 Xin Pan
Xin Pan Sihan Liu
Sihan Liu Li Liu1
Li Liu1 Hong Yao
Hong Yao Bo Tan
Bo Tan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 29 March 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.853028
This article is part of the Research TopicThe Genetics of Neurodevelopmental DisordersView all 11 articles
Au-Kline syndrome is a severe multisystemic syndrome characterized by several congenital defects, including intellectual disability. Loss-of-function and missense variants in the HNRNPK gene are associated with a range of dysmorphic features. This report describes an eleven-year-old Chinese boy with intellectual disability and developmental delays. Family-based whole-exome and Sanger sequencing identified a de novo missense variant in HNRNPK (NM_002140.3: c.143T > A, p. Leu48Val). In silico analysis predicted that this variant would be damaged in a highly conserved residue in the K homology 1 (KH1) domain. Bioinformatic analysis showed that the affinity change (ΔΔG) caused by this variant was -0.033 kcal/mol, indicating that it would have reduced affinity for RNA binding. Transcript analysis of the peripheral blood from this case found 42 aberrantly expressed and 86 aberrantly spliced genes (p-value <0.01). Functional enrichment analysis confirmed that the biological functions of these genes, including protein binding and transcriptional regulation, are associated with HNRNPK. In summary, this study identifies the first Chinese patient with a novel de novo heterozygous HNRNPK gene variant that contributes to Au-Kline syndrome and expands current knowledge of the clinical spectrum of HNRNPK variants.
Au-Kline syndrome (AKS) was first described in 2015 in two unrelated boys who presented a wide spectrum of abnormalities, including atypical facial features, developmental delays, and hypotonia with intellectual disability. AKS-associated facial features include long faces, ptosis, cleft palate, and oligodontia. Genetic alterations of the heterogeneous nuclear ribonucleoprotein K (HNRNPK) gene are responsible for the development of AKS.
HNRNPK is a member of the RNA-binding protein family and is involved in both physiological and pathological processes, including spermatogenesis, nervous system and ovary development, erythroid differentiation, organogenesis, and carcinogenesis (Barboro et al., 2014; Gallardo et al., 2016; Geuens et al., 2016). HNRNPK contains three repeat K homology domains, KH1, KH2, and KH3, which recognize target RNAs and play a central role in regulating gene expression, chromatin structure, and other genetic functions. To date, the genotypes and detailed clinical features of over 30 AKS patients have been well-characterized (Dentici et al., 2018; Gillentine et al., 2021). However, there are no reports of Chinese cases of AKS. In addition, more cases are needed to better understand the relationship between AKS and its associated pathogenic variants.
This study describes the clinical and molecular characteristics of the first Chinese AKS patient who had a novel de novo missense variant of HNRNPK (NM_002140.3: c.143T > A) and expands the current understanding of the genotypic spectrum of AKS.
The proband, a boy 11 years and 10 months of age, was the first child of nonconsanguineous Chinese parents. Prenatal ultrasounds were normal. The proband failed to raise his head and exhibited hypotonia at 6 months of age and a language delay with his first words spoken at 3 years of age. He had a moderate degree of intellectual disability. Dysmorphic features included a long face, long palpebral fissures, ptosis, and hypoplastic alar nasi (Figure 1A).
