 Veronika Todorovova
Veronika Todorovova Tereza Altschmiedova
Tereza Altschmiedova Michal Vrablik
Michal Vrablik Richard Ceska
Richard Ceska
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 28 February 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.849008
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New Insight - Volume IIView all 17 articles
Introduction: The cause of familial hypercholesterolemia (FH) is defect in LDL receptor or familial defect of apolipoprotein B-100 (FDB) or, rarely, defect in proprotein convertase subtilisin/kexin type 9. Identification and treatment of patients with FH improves their prognosis. Our data represent retrospective analysis of 50 years of specialised care in our center.
Patients and Methods: A group of 1236 FH patients (841 women, 395 men; 993 study subjects and 243 relatives; mean age 44.8 ± 16.7 years) included 154 FDB patients followed at the Lipid Clinic of the General University Hospital in Prague since the mid-1960s to the present. Clinical diagnosis was based on the Dutch Lipid Clinic Network Criteria. Genetic analysis was performed using PCR-RFLP to detect FDB and apolipoprotein E (APOE) polymorphism. Biochemical data were collected and statistically analysed.
Results: At baseline, mean LDL-C and total cholesterol (TC) levels of all FH patients combined were 6.49 ± 1.92 mmol/L and 8.95 ± 1.95 mmol/L, respectively. Their LDL-C levels decreased to 3.26 ± 1.57 mmol/L and TC levels to 5.43 ± 1.69 mmol/L during follow-up. In the subgroup of LDL receptor-mediated FH (non-FDB) patients, baseline LDL-C and TC levels of 6.61 ± 1.95 mmol/L and 9.09 ± 1.97 mmol/L declined to 3.21 ± 1.60 mmol/L and 5.39 ± 1.72 mmol/L, respectively, during follow-up. In the FDB subgroup of patients, baseline levels of LDL-C and TC were 5.57 ± 1.46 mmol/L and 7.88 ± 1.58 mmol/L decreasing to 3.45 ± 0.24 mmol/L and 5.58 ± 1.37 mmol/L, respectively, during follow-up. Differences were also found in the effects of various APOE isoforms on lipid lowering. A significant decrease in lipid parameters was observed with the E2E2 isoform whereas a minimal decrease was seen with the E4E4 and E3E3 isoforms.
Conclusion: Whereas, overall, non-FDB patients had higher baseline lipid levels, these levels declined more appreciably compared with FDB patients during follow-up. Our retrospective analysis also found different effects of APOE isoforms on the decrease in lipid levels.
Familial hypercholesterolemia (FH) is an autosomal dominant inherited disorder characterised by elevated levels of low-density lipoprotein cholesterol (LDL-C) whose accumulation leads to the development of atherosclerotic cardiovascular disease (ASCVD); moreover, if not treated properly, it may result in premature death (Watts et al., 2016). It is estimated that, while there are 30 million FH patients worldwide, most of them are unaware of their condition (The FH Foundation, 2021). The prevalence of heterozygous FH (HeFH) is 1 per 200 to 250, with their LDL-C levels ranging between 4 and 13 mmol/L (Cuchel et al., 2014; Benn et al., 2016). In homozygous FH patients, the levels of LDL-C are >13 mmol/L and the prevalence of this rare disease is approximately 1 per 160,000 to 300,000 (Cuchel et al., 2014). The diagnosis of FH is established based on the Dutch Lipid Clinic Network Criteria (DLCNC) categorizing patients into definite (>8 points), probable (6–8 points), possible (3–5 points) and unlikely (<3 points) FH groups. The patients are assigned to their respective categories based on each individual´s family history, clinical history, physical examination, levels of LDL-C and, possibly, genetic testing (Benn et al., 2016). The patients are first asked to change their eating habits and increase physical activity. However, lifestyle changes are not always enough and treatment has to be enhanced pharmacologically. Familial hypercholesterolemia patients are most often treated with statins, a class of drugs highly effective in lowering LDL-C levels, especially when combined with ezetimibe (Gagné et al., 2002). A breakthrough in the treatment of FH came with the discovery of PCSK9 inhibitors shown to decrease LDL-C by ≥50% (Watts et al., 2020).
Variants of three genes are a major cause of FH. The most common of these mutations occur in the LDL receptor (LDLR) gene and can lead to ligand-binding dysfunction, impaired LDL transport or internalization, recycling, or complete receptor deficiency (Soutar and Naoumova, 2007; Cuchel et al., 2014). Likewise, FH can be caused by mutations in the apolipoprotein B (APOB) and the proprotein convertase subtilisin/kexin type 9 (PCSK9) (Vrablik et al., 2020). To the best of our knowledge, no mutation in the PCSK9 gene in the Czech population has been reported to date. An important role is also played by the apolipoprotein E (APOE) gene, which affects the levels of LDL-C thus contributing to higher LDL-C levels in FH patients (Pirillo et al., 2017; Rashidi et al., 2017; Khalil et al., 2021).
