
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 08 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.848879
This article is part of the Research TopicThe Genetics of Human Mendelian Skin DisordersView all 11 articles
 Yu-Rong Lee1,2†
Yu-Rong Lee1,2† Yu-Chen Lin1,3†
Yu-Chen Lin1,3† Yi-Han Chang1,4
Yi-Han Chang1,4 Hsin-Yu Huang1Yi-Kai Hong1,3
Hsin-Yu Huang1Yi-Kai Hong1,3 Wilson Jr F. Aala5Wei-Ting Tu1
Wilson Jr F. Aala5Wei-Ting Tu1 Meng-Che Tsai6Yen-Yin Chou6
Meng-Che Tsai6Yen-Yin Chou6 Chao-Kai Hsu1,3,5*
Chao-Kai Hsu1,3,5*Rubinstein–Taybi Syndrome (RSTS) is a rare congenital disease with distinctive facial features, broadening of the thumbs and halluces, and developmental delay. RSTS is caused by de novo genetic alterations in CREBBP and the homologous EP300 genes. In this study, we established a genetic diagnostic protocol by integrating multiplex ligation-dependent probe amplification (MLPA) and whole-exome sequencing (WES). Five patients clinically diagnosed with RSTS were enrolled for genetic testing. Germline DNA was extracted from the peripheral blood of the patients and their families. One patient (case 1) was identified as harboring a large heterozygous deletion in the 16p13.3 region, spanning the CREBBP gene. Three patients (Cases 2–4) harbored different CREBBP variants (c.2608C>T:p.Gln870Ter,c.4404_4405del:p.Thr1468fs,c.3649C>T:p.Gln1217Ter). No causative variants were identified for the fifth RSTS patient (case 5). Here, we propose a molecular diagnostic protocol that identified causative genetic alterations in 4/5 of the patients, yielding a molecular diagnostic rate of 80%. Given the rarity of RSTS, more research is needed to explore its pathogenesis and mechanism.
Rubinstein–Taybi Syndrome (RSTS, Broad thumb–hallux syndrome, MIM 180849) is a rare congenital malformation syndrome that affects approximately 1 in 100,000–125,000 newborns. RSTS is characterized by the broadening of the thumbs and halluces, developmental delay, moderate to severe intellectual disability, proneness to keloids, and distinctive facial features such as a large beaked nose and a low-hanging columella (Roelfsema and Peters, 2007).
Genetic research on RSTS has mainly focused on mutations in the genes encoding CREB binding protein (CREBBP, MIM 600140) and its homolog, E1A-binding protein P300 (EP300, MIM 602700). CREBBP is located at 16p13.3, while EP300 is located at 22q13.2. Both CBP and p300 are highly homologous transcriptional coactivators sharing conserved protein–protein interaction domains including the enzymatic histone acetyltransferase (HAT) domain. Acetylation disrupts the DNA–histone interaction by neutralizing the positively charged lysine residues in histones and enabling increased accessibility for transcription factors to activate gene expression (Kalkhoven, 2004; Oliveira et al., 2006). Among patients with RSTS, roughly 50–60% harbor pathogenic variants in CREBBP (RSTS type I), while only 3–8% of the patients harbor mutations in EP300 (RSTS type II) (López et al., 2018). The vast majority (about 99%) of RSTS cases occur sporadically from de novo heterozygous CREBBP mutations, although vertical transmission has also been documented in a handful of cases (Bartsch et al., 2010). The spectrum of reported genetic alterations regarding RSTS includes point mutations (i.e., frameshift, nonsense, missense, and splice-site mutations), intragenic or large deletions, translocations and inversions (Negri et al., 2019).