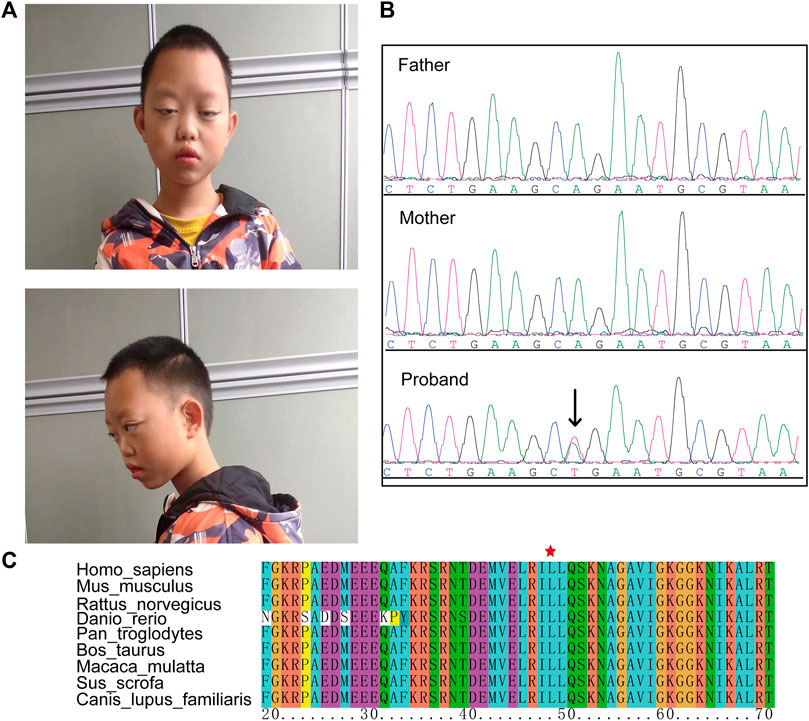
FIGURE 1. The clinical phenotype of the case. (A) Dysmorphic features including a long face, long palpebral fissures, ptosis, and hypoplastic alar nasi. (B) Sanger sequencing confirmed a de novo HNRNPK variant (NM_002140.3: c.143T > A) in the proband. (C) This variant caused an amino acid change (p. L48G) that in a highly conserved region.
An ultrasound confirmed mild hydronephrosis and cryptorchidism at 11 years of age (Supplementary Figure S1), and oligodontia was determined by panoramic radiographs. The proband’s hearing was normal, and no significant abnormalities were found by brain magnetic resonance imaging (MRI). Chromosomal karyotype and microarray analysis results were normal.
Informed consent was obtained from the patient’s parents. This study was approved by the ethics committee from the Second Affiliated Hospital of Chongqing Medical University.
Genomic DNA was isolated from each participant’s peripheral blood using a blood genomic DNA extraction kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s protocol. The gDNA was fragmented, captured, and sequenced using the MGI-2000 sequencing system (BGI China).
Raw sequencing reads were filtered to obtain clean reads using Fastp,(Chen et al., 2018) and FastQC (Trivedi et al., 2014) was used to evaluate the quality of sequencing data in each sample. Clean DNA sequencing reads were mapped to the human reference genome hg19 (GRCh37) using the BWA-MEM algorithm (Li and Durbin, 2009), and ambiguously mapped reads (MAPQ <10) and duplicated reads were removed using SAMtools (Li et al., 2009) and PicardTools [http://broadinstitute.github.io/picard/], respectively. SNPs and small insertions and deletions (INDEL) were identified according to the Genome Analysis Toolkit software’s best practices and variants were annotated using the Ensembl Variant Effect Predictor (VEP) (McLaren et al., 2016). According to guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG) (Richards et al., 2015), variants were classified as pathogenic (P), likely pathogenic (LP), benign (B), likely benign (LB), or variants of uncertain significance (VUS). Variant validation was performed using Sanger sequencing (ABI 3730xl Genetic Analyzer).
Total RNA was isolated from peripheral blood and enriched by oligo-dT bead capture and cDNA was synthesized according to the manufacturer’s protocol. cDNA libraries were constructed using the Illumina trueSeq stranded mRNA sample prep kit protocol (Illumina). Pooled samples were sequenced using a NovaSeq 6000 sequencing system.
Raw sequencing reads were filtered to obtain clean reads using Fastp, and FastQC was used to evaluate the quality of sequencing data based on several measures, including sequence quality per base, sequence duplication level, and quality score distribution for each sample. The average quality score for overall RNA sequences was >30, indicating that a large percentage of the sequences were high quality. The clean RNA-sequencing reads were mapped to the human reference genome (hg19) using STAR (2.4.2a) with the Gencode v19 annotation (Dobin et al., 2013).