A mutation in the APOB gene causes familial defective apolipoprotein B-100 (FDB), an autosomal dominant disease of lipid metabolism similar to LDL receptor-mediated FH (non-FDB) characterised by elevated plasma LDL-C levels (Vega and Grundy, 1986; Innerarity et al., 1987). The prevalence of FDB varies largely being, e.g., approximately 1 per 209 in Switzerland while the figure for Denmark is 1 per 883 (Miserez et al., 1994; Miserez and Muller, 2000; Benn et al., 2016). Familial defective apolipoprotein B-100 is caused by monogenic variants in the APOB gene where a single amino acid, arginine, at position 3527 is replaced, most frequently, by glutamine (p.R3527Q) and, rarely, by tryptophan (p.R3527W) or lysine (p.R3527L) or at position 3558 where arginine is replaced by cysteine (p. R3558C). This replacement leads to other protein conformations disrupting the binding of apolipoprotein B-100 (as a part of LDL particles) to LDLR (Brown and Goldstein, 1986; Whitfield et al., 2004).
The most common APOE isoform is E3E3 with the p.C112 and p.R158 variants (Ferrières et al., 1994; Eichner et al., 2002; Phillips, 2014). A less frequent isoform increasing LDL-C levels and contributing to the risk of developing Alzheimer’s disease is E4E4, i.e., the p.C112R and p.R158 variants (Huebbe and Rimbach, 2017; Muñoz et al., 2019). A rare isoform is E2E2 determined by the p.C112 and p.R158C variants.
Familial defective apolipoprotein B-100 is clinically almost indistinguishable from FH; it is easier to identify FDB genetically as a common monogenic variant R3527Q. Unlike FDB, FH can be caused by monogenic variants as well as polygenic forms encountered in approximately 20% of FH patients (Trinder et al., 2019). Although many studies have focused on numerous aspects of FH, data in the relevant literature about the individual FH subgroups and the differences between them are relatively scarce. Still, it is most likely that the clinical features, effect of treatment and inherent risks of the disease are significantly different between the non-FDB and FDB subgroups of patients.
The aim of this retrospective analysis was to analyse data of a large homogeneous group of patients diagnosed to have FH and followed in a single lipid center and, also, to show the benefits of therapy and the results obtained over the course of half of a century in specialised care. This large group was followed and processed in 2 different perspectives. This article (Part I) is focused on differences in the lipid profiles in subgroups of FH patients, i.e. FDB versus non-FDB patients, and in FH patients with different APOE genotypes. Concurrent article (Part II) by Altschmiedova et al. (2022) is focused on clinical symptomatology, i.e., on differences between the parameters in patients whose FH is already complicated by overt ASCVD and those without ASCVD in order to identify factors contributing to a complicated course of the disease.
A total of 1236 FH patients (841 women and 395 men; 993 study subjects, 243 relatives; mean age 44.8 ± 16.7 years) attending the Lipid Clinic of the General University Hospital in Prague, Czech Republic, were followed. The diagnosis of FH in our patients was based on the Dutch Lipid Clinic Network Criteria (DLCNC). Genetic analysis including FDB and APOE isoforms was performed in more than 76% of FH patients; however, a mutation in the LDLR gene was investigated in only ≥10% of these patients (Supplementary Figure S1). Risk factors and clinical complications are summarised in Table 1 and they are in more detail described in the article about clinical symptomatology by Altschmiedova et al. (2022) Enrolled in the retrospective analysis were patients, both pharmacotherapy-naïve and those treated pharmacologically, and their relatives.

TABLE 1. Clinical characteristics.
The first patients of this retrospective analysis have been followed since the mid-1960s when diagnosed with FH based on their clinical symptoms; complete biochemical and genetic data have been available here since 1974. The follow-up has continued to date with the latest biochemical values recorded in late 2020.
Blood samples were collected from study subjects. The serum levels of total cholesterol (TC), HDL-cholesterol (HDL-C) and triglycerides (TG) were measured enzymatically on automated analysers (Modular P800, Roche, Basel, Switzerland and UniCel DxC 880i Beckman Coulter, Brea, CA, United States). LDL-C was calculated using the Friedewald formula whereas apolipoprotein B (APOB) and lipoprotein (a) [Lp(a)] were measured by nephelometry and immunonephelometry.
Genomic DNA was isolated from peripheral blood collected into EDTA-anticoagulated tubes using the salting out method proposed by Miller et al., 1998. The concentration and purity of DNA were determined using a spectrophotometer (A260/A280; BioPhotometer Eppendorf 6131, Eppendorf, Germany).
The p.R3527Q (MluI) and p.R3558C (MspI) variants in the APOB gene were detected by polymerase chain reaction (PCR). Oligonucleotides 5′-CTT ACT TGA ATT CCA AGA GCA CCC-3′ and 5′-TGT ACT CCC AGA GGG AAT ATA CGC-3′ were used as the primer set. MluI and MspI as restriction enzymes were used in PCR-restriction fragment length polymorphism (PCR-RFLP) analysis. Fragments were subsequently separated and visualised by electrophoresis on GelRed-stained 4% agarose gel (MetaPhor-agarose: agarose = 3:1).
The E2, E3, E4 variants in the APOE gene were detected by PCR. Here, oligonucleotides 5′-TCC AAG GAG CTG CAG GCG GCG CA-3′ and 5′-ACA GAA TTC GCC CCG GCC TGG TAC ACT GCC A-3′ were used as the primer set. CfoI as a restriction enzyme was used in PCR-RFLP analysis. Fragments were subsequently separated and visualised by electrophoresis on GelRed-stained 10% polyacrylamide gel.
Variants in the LDLR gene were analysed by Sanger sequencing. The specific primers (Supplementary Material S1) were designed according to the sequence of 18 LDLR exons. The sequencing reaction was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit.