Whole-exome sequencing (WES), which utilizes the “shotgun-based” approach of next-generation sequencing, is known for its efficiency and effectiveness in detecting single-nucleotide polymorphisms (SNPs) and small insertions/deletions (indels). Structural variants and large deletions, on the other hand, remain challenging for WES due to its reliance on the short-read approach (Chong et al., 2015; Chiu et al., 2021). Multiplex ligation-dependent probe amplification (MLPA), on the other hand, although unable to recognize SNPs and indels, shows strengths in detecting genomic variations, such as copy number variations (CNVs) and large-spanning deletions by comparing the differences in PCR-amplified fluorescently labeled primers binding to the probes. Although molecular diagnosis can reveal the genetic alterations in most RSTS cases, a sizeable subset of patients (30%) remain undiagnosed when this method is used (Tajir et al., 2013). In this study, we leveraged the distinct advantages of WES and MLPA by combining the two methods in our genetic diagnostic protocol for RSTS. Here, we report three patients with pathogenic CREBBP mutations and one with a large deletion spanning CREBBP in a five-patient cohort of clinically diagnosed RSTS individuals.
The inclusion criteria included concurrent presentation of common RSTS clinical characteristics (distinctive facial features, broadening of the thumbs and halluces, short stature) unexplained by other systemic syndromes (Figure 2). Five patients (3 males and 2 females) fulfilled the criteria and were enrolled for further molecular genetic diagnosis. Detailed clinical data of the cases were documented. 3/5 (60%) of the cases also had microcephaly, another common presentation of RSTS. Additional clinical findings relevant to RSTS, including cyanotic heart disease, pulmonary hypertension, intellectual disability, and other neurological impairments, were found in some cases. The clinical manifestations of each case enrolled in this study are summarized in Table 1. Ethics approval was granted by the Ethics Committee of National Cheng Kung University Hospital (A-BR-104-052). Informed consent was obtained from the patients’ parents. Peripheral blood specimens were collected from the patients. DNA was extracted using the Qiagen FlexiGene DNA Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.

TABLE 1. Summary of causative genetic alterations found in this study.
MLPA was performed on DNA from each patients via the Affymetrix 750K array platform (Thermo Fisher Scientific, Waltham, United States) using a human microdeletion syndrome probe set (SALSA P096; MRC-Holland, Amsterdam, Netherlands). On the whole, microdeletions in the 4p, 5p15, 8p, 8q24, 11p13-14, 16p13.3, and 21q22.2 regions were detected with the probe set and were thus analyzed. MLPA reveals the relative quantification of the number of copies of targeted DNA sequences. Data were analyzed using GeneMarker software (SoftGenetics, State College, PA, United States). For the purpose of this study, heterozygous or homozygous deletions in the 16p13.3 region, which spans the CREBBP gene, were considered abnormal positives.
In one patient showing an abnormal large deletion spanning the CREBBP gene (case 1), MLPA was also done on the germline DNA from the parents of the proband to examine whether the mutation occurs de novo. As for the patients showing negative results for CREBBP large deletions, WES was performed on the DNA specimen to detect pathogenic SNPs and indels in the genes CREBBP and EP300.
Germline DNA extracted from the probands was used for paired-end library preparation using the SureSelect All Exon 50 Mb Version 4.0 kit (Agilent, Santa Clara, CA, United States) according to the manufacturer’s recommendations. Sequencing was carried out by massively parallel sequencing with 100-bp paired-end reads using the HiSeq-2000 platform (Illumina, CA, United States). The Novoalign software package (Novocraft Technologies Sdn Bhd) was used to align reads generated to the reference human genome. Reads mapping to multiple locations on the reference human genome were excluded from downstream analysis. The BedTools package was used to calculate the depth and breadth of sequence coverage (Quinlan and Hall, 2010). Single-nucleotide substitutions and small indels were detected with the SamTools package (Li et al., 2009). Sequence variants were annotated with the Annovar tool (Wang et al., 2010). To assess the pathogenicity of the candidate variants, an in-house variant-filtering pipeline was used. Nonsense variants or indels resulting in frame shifts in CREBBP or EP300 with minor allele frequencies (MAF) of less than 0.5% in the 1,000 Genomes Project (Siva, 2008) and Exome Aggregation Consortium (ExAC) were included. The damage prediction criteria for filtering the candidate variants included a Combined Annotation Dependent Depletion (CADD) score of above 15, a Deleterious Annotation of Genetic variants using Neural Networks (DANN) score of above 0.95, and a Polymorphism Phenotyping v2 (PolyPhen-2) score of above 0.95. Variants with MAF exceeding 0.5% or with damage prediction scores not fulfilling our criteria were excluded as non-pathogenic.