Aberrant gene expression, splicing, and monoallelic expression were detected using DROP (Yepez et al., 2021) with the default filter parameters. To increase the power to detect aberrantly expressed genes, in-house data with the same sequencing and analysis pipeline was included. Genes were defined as having aberrant expression, splicing, or monoallelic expression with a p-value <0.01. Functional enrichment of the aberrant genes was performed with KOBAS-i, a service that provides comprehensive pathway enrichment analysis using several databases, including GO, KEGG, Reactome, and GWAS catalogs (Bu et al., 2021). An adjusted p-value <0.05 was selected as the threshold for significant pathways.
After trio whole-exome sequencing (Trio-WES) was performed on each family member, the causal variants were evaluated using ACMG guidelines. Results identified a novel missense HNRNPK variant (NM_002140.3: c.143T > A) in the patient that was absent in the parents (Figure 1B). This variant was classified as LP with the following evidence (PS2_Moderate + PM2 +PP3). This variant was not reported in the dbSNP and gnomAD databases, and predicted as pathogenic using in silico prediction tools (SIFT = 0.001, Polyphen2 = 0.99, MutationTaster = 1, and EVE = 0.945) (Frazer et al., 2021). The HNRNPK variant caused a missense substitution (p. Leu48Gln) localized in the K homology domains of a highly conserved region, suggesting that the variant may disrupt the binding ability of the HNRNPK protein (Figure 1C).
To prove this hypothesis, we predicted the effect of this missense variant on protein affinity with mCSM-NA (Pires and Ascher, 2017) using the PDB (Protein Data Bank) file provided by AlphaFold. The predicted affinity change (ΔΔG) was -0.033 kcal/mol, indicating that HNRNPK had reduced affinity for RNA binding. In addition, the predicted stability effect of HNRNPK was -3.541 kcal/mol, indicating that the protein was destabilized. These results suggest that the de novo variants (NM_002140.3:c.143T > A) may lead to loss of function of HNRNPK.