Statistical analysis was performed using STATISTICA 13 software (TIBCO Software Inc., Palo Alto, CA, United States). Values in the text, tables and figures are means ± standard deviation (SD). The level of statistical significance was set at 5%. Comparison of baseline vs. follow-up was done using the paired t-test. The lipid parameters of the two subgroups were compared using the two-sample t-test or, when comparing three groups, using analysis of variance (ANOVA).
An appreciable decrease in LDL-C levels, from 6.49 ± 1.92 mmol/L to 3.26 ± 1.57 mmol/L (∼49.8%) was observed (Figure 1) in all patients. Likewise, a 39.3% decrease, from 8.95 ± 1.95 mmol/L to 5.43 ± 1.69 mmol/L, in TC levels was seen (Supplementary Figure S2). The mean levels of other 2 parameters (TG and HDL-C) are clearly shown in Table 2. Major reductions were noted in APOB (−38.1%) and TG (−23.8%) levels (Supplementary Figure S3, S4). The decrease in HDL-C levels was only a small one (−6.6%) (Supplementary Figure S5). The differences between the baseline and follow-up levels of the parameters investigated were statistically significant (p < 0.001) except for Lp(a) whose levels remained almost unaltered throughout the retrospective analysis (Supplementary Figure S6).
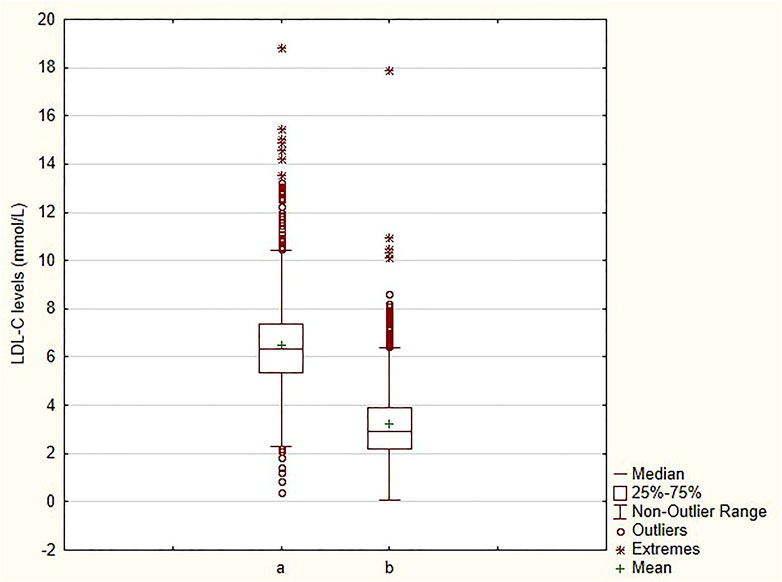
FIGURE 1. Comparison of LDL-C levels at baseline and at follow-up a—baseline; b—follow-up; LDL-C—low-density lipoprotein cholesterol.
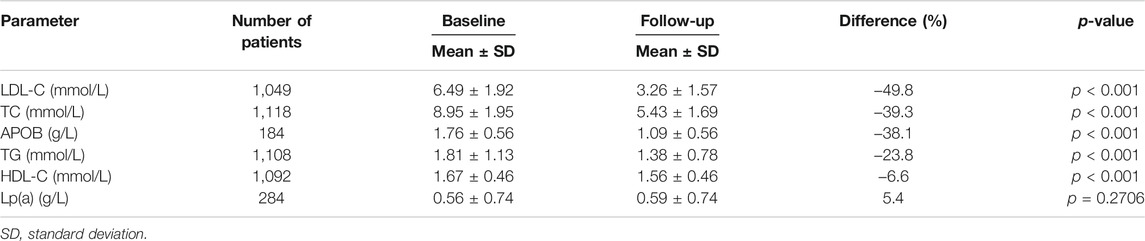
TABLE 2. Baseline and follow-up lipid levels of FH cohort.
Based on the division of our patients into subgroups according to the use (Y) or non-use (N) of therapy, statistically significant differences (p < 0.005) were found between the subgroups in the baseline levels of LDL-C, TC, APOB, TG, and Lp(a). However, statistically significant differences (p < 0.001) between the subgroups were found during follow-up in their LDL-C, TC, APOB, and HDL-C levels. When comparing the on-therapy levels of these parameters with baseline, the biggest decreases—except for Lp(a)—were noted in the groups not initiating their therapy until the start of the analysis (N/Y). In this particular group, LDL-C, TC and APOB levels dropped by as much as 55.7, 44.8 and 45.4%, respectively (Table 3). A smaller decline (−26.4%) in the N/Y group occurred in TG levels. Decreases in the levels of lipid parameters were likewise observed in the group of patients on pre-existing therapy (Y/Y) whose LDL-C, TC and APOB levels decreased by 49.6, 38.2 and 37.0%, respectively. The Y/Y group showed a smaller decrease in TG levels (−23.4%). As to HDL-C levels, similar to all study parameters except for Lp(a), there was an obvious decrease in the group of patients not receiving therapy throughout the analysis (N/N). The decreases in the levels of LDL-C, TC, APOB, TG and HDL-C were significant in all groups of patients (categorised by their therapy) throughout the analysis (p < 0.05).