BAM files of WES were visualized via Integrative Genomics Viewer (IGV) (Robinson et al., 2011). Confirmative polymerase chain reaction (PCR) and Sanger sequencing tests were performed on the DNA from the probands and their parents to validate the filtered variants detected by WES and for segregation analysis. Primers were designed using the Ensembl database (Howe et al., 2021) and Primer3 (Untergasser et al., 2012) online software. The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA806385.
Genetic alterations relevant to RSTS identified in this study by both MLPA and WES are summarized in Figure 1. The characteristic clinical manifestations of RSTS for all five cases are summarized in Figure 2. As a whole, among the patients enrolled, MLPA showed a large 16p13.3 DNA deletion spanning the CREBBP gene in 1/5 (20%) patient, while causative CREBBP variants were detected in 3/5 (60%) patients by WES and were confirmed by Sanger sequencing. Through MLPA and WES, causative variants were identified in 4/5 (80%) of the RSTS patients and are summarized in Table 1.
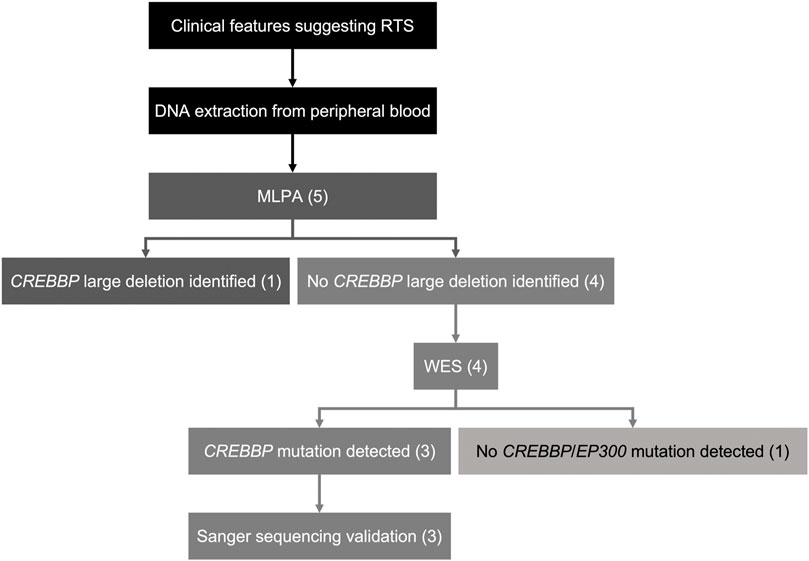
FIGURE 1. Schematic of the diagnostic workflow in this study. Five patients clinically diagnosed with RSTS were enrolled. MLPA was done on DNA samples of all the RSTS patients. 1/5 patient (20%) was found to harbor a large de novo deletion spanning CREBBP. WES was performed on DNA samples of the other four patients showing negative MLPA results. 3/5 patients (60%) were found to have novel CREBBP mutations with high pathogenicity scores. 1/5 patient (20%) showed negative results for MLPA and WES.