RNA sequencing was also performed and 42 and 86 genes with aberrant expression and splicing were identified, respectively (Supplementary Figures S2–S5, Supplementary Tables S1, S2). While the HNRNPK gene was not differentially expressed, two target genes regulated by HNRNPK, TUBB2A (p = 0.0093) and TUBB2B (p = 0.0092), were differentially expressed. Pathway enrichment analysis showed that the biological functions of these genes correlated with protein binding, transcriptional regulation, and nervous system regulation (Figure 2; Supplementary Tables S3, S4).
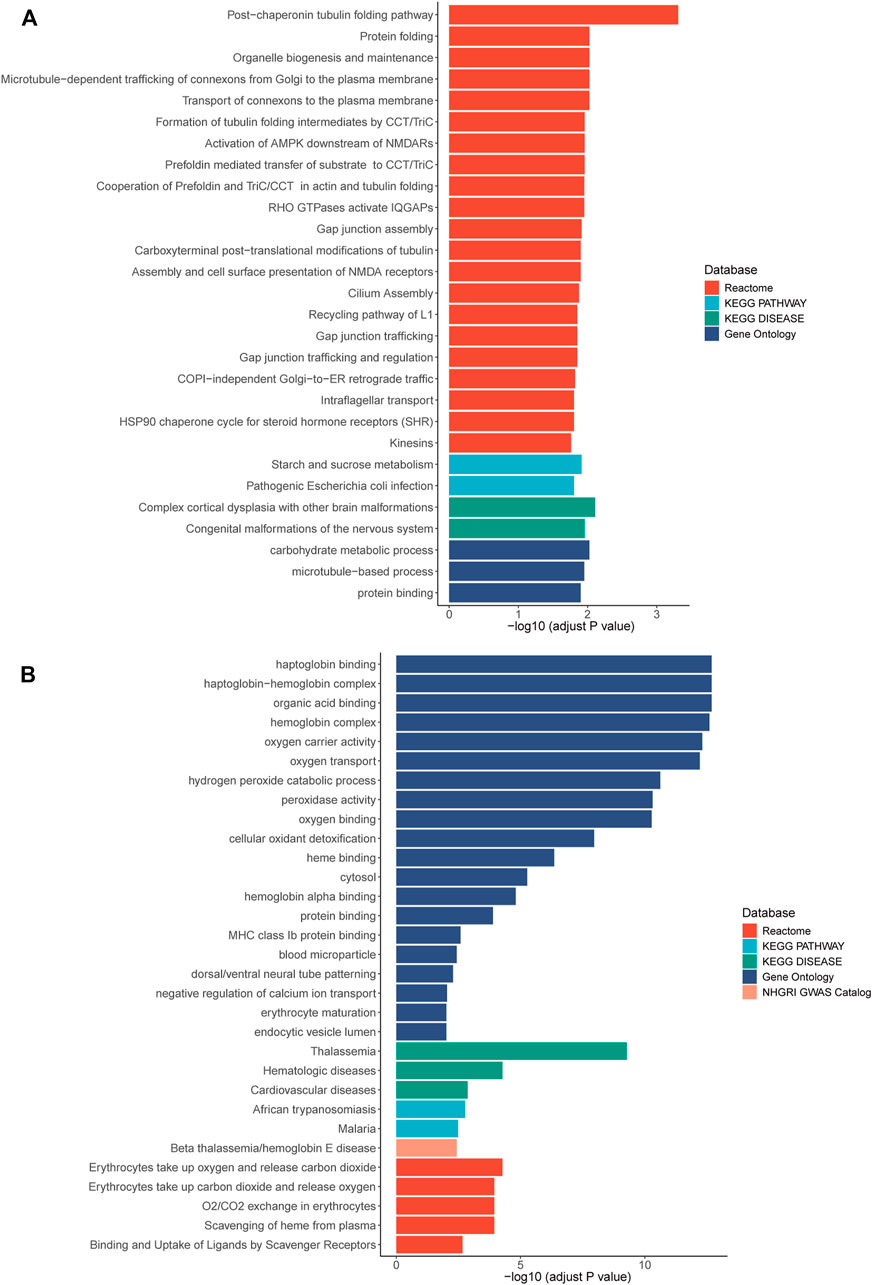
FIGURE 2. Pathway enrichment results of genes with aberrant expression (A) and with aberrant splicing (B). Colors stand for pathways items from different database. The X axis shows adjusted p-value with log transformed. Pathways with an adjusted p-value <0.05 was selected as significant pathways and plotted.
In this study, we reported a patient with AKS who had multiple system anomalies, including developmental delay, facial dysmorphism, and kidney and genital abnormalities. Exome sequencing and Sanger validation showed that these phenotypes may be explained by a novel de novo missense variant of the HNRNPK gene (NM_002140.3: c.143T > A). In addition, there is a missense variant (NM_002140.3:c.142C > G; p. Leu48Val) interpreted as uncertain significance in ClinVar database which affect the same site with different amino acid.
With the addition of our patient, a total of 33 pathogenic HNRNPK variants (21 LOF and 12 missense) that caused AKS have been reported in 35 patients (Table 1) (Miyake et al., 2017; Au et al., 2018; Gillentine et al., 2021). Three-quarters (75%) of the missense variants occurred within the KH domain (42% in the KH1 domain). In contrast, most LOF variants was in the interdomain. The most common phenotype in patients with AKS includes intellectual disability (85%), developmental delay (72%), genitourinary abnormalities (66%), and hand and foot abnormalities (63%). Phenotypic differences between individuals with loss-of-function and missense variants were observed in ptosis, wide nasal bridge, brain imaging abnormalities, cardiac defects, and skeletal and gastrointestinal abnormalities. The patient reported here had a unique phenotype of craniofacial features and oligodontia (Figure 3; Supplementary Table S5).