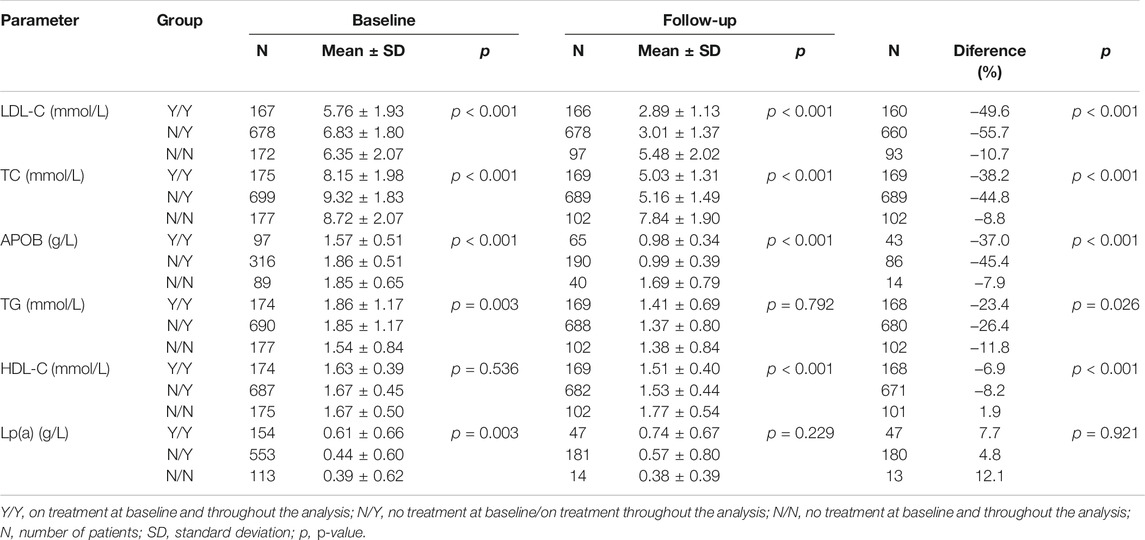
Table 3. Distribution of FH patients by treatment and effect of treatment on lipid levels.
Another division of our population of FH patients was based on genetic analysis of APOE polymorphisms by individual APOE isoforms, where differences in the levels of individual lipid parameters throughout the analysis were compared (Table 4). The biggest decreases in the levels of lipid parameters were seen in patients with the E2E2 isoform, being 75.3, 59.0, 53.5, 56.2, 17.6, and 36.7% in LDL-C, TC, APOB, TG, HDL-C and Lp(a) levels, respectively. In patients with the other isoforms, the decreases in LDL-C, TC, APOB and TG levels were within the ranges of 49.0–57.8%, 36.8–43.9%, 32.3–57.3%, and 22.4–39.4%, respectively, while HDL-C levels remained almost unchanged. A decline in Lp(a) levels was only seen in patients with the E2E2 isoform. The decrease was significant (p < 0.001) in only TG levels during follow-up.
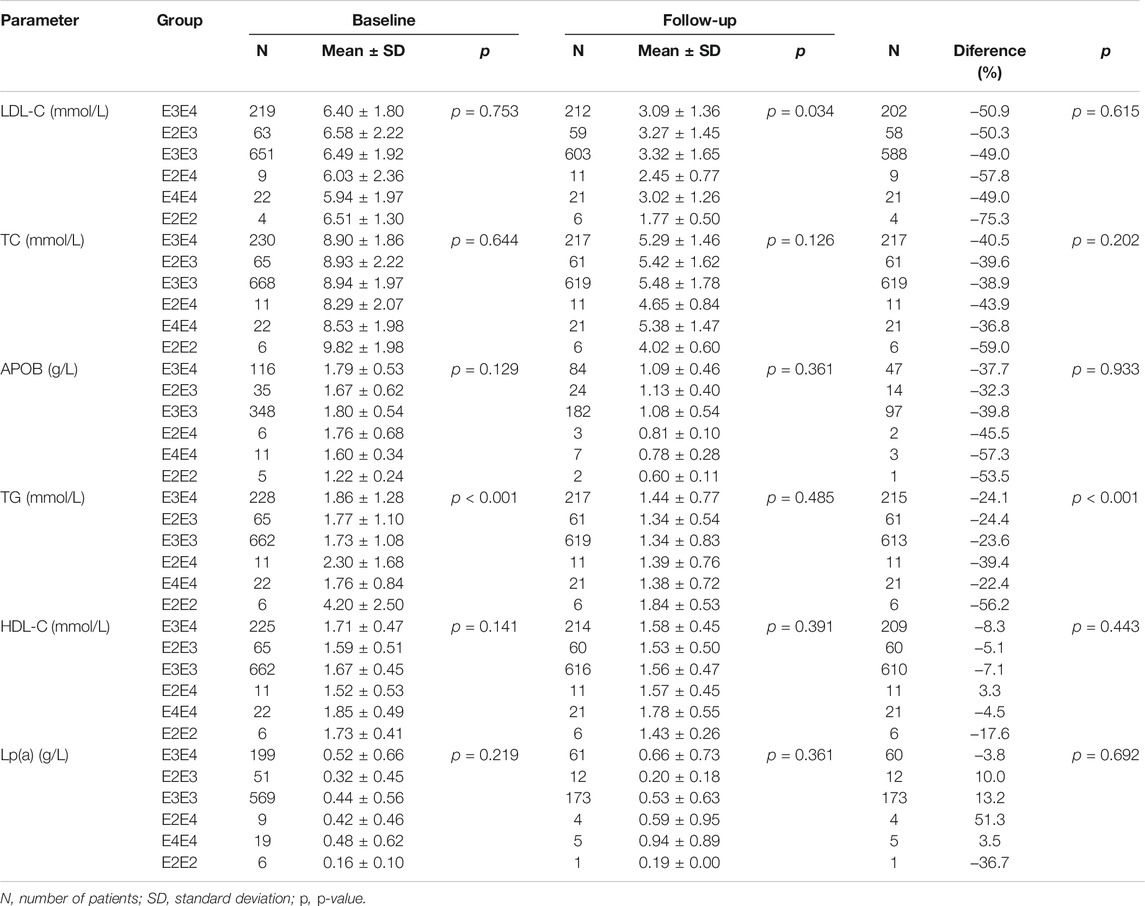
TABLE 4. Patients with specific APOE isoforms and effect of treatment on lipid levels.
