatment data from 5–7 years old) was higher than that of normal Chinese male children by 6.35 cm/year (5–7 years old) (Li et al., 2009), so GH treatment was beneficial for this child. If GH treatment was not taken at that time, the child’s height would be shorter, just as he suspended GH treatment between 7–7.5 years old, and the height increased by only 1.5 cm in half a year. The IGF1 level of 3.25 nmol/L (at initial diagnosis) and the change in IGF1 levels during GH therapy afterwards (Figure 2B), although low, were within the normal range (4–6 years: 2.86-27.04 nmol/L; 7–9 years: 5.2-33.15 nmol/L) (Xu et al., 2010).
Moreover, a genetic testing method combining NGS-based CNV analysis and SNP array-based haplotyping could become a powerful tool to precisely identify the location of a chromosomal deletion and trace its parent of origin. There are also some limitations to our research, due to the limitation of sample collection, the relationship between methylation status in this region and phenotype needs further study in the future.
In conclusion, here we presented a Chinese patient with a novel interstitial deletion around 20q13.2-13.32, containing GNAS locus. He exhibited high concordance with previously reported phenotypes. However, none of the previous reports presented any treatment information on the patients. Therefore, this case report was the first time to provide clinical treatment data on a patient with such rare interstitial deletion on chromosome 20, which shows GH treatment did have a positive effect on his growth.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Guiyang Maternal and Child Health Care Hospital, Guiyang Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YL, YY, AG, JW, WD, and YL collected clinical data and blood samples and performed DNA extraction. LC, SR, and YL conducted CNV analysis and SNP array. YL, YY, and LC collected the literature and wrote the manuscript. LK, BL, and XS were responsible for the study design and guiding of the study implementation and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
LC, SR, YL, and LK were employed by Basecare Medical Device Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patient and his family members for participating in this study. We also acknowledge Huihan Zhi, for her suggestions in manuscript writing and data collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.859185/full#supplementary-material
References
Aldred, M. A., Aftimos, S., Hall, C., Waters, K. S., Thakker, R. V., Trembath, R. C., et al. (2002). Constitutional Deletion of Chromosome 20q in Two Patients Affected with Albright Hereditary Osteodystrophy. Am. J. Med. Genet. 113 (2), 167–172. doi:10.1002/ajmg.10751
Aldred, M. A., Bagshaw, R. J., Macdermot, K., Casson, D., Murch, S. H., Walker-Smith, J. A., et al. (2000). Germline Mosaicism for a GNAS1 Mutation and Albright Hereditary Osteodystrophy. J. Med. Genet. 37 (11), 35e–35. doi:10.1136/jmg.37.11.e35
Balasubramanian, M., Atack, E., Smith, K., and Parker, M. J. (2015). A Novel De Novo 20q13.32-q13.33 Deletion in a 2-Year-Old Child with Poor Growth, Feeding Difficulties and Low Bone Mass. J. Hum. Genet. 60 (6), 313–317. doi:10.1038/jhg.2015.22
Bastepe, M., and Jüppner, H. (2005). GNAS Locus and Pseudohypoparathyroidism. Horm. Res. Paediatr. 63 (2), 65–74. doi:10.1159/000083895
Bastepe, M. (2007). The GNAS Locus: Quintessential Complex Gene Encoding Gsα, XLαs, and Other Imprinted Transcripts. Cg 8 (6), 398–414. doi:10.2174/138920207783406488
Butler, M. G., Usrey, K. M., Roberts, J. L., Manzardo, A. M., and Schroeder, S. R. (1320). 20q13.2-q13.33 Deletion Syndrome: A Case Report. J. Pediatr. Genet. 2 (3), 157–161. doi:10.3233/PGE-13065
Cabrera-Vera, T. M., Vanhauwe, J., Thomas, T. O., Medkova, M., Preininger, A., Mazzoni, M. R., et al. (2003). Insights into G Protein Structure, Function, and Regulation. Endocr. Rev. 24 (6), 765–781. doi:10.1210/er.2000-0026
Davies, S. J., and Hughes, H. E. (1993). Imprinting in Albright's Hereditary Osteodystrophy. J. Med. Genet. 30 (2), 101–103. doi:10.1136/jmg.30.2.101
Geneviève, D., Sanlaville, D., Faivre, L., Kottler, M.-L., Jambou, M., Gosset, P., et al. (2005). Paternal Deletion of the GNAS Imprinted Locus (Including Gnasxl) in Two Girls Presenting with Severe Pre- and post-natal Growth Retardation and Intractable Feeding Difficulties. Eur. J. Hum. Genet. 13 (9), 1033–1039. doi:10.1038/sj.ejhg.5201448
He, Q., Zhu, Y., Corbin, B. A., Plagge, A., and Bastepe, M. (2015). The G Protein α Subunit Variant XLα S Promotes Inositol 1,4,5-trisphosphate Signaling and Mediates the Renal Actions of Parathyroid Hormone In Vivo. Sci. Signal. 8 (391), ra84. doi:10.1126/scisignal.aaa9953
Levine, M. (2002). Pseudohypoparathyroidism. Principles bone Biol. 1, 1137–1163. doi:10.1016/b978-012098652-1/50166-9
Li, H., Ji, C. Y., Zong, X. N., and Zhang, Y. Q. (2009). [Height and Weight Standardized Growth Charts for Chinese Children and Adolescents Aged 0 to 18 Years]. Zhonghua Er Ke Za Zhi 47 (7), 487–492.