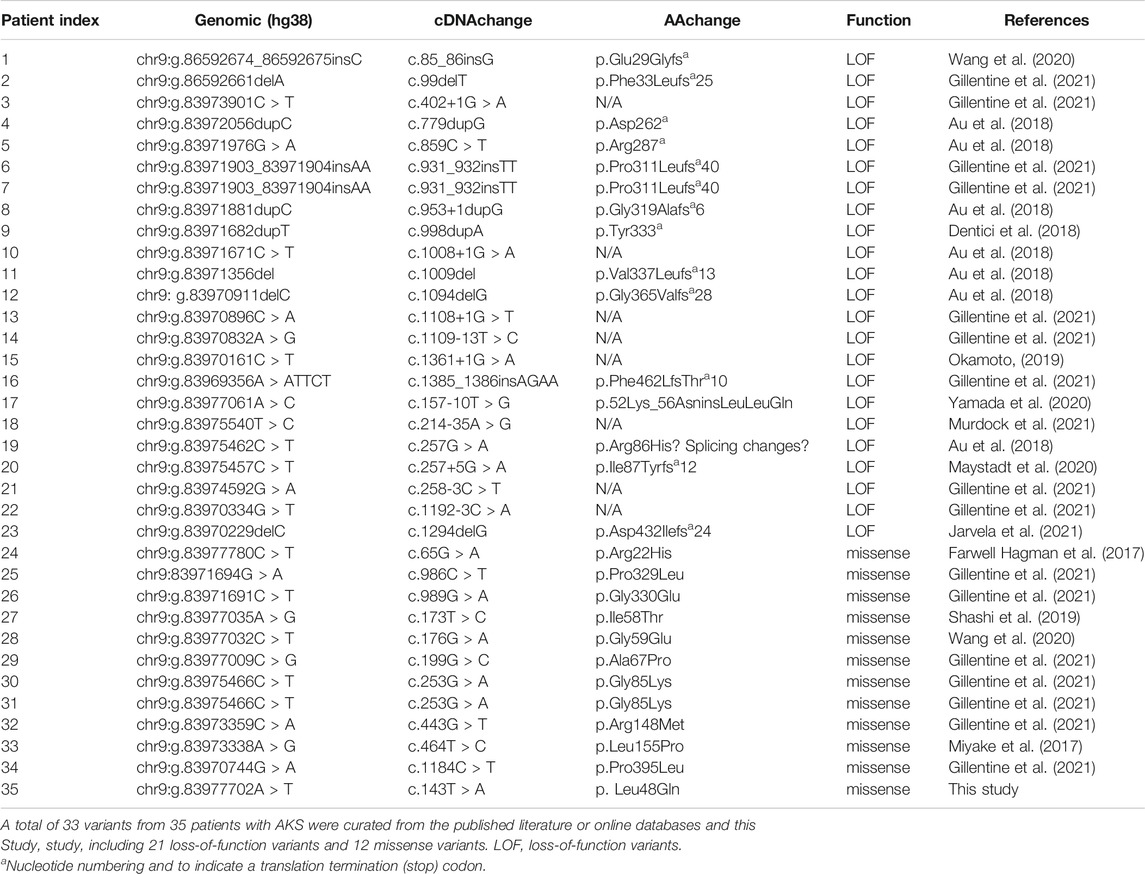
TABLE 1. Pathogenic variants identified in HNRNPK gene (NM_002140.3).