A total of 1008 FH patients were genetically tested. Familial defective apolipoprotein B-100 was detected in 154 patients (mean age 40.8 ± 18.1 years; 107 women and 47 men; 117 study subjects and 37 relatives). One of these patients was diagnosed as FDB homozygote. The rest of genetically tested FH patients group consisted of 854 patients (557 women and 277 men; 686 study subjects and 168 relatives) supposed to have mutation in LDLR gene (non-FDB) (mean age 44.8 ± 16.0 years). Five patients were diagnosed with homozygous FH due to a mutation in the LDLR gene. Overall, another 228 study subjects met the DLCN criteria for FH but genetic testing was not performed and, thus, these subjects were excluded from further analysis.
One of the main subgroups within our study participants was that of non-FDB patients where significant decreases in the levels of LDL-C were noted in 754 patients (−51.1%); TC, in 795 patients (−40.7%); APOB, in 132 patients (−40.7%) and TG, in 788 patients (−24.4%). In 781 non-FDB patients, HDL-C levels declined by a mere 7.6% whereas Lp(a) levels remained almost unaltered. The second subgroup consisted of 154 patients with FDB. Their mean values of lipid parameters, both baseline and follow-up, are given in more detail in Table 5. Among the FDB patients, appreciable decreases in LDL-C were seen in 120 patients (−37.7%), TC in 131 patients (−30.3%), APOB in 26 patients (−29.2%), and TG in 130 patients (−24.0%). In 127 patients, HDL-C levels decreased by 5.9% during follow-up.

TABLE 5. FDB and non-FDB patients and effect of treatment on lipid levels.
A comparison of non-FDB and FDB patients revealed significant differences (p < 0.001) in their baseline levels of LDL-C, TC, APOB and TG whereas the difference versus follow-up levels was significant (p < 0.001) only in TG levels. Statistically significant differences (p < 0.05) were found between the baseline and follow-up levels of LDL-C, TC, APOB and Lp(a) in both, non-FDB and FDB, subgroups of patients.
At the end of the day, we would like to present the least favourable mean levels of Mr. and Mrs. FH and FDB in our analysis.
• Those of Mr. and Mrs. FH in our group are as follows: LDL-C 6.61 mmol/L, TC 9.09 mmol/L, APOB 1.83 g/L, TG 1.86 mmol/L, HDL-C 1.68 mmol/L and Lp(a) 0.46 g/L and
• Those of Mr. and Mrs. FDB in our group are as follows: LDL-C 5.57 mmol/L, TC 7.88 mmol/L, APOB 1.53 g/L, TG 1.40 mmol/L, HDL-C 1.68 mmol/L and Lp(a) 0.40 g/L.
What makes our retrospective analysis actually important is that our data were collected from a large group of more than 1,000 patients with FH attending a single lipid clinic. A positive finding of the long-term follow-up of patients in our center were decreases in the levels of LDL-C by more than 50%, which were not only statistically significant, but, also, clinically beneficial. Of no less importance was the decrease (by as much as 38%) in LDL-C levels in our FDB patients. As suggested by earlier reports, the levels of lipid parameters in FBD patients are generally lower than in those with LDL receptor-mediated FH (Gaffney et al., 2002; Vohnout et al., 2003; Fouchier et al., 2004). Similarly, the disorder diagnosed in our FDB homozygous patient was not as severe as that seen in homozygous individuals with receptor-mediated disorder. This may be explained by the APOE-regulated clearance of very low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) particles in FDB patients and the interaction between APOB and LDLR, important for the conversion of IDL to LDL-C (Gaffney et al., 2002; Vohnout et al., 2003).
Our analysis is a retrospective one whose first participants were receiving specialised care in a Prague-based clinic headed by Josef Šobra, with their data collection starting as early as 1960s (Šobra, 1970). Long-term care of these patients succeeded in reducing their cardiovascular risk and the clinic continues to provide individual care to each FH patient. While some of the patients have been taken care of for over 30 years, others have been attending the facility for less than 2 years; nonetheless, their personalised treatment plans have been shown to be beneficial in the long term. This explains the absence of statistically analysed data from the above period. This is partly due to the different numbers of patients and amount of analysed data in the individual subgroups of patients, with some of them referred to other physicians using different procedures, approaches and requirements for lipid parameter determination. However, the differences in the amount of data analysed and presented here are mainly due to FH patients referred from general practitioners to specialised centers; the result is some patients had incomplete baseline data while baseline blood sampling had not been performed in others. Another reason for the incompleteness data of some patients is only one value of some of the lipid parameters was obtained before the patient decided to discontinue follow-up.