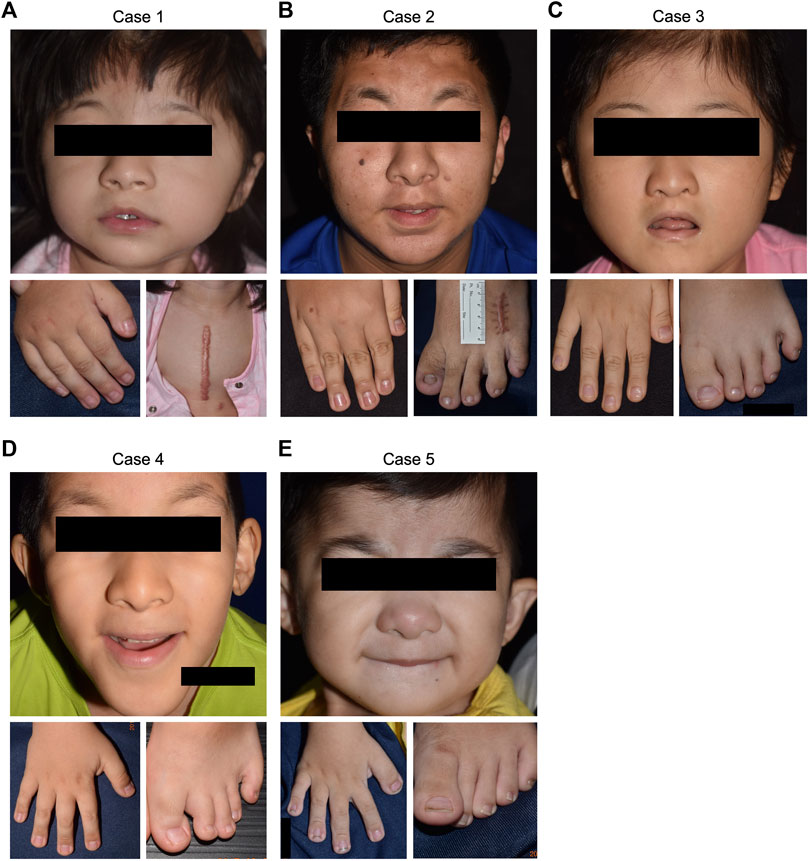
FIGURE 2. Clinical manifestations of the five RSTS cases. Positive findings of common RSTS characteristics, including distinctive facial features, and broadened thumbs and halluces, were noted in all of the enrolled cases. (A) case 1. (B) case 2. (C) case 3. (D) case 4. (E) case 5.
Case 1. was a 4-year-old female showing typical clinical features, including a high-arched palate, broadened thumbs and halluces, and a tendency to keloid development (Figure 2A). Congenital cyanotic heart disease were also found, including hypoplastic aortic isthmus with mild coarctation of the aorta, perimembranous ventricular septal defect, large right patent ductus arteriosus (PDA), and anomalous left pulmonary artery arising from left side PDA. MLPA of DNA from the proband and the parents revealed a de novo heterozygous 16p13.3:3,728,096-3,962,938 deletion that spanned the loci of TRAP1 and CREBBP (Figures 3A, 4A). To validate the results, we also performed WES on the proband’s DNA, and a visualization of the WES results in IGV revealed low read coverage across the mutation span (Figure 4B). Surgical repair of the patient’s congenital cyanotic heart disease was performed to alleviate the cardiac symptoms, and diflucortolone ointment was prescribed for the treatment of the developed keloid. The developmental delay was managed by follow-up treatments in the outpatient department and diet education.
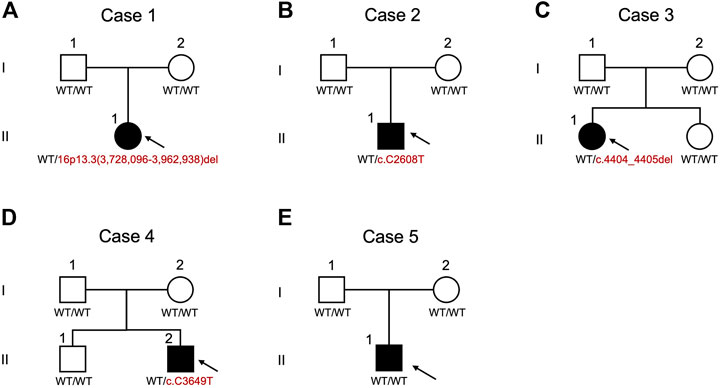
FIGURE 3. Pedigrees and schematic summary of genetic alteration discoveries in this study. (A) case 1. (B) case 2. (C) case 3. (D) case 4. (E) case 5. WT, wild type.