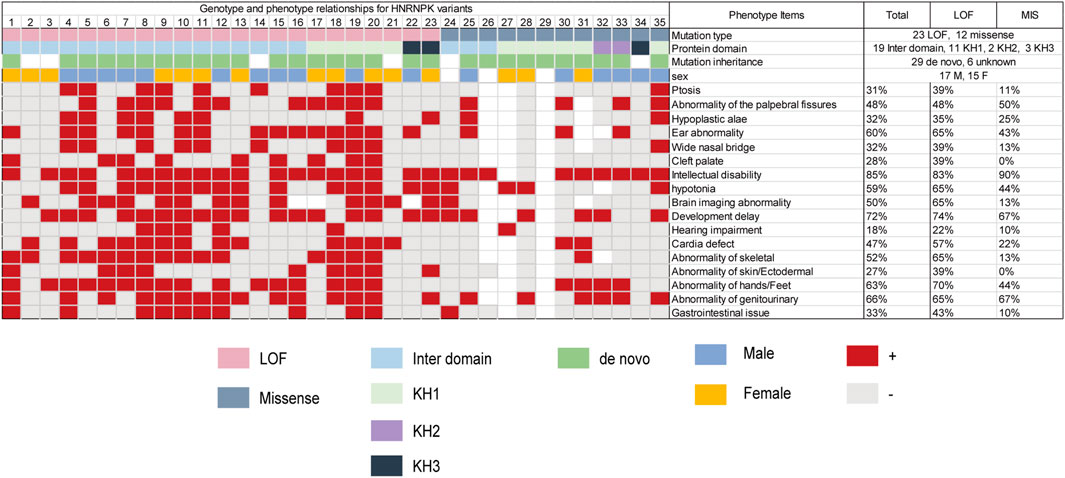
FIGURE 3. Genotype and phenotype relationships for HNRNPK variants. Pink boxes represent loss-of-function variants; Navy blue boxes represent missense variants; Green boxes represent de novo; blue boxes represent male; yellow boxes represent female; red boxes represent present. Light gray boxes represent absent; white boxes represent unreported. Total (%) represents the number of patients who were reported to have the specific phenotype (numerator) divided by the number of patients. LOF (%) represents the number of patients with LOF variants who were reported to have the specific phenotype (numerator) divided by the number of patients with LOF variants. MIS (%) represents the number of patients with missense variants who were reported to have the specific phenotype (numerator) divided by the number of patients with missense variants.
We hypothesized that missense variants affect RNA binding and thus cause dysfunction of related biological pathways that lead to the development of AKS. The de novo missense variant identified in this study was in the KH1 domain, and the predicted affinity change (ΔΔG) showed a reduced affinity for RNA binding. Transcriptome results identifying 42 aberrantly expressed and 86 spliced genes in the patient provided additional evidence to support the hypothesis. These genes were significantly associated with protein binding, transcriptional regulation, and nervous system function. Two of the downregulated genes, TUBB2A (p = 0.0093) and TUBB2B (p = 0.0092), are known to interact with HNRNPK at the protein level (Cerami et al., 2011). Both genes encode for the tubulin protein, which plays a critical role in neuronal function, neuronal migration, and postmigration development. Prior studies have shown that reduced expression of tubulin can lead to intellectual disability, matching the phenotype of the case in this study (Breuss et al., 2017; Jimenez et al., 2019; Schmidt et al., 2021). Further experiments are needed to assess the mechanism by which HNRNPK variants impact the development of AKS.
In summary, by integrating Trio-WES and RNA-seq analyses, we were able to better understand the role of an HNRNPK variant in a patient with AKS. Our findings expand the current understand of the clinical spectrum of HNRNPK variants.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the ethics committee of the Second Affiliated Hospital of Chongqing Medical University in China. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
XP performed the experiments and prepared the figures. SL performed RNA-seq analysis and wrote the manuscript. HY, XZ and LL provided intellectual and material support. BT designed and supervised the study and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Chongqing Natural Science Foundation (cstc2019jcyj-msxmx0318); “Kuanren talents” project of the Second Affiliated Hospital of Chongqing Medical University (13-003-003) and 2020 Nan’an District Science and Health Joint Medical Scientific Research Project (2020-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the patient and the family members for their participation in this study. We thank professor Fengxiao Bu for his helpful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.853028/full#supplementary-material