Manfredi, R., Zucchini, A., Azzaroli, L., and Manfredi, G. (1993). Pseudopseudohypoparathyroidism Associated with Idiopathic Growth Hormone Deficiency. Role of Treatment with Biosynthetic Growth Hormone. J. Endocrinol. Invest. 16 (9), 709–713. doi:10.1007/BF03348916
Mantovani, G., Bastepe, M., Monk, D., de Sanctis, L., Thiele, S., Ahmed, S. F., et al. (2020). Recommendations for Diagnosis and Treatment of Pseudohypoparathyroidism and Related Disorders: An Updated Practical Tool for Physicians and Patients. Horm. Res. Paediatr. 93 (3), 182–196. doi:10.1159/000508985
Pereira, S. S., Lobato, C. B., and Monteiro, M. P. (2020). Cell Signaling within Endocrine Glands: Thyroid, Parathyroids and Adrenal Glands. Tissue-Specific Cell Signaling 1, 63–91. doi:10.1007/978-3-030-44436-5_3
Porfirio, B., Valorani, M. G., Giannotti, A., Sabetta, G., and Dallapiccola, B. (1987). Ring 20 Chromosome Phenotype. J. Med. Genet. 24 (6), 375–377. doi:10.1136/jmg.24.6.375
Soliman, A., Rogol, A. D., Elsiddig, S., Khalil, A., Alaaraj, N., Alyafie, F., et al. (2021). Growth Response to Growth Hormone (GH) Treatment in Children with GH Deficiency (GHD) and Those with Idiopathic Short Stature (ISS) Based on Their Pretreatment Insulin-like Growth Factor 1 (IGFI) Levels and at Diagnosis and IGFI Increment on Treatment. J. Pediatr. Endocrinol. Metab. 34 (10), 1263–1271. doi:10.1515/jpem-2021-0389
Solomon, B. D., Pineda-Alvarez, D. E., Hadley, D. W., Keaton, A. A., Agochukwu, N. B., Raam, M. S., et al. (2011). De Novo deletion of Chromosome 20q13.33 in a Patient with Tracheo-Esophageal Fistula, Cardiac Defects and Genitourinary Anomalies Implicates GTPBP5 as a Candidate Gene. Birth Defects Res. A: Clin. Mol. Teratology 91 (9), 862–865. doi:10.1002/bdra.20821
Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, the Society of Pediatrics (2013). [Recommendations for the Clinical Use of Recombinant Human Growth Hormone in Children]. Zhonghua Er Ke Za Zhi 51 (6), 426–432.
Tannorella, P., Minervino, D., Guzzetti, S., Vimercati, A., Calzari, L., Patti, G., et al. (2021). Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases. Genes 12 (4), 588. doi:10.3390/genes12040588
Toumba, M., Albanese, A., Azcona, C., and Stanhope, R. (2010). Effect of Long-Term Growth Hormone Treatment on Final Height of Children with Russell-Silver Syndrome. Horm. Res. Paediatr. 74 (3), 212–217. doi:10.1159/000295924
Turan, S., and Bastepe, M. (2015). GNAS Spectrum of Disorders. Curr. Osteoporos. Rep. 13 (3), 146–158. doi:10.1007/s11914-015-0268-x
Turan, S., and Bastepe, M. (2013). TheGNASComplex Locus and Human Diseases Associated with Loss-Of-Function Mutations or Epimutations within This Imprinted Gene. Horm. Res. Paediatr. 80 (4), 229–241. doi:10.1159/000355384
Weinstein, L. S., Yu, S., Warner, D. R., and Liu, J. (2001). Endocrine Manifestations of Stimulatory G Protein α-Subunit Mutations and the Role of Genomic Imprinting. Endocr. Rev. 22 (5), 675–705. doi:10.1210/edrv.22.5.0439
Xia, C.-L., Lyu, Y., Li, C., Li, H., Zhang, Z.-T., Yin, S.-W., et al. (2019). Rare De Novo IGF2 Variant on the Paternal Allele in a Patient with Silver-Russell Syndrome. Front. Genet. 10, 1161. doi:10.3389/fgene.2019.01161
Xu, S., Gu, X., Pan, H., Zhu, H., Gong, F., Li, Y., et al. (2010). Reference Ranges for Serum IGF-1 and IGFBP-3 Levels in Chinese Children during Childhood and Adolescence. Endocr. J. 57 (3), 221–228. doi:10.1507/endocrj.k09e-200
Keywords: short stature, growth retardation, 20q13.2-13.32, growth hormone, gnas
Citation: Liu Y, Yang Y, Chu L, Ren S, Li Y, Gao A, Wen J, Deng W, Lu Y, Kong L, Liang B and Shao X (2022) Case Report: A Paternal 20q13.2-q13.32 Deletion Patient With Growth Retardation Improved by Growth Hormone. Front. Genet. 13:859185. doi: 10.3389/fgene.2022.859185
Received: 21 January 2022; Accepted: 09 March 2022;
Published: 24 March 2022.
Edited by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyReviewed by:
Andrea Riccio, University of Campania Luigi Vanvitelli, ItalyMurat Bastepe, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2022 Liu, Yang, Chu, Ren, Li, Gao, Wen, Deng, Lu, Kong, Liang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Liang, Ym9saWFuZzg4MEBzanR1LmVkdS5jbg==; Xiaoshan Shao, c3hzMjJAcXEuY29t
† These authors have contributed equally to this work and share first authorship