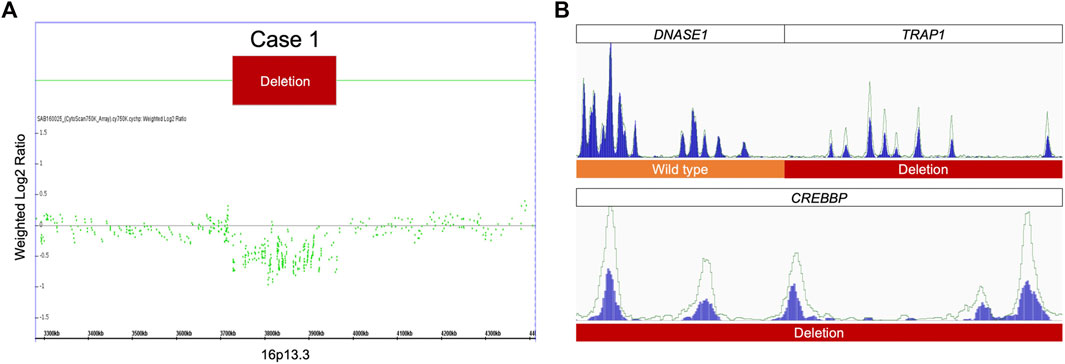
FIGURE 4. MLPA and WES results for case 1. (A) MLPA of the proband showing heterozygous deletion at 16p13.3. (B) A visualization of the WES results for case 1 show fewer reads in the proband than in the healthy control, confirming the heterozygous deletion detected by MLPA.
Case 2. was a 15-year-old male with typical clinical features suggesting RSTS (Figure 2B). MLPA of case 2 showed no detectable deletions in CREBBP. WES of the proband’s germline DNA revealed a novel CREBBP heterozygous nonsense mutation (NM_004380:c.C2608T:p.Gln870Ter) with a significant pathogenicity score (CADD:21.4, DANN:0.9986). Further segregation analysis by Sanger sequencing confirmed the variant detected through WES and revealed both parents to harbor homozygous wild-type alleles at the mutation site, confirming that the proband’s mutation had occurred de novo (Figures 3B, 5A). Since that the patient also presented with intellectual disability and mental retardation, the patient was referred to neurologists and psychiatrists at our hospital for diagnostic interviews and further psychological assessments.
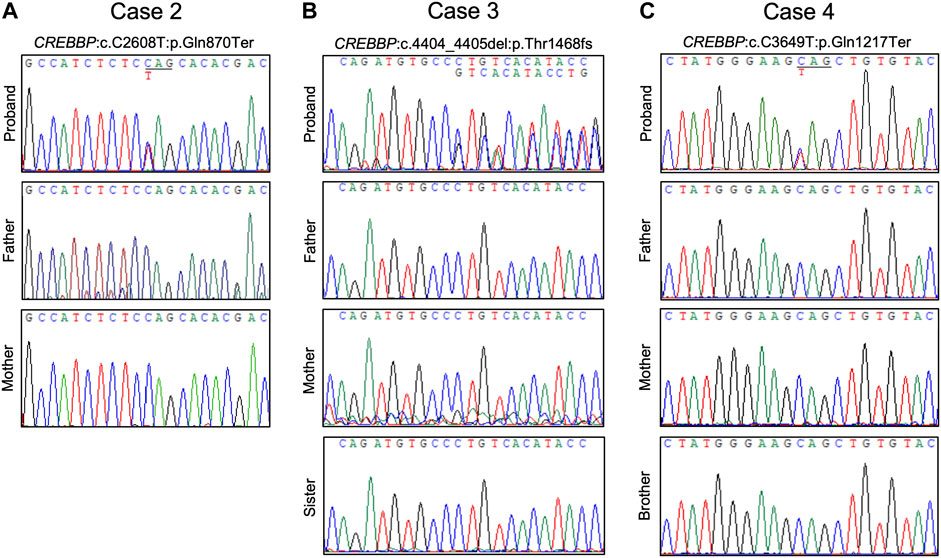
FIGURE 5. Chromatograms of confirmative PCR-based Sanger sequencing and segregation analysis results. (A) A de novo nonsense CREBBP mutation in case 2. (B) A de novo CREBBP 2-bp deletion in case 3. (C) The CREBBP nonsense mutation was detected in both case 4 and the proband’s mother.