Au, P. Y. B., Goedhart, C., Goedhart, C., Ferguson, M., Breckpot, J., Devriendt, K., et al. (2018). Phenotypic Spectrum of Au-Kline Syndrome: a Report of Six New Cases and Review of the Literature. Eur. J. Hum. Genet. 26 (9), 1272–1281. doi:10.1038/s41431-018-0187-2
Barboro, P., Ferrari, N., and Balbi, C. (2014). Emerging Roles of Heterogeneous Nuclear Ribonucleoprotein K (hnRNP K) in Cancer Progression. Cancer Lett. 352 (2), 152–159. doi:10.1016/j.canlet.2014.06.019
Breuss, M. W., Nguyen, T., Srivatsan, A., Leca, I., Tian, G., Fritz, T., et al. (2017). Uner Tan Syndrome Caused by a homozygousTUBB2Bmutation Affecting Microtubule Stability. Hum. Mol. Genet. 26 (2), ddw383. doi:10.1093/hmg/ddw383
Bu, D., Luo, H., Huo, P., Wang, Z., Zhang, S., He, Z., et al. (2021). KOBAS-i: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 49 (W1), W317–W325. doi:10.1093/nar/gkab447
Cerami, E. G., Gross, B. E., Demir, E., Rodchenkov, I., Babur, O., Anwar, N., et al. (2011). Pathway Commons, a Web Resource for Biological Pathway Data. Nucleic Acids Res. 39 (Database issue), D685–D690. doi:10.1093/nar/gkq1039
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Dentici, M. L., Barresi, S., Niceta, M., Pantaleoni, F., Pizzi, S., Dallapiccola, B., et al. (2018). Clinical Spectrum of Kabuki-like Syndrome Caused by HNRNPK Haploinsufficiency. Clin. Genet. 93 (2), 401–407. doi:10.1111/cge.13029
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
Farwell Hagman, K. D., Shinde, D. N., Mroske, C., Smith, E., Radtke, K., Shahmirzadi, L., et al. (2017). Candidate-gene Criteria for Clinical Reporting: Diagnostic Exome Sequencing Identifies Altered Candidate Genes Among 8% of Patients with Undiagnosed Diseases. Genet. Med. 19 (2), 224–235. doi:10.1038/gim.2016.95
Frazer, J., Notin, P., Dias, M., Gomez, A., Min, J. K., Brock, K., et al. (2021). Disease Variant Prediction with Deep Generative Models of Evolutionary Data. Nature 599 (7883), 91–95. doi:10.1038/s41586-021-04043-8
Gallardo, M., Hornbaker, M. J., Zhang, X., Hu, P., Bueso-Ramos, C., and Post, S. M. (2016). Aberrant hnRNP K Expression: All Roads lead to Cancer. Cell Cycle 15 (12), 1552–1557. doi:10.1080/15384101.2016.1164372
Geuens, T., Bouhy, D., and Timmerman, V. (2016). The hnRNP Family: Insights into Their Role in Health and Disease. Hum. Genet. 135 (8), 851–867. doi:10.1007/s00439-016-1683-5
Gillentine, M. A., Wang, T., Wang, T., Hoekzema, K., Rosenfeld, J., Liu, P., et al. (2021). Rare Deleterious Mutations of HNRNP Genes Result in Shared Neurodevelopmental Disorders. Genome Med. 13 (1), 63. doi:10.1186/s13073-021-00870-6
Järvelä, I., Määttä, T., Acharya, A., Leppälä, J., Jhangiani, S. N., Arvio, M., et al. (2021). Exome Sequencing Reveals Predominantly De Novo Variants in Disorders with Intellectual Disability (ID) in the Founder Population of Finland. Hum. Genet. 140 (7), 1011–1029. doi:10.1007/s00439-021-02268-1
Jimenez, J., Herrera, D. A., Vargas, S. A., Montoya, J., and Castillo, M. (2019). β-Tubulinopathy Caused by a Mutation of the TUBB2B Gene: Magnetic Resonance Imaging Findings of the Brain. Neuroradiol J. 32 (2), 148–150. doi:10.1177/1971400919828142
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Maystadt, I., Deprez, M., Moortgat, S., Benoît, V., and Karadurmus, D. (2020). A Second Case of Okamoto Syndrome Caused by HNRNPK Mutation. Am. J. Med. Genet. 182 (6), 1537–1539. doi:10.1002/ajmg.a.61568