A limitation of our analysis is the composition of our entire FH group consisting predominantly of patients attending a lipid clinic with only a small proportion being their family members. While not usual in other countries (Bhatnagar et al., 2000; Jarauta et al., 2016), a small number of relatives is a typical feature in the Czech Republic.
As expected, the differences in the decreases in lipid parameters between the untreated versus treated groups seen during the analysis in our lipid clinic were statistically significant. Nonetheless, the group of patients receiving treatment from practitioners prior to initiation of therapy by a lipid specialist also showed an appreciable decrease in their lipid levels, similar to that seen in patients not starting therapy before admission to our center. The implication is that targeted and proper management of FH patients is of crucial importance and, despite the undeniable role of general practitioners, tailored and specific care provided in lipid clinics is more effective and beneficial.
While it is difficult to identify a specific therapeutic strategy for over more than 50 years, generally, the treatment copied the availability and development of pharmacotherapy. It can be clearly stated that, until 1990, the mainstay of therapy of FH were cholestyramine and colestipol. Since 1990, treatment of FH has been based on statins (always the most efficient statin available, i.e., lovastatin, simvastatin, atorvastatin and rosuvastatin). A combination with ezetimibe has been used since the beginning of the 21st century and the monoclonal antibodies evolocumab and alirocumab have been available since 2018.
Patients with the rare APOE E2E2 genotype showed an obviously major drop in the levels of LDL-C, TC and TG corresponding the metabolic processing of the E2E2 isoform, where particle clearance does not occur through binding to LDLR but through the LDL-related receptor and heparin sulfate proteoglycans (Phillips, 2014). A similar major decrease was observed in patients with the E2E4 isoform, processed partly in the same way as the above E2E2 isoform. In FH patients, the decreases seen with the E3 and E4 isoforms were smaller, a fact possibly attributable to the clearance of APOE via LDLR where binding may be impaired due to the high frequency of LDLR gene mutations in FH patients.
Another parameter assessed in our study were Lp(a) levels not showing significant changes in some of our study subgroups. This may be partly explained by the fact that analysis of Lp(a) levels was undertaken in a period when no therapy to modify Lp(a) levels was available yet.
The relationship between high Lp(a) levels and proprotein convertase subtilisin/kexin type 9 (PCSK9) was not investigated until 2018, when Sun et al. reported their data obtained from patients with heterozygous FH; it has been shown only recently that Lp(a) levels can be decreased with the use of PCSK9 inhibitors (Sun et al., 2018). PCSK9 inhibitors were approved for clinical use in the Czech Republic in 2018; hence, the introduction of PCSK9 inhibitors is not significantly reflected in our analysis.
Despite their long-term follow-up, a small group of patients has not had genetic testing, with their diagnosis established solely using the DLCNC. As a result, some of these patients could not be conclusively identified as actually being or not being FDB patients. The relatively small number of mutations detected in the LDLR gene is due to the fact that the sequencing technique developed by Sanger was adopted by our lipid clinic only recently. Besides, the technique is also more time-consuming than those of PCR restriction fragment length polymorphism (RFLP) or real-time PCR detecting point mutations. The analysed LDLR gene region contains 18 exons which have to be sequenced separately when using Sanger’s technique. The proportion of LDLR gene mutation analyses is likely to increase in our clinic with the introduction of new generation sequencing (NGS) techniques in the years to come.
Our retrospective analysis provides initial data obtained from a large group of patients attending a single lipid clinic and analysed in terms of the biochemical and genetic characteristics.
Using a large group of patients with familial hypercholesterolemia, the present analysis reports data related to lipid and lipoprotein metabolism. The project was designed to assess changes in the levels of these parameters between baseline and follow-up in patients receiving personalised care admitted to our clinic. Our experience gained within the international ScreenPro FH project shows that patient surveillance and long-term follow-up are most beneficial as documented by Ceska et al., 2019. As an extension to the outcome of the present retrospective analysis, clinical data of our FH cohort are reported in Part II by Altschmiedova et al. (2022)
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Ethics Committee of General University Hospital, Prague. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
VT processed the data, wrote the article and participated in the interpretation of the analyzed data. TA contributed to the data processing and TA and MV contributed to the writing and interpretation of the data. RC designed the project, interpreted the data and participated in the writing of the article.
The conduct of this retrospective analysis was funded by the ScreenPro FH project and Specific University Research project 260525 of the First Faculty of Medicine, Charles University, Prague, Czech Republic.