Case 3. was a 9-year-old female with typical clinical features suggesting RSTS (Figure 2C). MLPA did not reveal any large deletions spanning CREBBP. WES showed a 2-bp deletion leading to a frame-shift in the CREBBP locus (NM_004380:c.4404_4405del:p.Gly1469AlafsTer9), which had been reported in another RSTS patient (Murata, 2001). Segregation analysis showed a wild-type genotype for both parents at the mutation site (Figures 3C, 5B), indicating that the mutation had occurred de novo.
Case 4. a 10-year-old male, also showed features of RSTS (Figure 2D). No large deletions were found in the patient’s DNA by MLPA, but WES revealed a CREBBP nonsense mutation (NM_004380:c.C3649T:p.Gln1217Ter) with a significant pathogenicity score (CADD:44, DANN:0.9987) that had been reported in a previous RSTS patient (Saettini et al., 2020). Further segregation analysis showed that the mutation had occurred de novo (Figure 5C). The patient also showed microcephaly and white matter hyperintensities under magnetic resonance imaging (MRI) examination. Rehabilitation programs and psychological assessments were arranged on a regular time course for the neurological symptoms.
Case 5. was a 4-year-old boy also with typical features of RSTS (Figure 2E). MLPA showed no large deletions in CREBBP. Unexpectedly, WES also failed to identify pathogenic variants within the CREBBP and EP300 loci (Figure 3E). Therefore, the causative genetic or genomic variant for case 5 remains unknown. Since the patient also manifested congenital glaucoma, trabeculectomy including peripheral iridectomy under the microscope was performed.
Of the five patients whose clinical presentation suggested RSTS, pathogenic variants in CREBBP were identified in four patients (80%) using an MLPA–WES genetic diagnostic workflow. However, the causative variant for 1/5 (20%) patient, remains uncertain. Among the sequencing methods used in this study, although WES efficiently detects small indels in protein-coding (exonic) DNA regions, the detection of copy number variations (CNVs), structural variants, and homologous regions of the genome remains challenging for WES. Given that pathogenic variants in RSTS comprise single-nucleotide or oligonucleotide variations as well as large deletions, genomic assays that can screen for large genomic variants (e.g., MLPA) are highly desirable. In this study, we integrated MLPA and WES in our molecular diagnostic protocol. PCR-based Sanger sequencing was also utilized to confirm the variants identified by WES. Indeed, a similar workflow has been applied to a Korean cohort (Choi et al., 2021), leading to the detection of causative variants in 80% of their patients, which is similar to our diagnostic yield.
Given that RSTS is a rare disorder arising from de novo mutations, the number of reported cases remain limited. In the Taiwanese population, one study reported the clinical and molecular data of 10 RSTS patients (Hou, 2005). In that study, chromosomal deletions over the 16p13 region (responsible for the CREBBP gene) were detected by fluorescence in situ hybridization (FISH). Only 30% of the RSTS patients were detected as having interstitial submicroscopic deletions in the 16p13 region. The low diagnostic rate from the use of FISH alone might lie in its ability to resolve only large DNA deletions. Thus, pathogenic single-nucleotide variants would be neglected. Similarly, among the five RSTS patients collected in our study, only 1/5 (20%) patient showed a large DNA deletion spanning CREBBP (case 1). Our study highlights the potential gains from harnessing various genetic assays.
Mutation hotspots in the CREBBP and EP300 loci have yet to be documented. In a previous report, mutations in the highly conserved HAT domain were regarded as leading to high pathogenicity (López et al., 2018). However, no mutations resided within the HAT domain among our patients. Previous studies have further reported that frameshift mutations are the most prevalent type of mutation in RSTS patients (López et al., 2018; Choi et al., 2021). Although limited in number, more nonsense mutations (cases 2, 4) than frameshift mutations (case 3) were observed in the current study.