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., et al. (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17 (1), 122. doi:10.1186/s13059-016-0974-4
Miyake, N., Inaba, M., Mizuno, S., Shiina, M., Imagawa, E., Miyatake, S., et al. (2017). A Case of Atypical Kabuki Syndrome Arising from a Novel Missense Variant in HNRNPK. Clin. Genet. 92 (5), 554–555. doi:10.1111/cge.13023
Murdock, D. R., Dai, H., Burrage, L. C., Rosenfeld, J. A., Ketkar, S., Müller, M. F., et al. (2021). Transcriptome-directed Analysis for Mendelian Disease Diagnosis Overcomes Limitations of Conventional Genomic Testing. J. Clin. Invest. 131 (1), e141500. doi:10.1172/JCI141500
Okamoto, N. (2019). Okamoto Syndrome Has Features Overlapping with Au-Kline Syndrome and Is Caused by HNRNPK Mutation. Am. J. Med. Genet. 179 (5), 822–826. doi:10.1002/ajmg.a.61079
Pires, D. E. V., and Ascher, D. B. (2017). mCSM-NA: Predicting the Effects of Mutations on Protein-Nucleic Acids Interactions. Nucleic Acids Res. 45 (W1), W241–W246. doi:10.1093/nar/gkx236
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schmidt, L., Wain, K. E., Hajek, C., Estrada-Veras, J. I., Guillen Sacoto, M. J., Wentzensen, I. M., et al. (2021). Expanding the Phenotype of TUBB2A-Related Tubulinopathy: Three Cases of a Novel, Heterozygous TUBB2A Pathogenic Variant p.Gly98Arg. Mol. Syndromol 12 (1), 1–8. doi:10.1159/000512160
Shashi, V., Schoch, K., Spillmann, R., Cope, H., Tan, Q. K., Walley, N., et al. (2019). A Comprehensive Iterative Approach Is Highly Effective in Diagnosing Individuals Who Are Exome Negative. Genet. Med. 21 (1), 161–172. doi:10.1038/s41436-018-0044-2
Trivedi, U. H., Cã©zard, T. e., Bridgett, S., Montazam, A., Nichols, J., Blaxter, M., et al. (2014). Quality Control of Next-Generation Sequencing Data without a Reference. Front. Genet. 5, 111. doi:10.3389/fgene.2014.00111
Wang, T., Hoekzema, K., Vecchio, D., Wu, H., Sulovari, A., Coe, B. P., et al. (2020). Large-scale Targeted Sequencing Identifies Risk Genes for Neurodevelopmental Disorders. Nat. Commun. 11 (1), 4932. doi:10.1038/s41467-020-18723-y
Yamada, M., Shiraishi, Y., Uehara, T., Suzuki, H., Takenouchi, T., Abe‐Hatano, C., et al. (2020). Diagnostic Utility of Integrated Analysis of Exome and Transcriptome: Successful Diagnosis of Au‐Kline Syndrome in a Patient with Submucous Cleft Palate, Scaphocephaly, and Intellectual Disabilities. Mol. Genet. Genomic Med. 8 (9), e1364. doi:10.1002/mgg3.1364
Keywords: missense variant, Au-Kline syndrome, RNA-seq, hnRNPK, clinical diagnosis
Citation: Pan X, Liu S, Liu L, Zhang X, Yao H and Tan B (2022) Case Report: Exome and RNA Sequencing Identify a Novel de novo Missense Variant in HNRNPK in a Chinese Patient With Au-Kline Syndrome. Front. Genet. 13:853028. doi: 10.3389/fgene.2022.853028
Received: 12 January 2022; Accepted: 14 March 2022;
Published: 29 March 2022.
Edited by:
Tianyun Wang, University of Washington, United StatesReviewed by:
Madelyn Gillentine, Seattle Children’s Hospital, United StatesCopyright © 2022 Pan, Liu, Liu, Zhang, Yao and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Tan, dGFuYm9AaG9zcGl0YWwuY3FtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.