TA is a paid speaker for Amgen and Sanofi. RC is a consultant to Amgen, Astra Zeneca, Bayer, Boehringer Ingelheim, MSD, Sanofi, Zentiva, and a board member of Amgen, Novartis, Novatin, Pfizer, Promed, Roche, Sanofi, Servier, Viatris; he also serves as a paid speaker for Amgen, Astra Zeneca, Bayer, Boehringer Ingelheim, MSD, Novartis, Novatin, NovoNordisk, Pfizer, Promed, Roche, Sanofi, Servier and Zentiva. RC has received honoraria from Amgen, Esperion, Kowa, Regeneron and Sanofi (PI, NC). MV reports fees for clinical trials, consultancy and presentations from Amgen, Astrazeneca, Bayer, Boehringer Ingelheim, Lilly, Krka, Mylan, MSD, Novartis, Novo Nordisk, Sanofi and Zentiva.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank collaborators of Lipid Clinic of the General University Hospital in Prague, Marian Rybar, Jaroslav A. Hubacek and Tomas Freiberger for consultations and help with processing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.849008/full#supplementary-material
Altschmiedova, T., Todorovova, V., Vrablik, M., and Ceska, R. (2022). Familial hypercholesterolemia: Real-world data of 1236 patients attending a Czech lipid clinic. A retrospective analysis of experience in more than 50 years. Part II. Clinical characteristics. [Preprint]. Available at: https://www.frontiersin.org/articles/10.3389/fgene.2022.849267/abstract (Accessed February 9, 2022).
Benn, M., Watts, G. F., Tybjærg-Hansen, A., and Nordestgaard, B. G. (2016). Mutations Causative of Familial Hypercholesterolaemia: Screening of 98 098 Individuals from the Copenhagen General Population Study Estimated a Prevalence of 1 in 217. Eur. Heart J. 37, 1384–1394. doi:10.1093/eurheartj/ehw028
Bhatnagar, D., Morgan, J., Siddiq, S., Mackness, M. I., Miller, J. P., and Durrington, P. N. (2000). Outcome of Case Finding Among Relatives of Patients with Known Heterozygous Familial. Br. Med. J. 321, 1497. doi:10.1136/bmj.321.7275.1497
Brown, M. S., and Goldstein, J. L. (1986). A Receptor-Mediated Pathway for Cholesterol Homeostasis. Science 232, 34–47. doi:10.1126/science.3513311
Ceska, R., Latkovskis, G., Ezhov, M. V., Freiberger, T., Lalic, K., Mitchenko, O., et al. (2019). The Impact of the International Cooperation on Familial Hypercholesterolemia Screening and Treatment: Results from the ScreenPro FH Project. Curr. Atheroscler. Rep. 21, 36. doi:10.1007/s11883-019-0797-3
Cuchel, M., Bruckert, E., Ginsberg, H. N., Raal, F. J., Santos, R. D., Hegele, R. A., et al. (2014). Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 35, 2146–2157. doi:10.1093/eurheartj/ehu274
Eichner, J. E., Dunn, S. T., Perveen, G., Thompson, D. M., Stewart, K. E., and Stroehla, B. C. (2002). Apolipoprotein E Polymorphism and Cardiovascular Disease: a HuGE Review. Am. J. Epidemiol. 155, 487–495. doi:10.1093/aje/155.6.487
Ferrières, J., Sing, C. F., Roy, M., Davignon, J., and Lussier-Cacan, S. (1994). Apolipoprotein E Polymorphism and Heterozygous Familial Hypercholesterolemia. Sex-specific Effects. Arterioscler. Thromb. 14, 1553–1560. doi:10.1161/01.atv.14.10.1553
Fouchier, S. W., Defesche, J. C., Kastelein, J. J., and Sijbrands, E. J. (2004). Familial Defective Apolipoprotein B versus Familial Hypercholesterolemia: an Assessment of Risk. Semin. Vasc. Med. 4, 259–264. doi:10.1055/s-2004-861493
Gaffney, D., Forster, L., Caslake, M. J., Bedford, D., Stewart, J. P., Stewart, G., et al. (2002). Comparison of Apolipoprotein B Metabolism in Familial Defective Apolipoprotein B and Heterogeneous Familial Hypercholesterolemia. Atherosclerosis 162, 33–43. doi:10.1016/s0021-9150(01)00679-7
Gagné, C., Gaudet, D., and Bruckert, E. (2002). Efficacy and Safety of Ezetimibe Coadministered with Atorvastatin or Simvastatin in Patients with Homozygous Familial Hypercholesterolemia. Circulation 105, 2469–2475. doi:10.1161/01.CIR.0000018744.58460.62
Huebbe, P., and Rimbach, G. (2017). Evolution of Human Apolipoprotein E (APOE) Isoforms: Gene Structure, Protein Function and Interaction with Dietary Factors. Ageing Res. Rev. 37, 146–161. doi:10.1016/j.arr.2017.06.002
Innerarity, T. L., Weisgraber, K. H., Arnold, K. S., Mahley, R. W., Krauss, R. M., Vega, G. L., et al. (1987). Familial Defective Apolipoprotein B-100: Low Density Lipoproteins with Abnormal Receptor Binding. Proc. Natl. Acad. Sci. 84, 6919–6923. doi:10.1073/pnas.84.19.6919
Jarauta, E., Pérez-Ruiz, M. R., Pérez-Calahorra, S., Mateo-Gallego, R., Cenarro, A., Cofán, M., et al. (2016). Lipid Phenotype and Heritage Pattern in Families with Genetic Hypercholesterolemia Not Related to LDLR, APOB, PCSK9, or APOE. J. Clin. Lipidol. 10, 1397–1405. e2. doi:10.1016/j.jacl.2016.09.011
Khalil, Y. A., Rabès, J.-P., Boileau, C., and Varret, M. (2021). APOE Gene Variants in Primary Dyslipidemia. Atherosclerosis 328, 11–22. doi:10.1016/j.atherosclerosis.2021.05.007
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16(3), 1215.