In our diagnostic workflow, the causative genetic alteration in 1/5 (20%) RSTS patient was not identified. Given that the pathogenesis of RSTS is not yet fully understood, the possibility remains that mutations occurring outside the CREBBP and EP300 loci may contribute to the RSTS phenotype. CREBBP and EP300 are both epigenetics-associated factors that alter acetyltransferase activity. In a previous study, experiments on a developmental animal model for RSTS were conducted using mice deficient in CREB-binding protein (CBP), and these CBP+/− mice were found to exhibit an abnormal RSTS skeletal pattern (Tanaka et al., 1997). Furthermore, transgenic mice generated by Korzus et al. expressing reversible CBP that lacked HAT activity showed deficits in long-term memory, indicating that the HAT activity of CBP is essential for brain information processing and cognitive function (Korzus et al., 2004; Korzus, 2017). Although the genetic and epigenetic functions of CREBBP genes in rodents have been thoroughly investigated, the complete pathogenicity of RSTS remains to be explored.
Here, we summarize the molecular genetic diagnostic progress of five patients clinically diagnosed with RSTS. Diagnostic workflow combining MLPA and WES was carried out to identify causative genetic alterations in CREBBP and EP300. Four of the five patients were found to harbor causative mutations in CREBBP. As the exact pathogenesis of RSTS has yet to be completely elucidated and genetic alterations in roughly 20% of the patients have yet to be found, more research is needed on the roles of genetic and epigenetic alterations in RSTS.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA806385.
The studies involving human participants were reviewed and approved by National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Y-RL: experiment performing, case collection, project managing. Y-CL: original manuscript drafting, figures arrangement, manuscript revision. Y-HC: figures arrangement, project design. H-YH: bioinformatic processing and interpretation. Y-KH: experiment performing, project design. WA: manuscript revision. W-TT: project design, case collection. M-CT: project design, case and clinical data collection. Y-YC: project design, case and clinical data collection. CH: project design, case and clinical data collection, supervision.
The authors gratefully acknowledge the Summer Research Project Grant no. NCKUMCS2020038 from College of Medicine at National Cheng Kung University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bartsch, O., Kress, W., Kempf, O., Lechno, S., Haaf, T., and Zechner, U. (2010). Inheritance and Variable Expression in Rubinstein-Taybi Syndrome. Am. J. Med. Genet. 152A, 2254–2261. doi:10.1002/ajmg.a.33598
Chiu, F. P. C., Doolan, B. J., McGrath, J. A., and Onoufriadis, A. (2021). A Decade of Next‐generation Sequencing in Genodermatoses: the Impact on Gene Discovery and Clinical Diagnostics*. Br. J. Dermatol. 184, 606–616. doi:10.1111/bjd.19384doi:19384
Choi, N., Kim, H. Y., Lim, B. C., Chae, J. H., Kim, S. Y., and Ko, J. M. (2021). Genetic and Clinical Heterogeneity in Korean Patients with Rubinstein-Taybi Syndrome. Mol. Genet. Genomic Med. 9. doi:10.1002/mgg3.1791
Chong, J. X., Buckingham, K. J., Jhangiani, S. N., Boehm, C., Sobreira, N., Smith, J. D., et al. (2015). The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 97, 199–215. doi:10.1016/j.ajhg.2015.06.009
Hou, J. W. (2005). Rubinstein-Taybi Syndrome: Clinical and Molecular Cytogenetic Studies. Acta Paediatr. Taiwan 46, 143–148. doi:10.7097/APT.200506.0143
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2021). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. doi:10.1093/nar/gkaa942
Kalkhoven, E. (2004). CBP and P300: HATs for Different Occasions. Biochem. Pharmacol. 68, 1145–1155. doi:10.1016/j.bcp.2004.03.045
Korzus, E., Rosenfeld, M. G., and Mayford, M. (2004). CBP Histone Acetyltransferase Activity Is a Critical Component of Memory Consolidation. Neuron 42, 961–972. doi:10.1016/j.neuron.2004.06.002
Korzus, E. (2017). Rubinstein-Taybi Syndrome and Epigenetic Alterations. Adv. Exp. Med. Biol. 978, 39–62. doi:10.1007/978-3-319-53889-1_3
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
López, M., García-Oguiza, A., Armstrong, J., García-Cobaleda, I., García-Miñaur, S., Santos-Simarro, F., et al. (2018). Rubinstein-Taybi 2 Associated to Novel EP300 Mutations: Deepening the Clinical and Genetic Spectrum. BMC Med. Genet. 19, 36. doi:10.1186/s12881-018-0548-2
Murata, T. (2001). Defect of Histone Acetyltransferase Activity of the Nuclear Transcriptional Coactivator CBP in Rubinstein-Taybi Syndrome. Hum. Mol. Genet. 10, 1071–1076. doi:10.1093/hmg/10.10.1071
Negri, G., Magini, P., Milani, D., Crippa, M., Biamino, E., Piccione, M., et al. (2019). Exploring by Whole Exome Sequencing Patients with Initial Diagnosis of Rubinstein-Taybi Syndrome: the Interconnections of Epigenetic Machinery Disorders. Hum. Genet. 138, 257–269. doi:10.1007/s00439-019-01985-y
Oliveira, A. M. M., Abel, T., Brindle, P. K., and Wood, M. A. (2006). Differential Role for CBP and P300 CREB-Binding Domain in Motor Skill Learning. Behav. Neurosci. 120, 724–729. doi:10.1037/0735-7044.120.3.724
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a Flexible Suite of Utilities for Comparing Genomic Features. Bioinforma. Oxf. Engl. 26, 841–842. doi:10.1093/bioinformatics/btq033
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26. doi:10.1038/nbt.1754
Roelfsema, J. H., and Peters, D. J. M. (2007). Rubinstein-Taybi Syndrome: Clinical and Molecular Overview. Expert Rev. Mol. Med. 9, 1–16. doi:10.1017/S1462399407000415
Saettini, F., Herriot, R., Prada, E., Nizon, M., Zama, D., Marzollo, A., et al. (2020). Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients. J. Clin. Immunol. 40, 851–860. doi:10.1007/s10875-020-00808-4
Tajir, M., Fergelot, P., Lancelot, G., Elalaoui, S. C., Arveiler, B., Lacombe, D., et al. (2013). Germline Mosaicism in Rubinstein-Taybi Syndrome. Gene 518, 476–478. doi:10.1016/j.gene.2012.12.105
Tanaka, Y., Naruse, I., Maekawa, T., Masuya, H., Shiroishi, T., and Ishii, S. (1997). Abnormal Skeletal Patterning in Embryos Lacking a Single Cbp Allele: A Partial Similarity with Rubinstein-Taybi Syndrome. Proc. Natl. Acad. Sci. U.S.A. 94, 10215–10220. doi:10.1073/pnas.94.19.10215
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new Capabilities and Interfaces. Nucleic Acids Res. 40, e115. doi:10.1093/nar/gks596
Keywords: rubinstein-taybi syndrome, multiplex ligation-dependent probe amplification, whole-exome sequencing, next-generation sequencing, genetic diagnosis, novel variant, CREBBP (Crebb binding protein)
Citation: Lee Y-R, Lin Y-C, Chang Y-H, Huang H-Y, Hong Y-K, Aala WJF, Tu W-T, Tsai M-C, Chou Y-Y and Hsu C-T (2022) Genetic Diagnosis of Rubinstein–Taybi Syndrome With Multiplex Ligation-Dependent Probe Amplification (MLPA) and Whole-Exome Sequencing (WES): Case Series With a Novel CREBBP Variant. Front. Genet. 13:848879. doi: 10.3389/fgene.2022.848879
Received: 05 January 2022; Accepted: 15 March 2022;
Published: 08 April 2022.
Edited by:
Ming Li, Shanghai Jiaotong University, ChinaReviewed by:
Nadia Akawi, United Arab Emirates University, United Arab EmiratesCopyright © 2022 Lee, Lin, Chang, Huang, Hong, Aala, Tu, Tsai, Chou and Hsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao-Kai Hsu, a3lsZWhzdUBtYWlsLm5ja3UuZWR1LnR3
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.