Miserez, A. R., Laager, R., Chiodetti, N., and Keller, U. (1994). High Prevalence of Familial Defective Apolipoprotein B-100 in Switzerland. J. Lipid Res. 35, 574–583. doi:10.1016/s0022-2275(20)41171-x
Miserez, A. R., and Muller, P. Y. (2000). Familial Defective Apolipoprotein B-100: a Mutation Emerged in the Mesolithic Ancestors of Celtic Peoples? Atherosclerosis 148, 433–436. doi:10.1016/s0021-9150(99)00470-0
Muñoz, S. S., Garner, B., and Ooi, L. (2019). Understanding the Role of ApoE Fragments in Alzheimer's Disease. Neurochem. Res. 44, 1297–1305. doi:10.1007/s11064-018-2629-1
Phillips, M. C. (2014). Apolipoprotein e isoforms and lipoprotein metabolism. IUBMB Life 66, 616–623. doi:10.1002/iub.1314
Pirillo, A., Garlaschelli, K., Arca, M., Averna, M., Bertolini, S., Calandra, S., et al. (2017). Spectrum of Mutations in Italian Patients with Familial Hypercholesterolemia: New Results from the LIPIGEN Study. Atheroscler. Supplements 29, 17–24. doi:10.1016/j.atherosclerosissup.2017.07.002
Rashidi, O. M., H.Nazar, F. A., Alama, M. N., and Awan, Z. A. (2017). Interpreting the Mechanism of APOE (p.Leu167del) Mutation in the Incidence of Familial Hypercholesterolemia; An In-silico Approach. Tocmj 11, 84–93. doi:10.2174/1874192401711010084
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of Disease: Genetic Causes of Familial Hypercholesterolemia. Nat. Rev. Cardiol. 4, 214–225. doi:10.1038/ncpcardio0836
Sun, D., Li, S., Zhao, X., Wu, N.-Q., Zhu, C.-G., Guo, Y.-L., et al. (2018). Association between Lipoprotein (A) and Proprotein Convertase Substilisin/kexin Type 9 in Patients with Heterozygous Familial Hypercholesterolemia: A Case‐control Study. Metabolism 79, 33–41. doi:10.1016/j.metabol.2017.11.004
The FH Foundation (2021). FH Awareness Day. Available at: https://thefhfoundation.org/fh-awareness-day/about-fhad (Accessed September 13, 2021).
Trinder, M., Li, X., DeCastro, M. L., Cermakova, L., Sadananda, S., Jackson, L. M., et al. (2019). Risk of Premature Atherosclerotic Disease in Patients with Monogenic versus Polygenic Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 74, 512–522. doi:10.1016/j.jacc.2019.05.043
Vega, G. L., and Grundy, S. M. (1986). In Vivo evidence for Reduced Binding of Low Density Lipoproteins to Receptors as a Cause of Primary Moderate Hypercholesterolemia. J. Clin. Invest. 78, 1410–1414. doi:10.1172/JCI112729
Vohnout, B., Rašlová, K., Gašparovič, J., Franeková, J., Fábryová, L., Belošovičová, M., et al. (2003). Lipid Levels and Their Genetic Regulation in Patients with Familial Hypercholesterolemia and Familial Defective Apolipoprotein B-100: The MEDPED Slovakia Project. Atheroscler. Supplements 4, 3–5. doi:10.1016/S1567-5688(03)00023-0
Vrablik, M., Tichý, L., Freiberger, T., Blaha, V., Satny, M., and Hubacek, J. A. (2020). Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 11, 574474. doi:10.3389/fgene.2020.574474
Watts, G. F., Chan, D. C., Pang, J., Ma, L., Ying, Q., Aggarwal, S., et al. (2020). PCSK9 Inhibition with Alirocumab Increases the Catabolism of Lipoprotein(a) Particles in Statin-Treated Patients with Elevated Lipoprotein(a). Metabolism 107, 154221. doi:10.1016/j.metabol.2020.154221
Watts, G. F., Ding, P. Y., George, P., Hagger, M. S., Hu, M., Lin, J., et al. (2016). Translational Research for Improving the Care of Familial Hypercholesterolemia: The “Ten Countries Study” and beyond. Jat 23, 891–900. doi:10.5551/jat.35949
Keywords: familial hypercholesterolemia, familial defective apolipoprotein B-100, LDL-C, ApoB, ApoE isoform, Lp(a), statin, ASCVD
Citation: Todorovova V, Altschmiedova T, Vrablik M and Ceska R (2022) Familial Hypercholesterolemia: Real-World Data of 1236 Patients Attending a Czech Lipid Clinic. A Retrospective Analysis of Experience in More than 50 years. Part I: Genetics and Biochemical Parameters. Front. Genet. 13:849008. doi: 10.3389/fgene.2022.849008
Received: 05 January 2022; Accepted: 02 February 2022;
Published: 28 February 2022.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Vanessa Bianconi, University of Perugia, ItalyCopyright © 2022 Todorovova, Altschmiedova, Vrablik and Ceska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tereza Altschmiedova, dGVyZXphLmFsdHNjaG1pZWRvdmFAdmZuLmN6
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.