
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 16 March 2022
Sec. Genomics of Plants and the Phytoecosystem
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.845619
 Feng Li1,2
Feng Li1,2 Ying Liu1Junhui Wang1
Ying Liu1Junhui Wang1 Peiyao Xin2Jiangtao Zhang3Kun Zhao4Minggang Zhang5Huiling Yun6Wenjun Ma1*
Peiyao Xin2Jiangtao Zhang3Kun Zhao4Minggang Zhang5Huiling Yun6Wenjun Ma1*Species within the Genus Catalpa are mostly semievergreen or deciduous trees with opposite or whorled leaves. C. bungei, C. fargesii f. duclouxii and C. fargesii are sources of traditional precious wood in China, known as the “kings of wood”. Due to a lack of phenotypic and molecular studies and insufficient sequence information, intraspecific morphological differences, common DNA barcodes and partial sequence fragments cannot clearly reveal the phylogenetic or intraspecific relationships within Catalpa. Therefore, we sequenced the complete chloroplast genomes of six taxa of the genus Catalpa and analyzed their basic structure and evolutionary relationships. The chloroplast genome of Catalpa shows a typical tetrad structure with a total length ranging from 157,765 bp (C. fargesii) to 158,355 bp (C. ovata). The length of the large single-copy (LSC) region ranges from 84,599 bp (C. fargesii) to 85,004 bp (C. ovata), that of the small single-copy (SSC) region ranges from 12,662 bp (C. fargesii) to 12,675 bp (C. ovata), and that of the inverted repeat (IR) regions ranges from 30,252 bp (C. fargesii) to 30,338 bp (C. ovata). The GC content of the six chloroplast genomes were 38.1%. In total, 113 unique genes were detected, and there were 19 genes in IR regions. The 113 genes included 79 protein-coding genes, 30 tRNA genes and four rRNA genes. Five hypervariable regions (trnH-psbA, rps2-rpoC2, rpl22, ycf15-trnl-CAA and rps15) were identified by analyzing chloroplast nucleotide polymorphisms, which might be serve as potential DNA barcodes for the species. Comparative analysis showed that single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs) were highly diverse in the six species. Codon usage patterns were highly similar among the taxa included in the present study. In addition to the stop codons, all codons showed a preference for ending in A or T. Phylogenetic analysis of the entire chloroplast genome showed that all taxa within the genus Catalpa formed a monophyletic group, clearly reflecting the relationships within the genus. This study provides information on the chloroplast genome sequence, structural variation, codon bias and phylogeny of Catalpa, which will facilitate future research efforts.
Chloroplasts are important organelles for most higher plants and algae, allowing them to photosynthesize and convert light energy into chemical energy, and are responsible for the production of organic matter and energy storage (Nazareno et al., 2015). The chloroplast (cp) genome shows a variety of structures in cells. It is generally double-stranded and circular but may be linear, unbound to proteins, and accompanied by a complete set of replication, transcription and translation systems (Xin et al., 2020). The chloroplast genome is mainly composed of four independent structures: a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions (IRA/B) (Liu et al., 2020; Abdullah et al., 2021a; Lee et al., 2021). The LSC and SSC regions are separated by the IR regions (Daniell et al., 2016; Gu et al., 2017). Since the chloroplast genomes of Nicotiana tabacum (Ohyama et al., 1986) and Marchantia polymorpha (Shinozaki et al., 1986) were first obtained in 1986, as of 2021, a total of approximately 4,650 sequencing records of higher plants had been added to the NCBI database. The size of the genome is generally between 120 and 160 kb, and the GC content is usually 35–40% (Cheng et al., 2017). Compared with mitochondrial and nuclear genomes, plant chloroplast genomes are more conserved in terms of structure, gene number, and gene composition, and their evolution is relatively slow, intermediate to the evolutionary rates of nuclear genomes and mitochondrial genomes (Wu et al., 2011; Dong et al., 2013). Complete chloroplast genomes are widely used for phylogenetic analysis and species identification due to their lack of recombination, small size, and high copy number per cell (Li et al., 2012; Twyford and Ness 2017; Abdullah et al., 2020a; Bi et al., 2020). Because the chloroplast genome is small and its sequence and gene composition are conserved, it is highly suitable for analyzing the systematic evolution of complex plant groups (Jansen et al., 2008; Daniell et al., 2016; Cui et al., 2019; Abdullah et al., 2020b). Studies have shown that the chloroplast genome contains additional information that can improve phylogenetic inference (Cros et al., 1998; Tong et al., 2016; Yang et al., 2018). Comparing chloroplast genome sequences provides an opportunity to discover sequence variations and identify mutation hotspots. The mutation hotspots and simple sequence repeats (SSRs) obtained from a chloroplast genome sequence can be used as effective molecular markers for identifying species and inferring population inheritance patterns (Wu et al., 2012).
Catalpa Scop (Bignoniaceae), an intercontinental disjunct genus, consists of ten species, with two species in eastern North America (ENA), four in eastern Asia (EAS), and four in the West Indies (WI) (Li 1952; Paclt 1952). Catalpa species are mostly semievergreen or deciduous trees with opposite or whorled leaves. These trees are traditional high-quality precious timber tree species in China, known as the “kings of wood”. In addition, the leaves and roots of Catalpa species can also be used as medicines for stomach ailments, cough, and rheumatic pain. Therefore, the development and utilization of Catalpa species are economically important. However, some species obtained commercially or noncommercially are mistakenly regarded as Catalpa species (Olsen and Kirkbride 2017). Some genes, including the inner transcribed spacer of ribosomal DNA (nrDNA ITS) and the chloroplast ndhF gene, have shown that in Bignoniaceae, Catalpa Scop is closely related to Chilopsis D. Don (Li 2008). However, due to the limited numbers of DNA fragments and variant markers in Catalpa, its phylogenetic relationships remain unclear (Li 2008). Further research is urgently needed to clarify the relationships between Catalpa species and lay a foundation for cross-breeding and drought resistance mechanism analysis. Molecular systematics has become an important method for species identification. As a source of molecular markers with more genetic information than a single gene, the chloroplast genome has been widely used in species identification (Zhao et al., 2016; Yang et al., 2020). To date, research on the chloroplast genome of Catalpa species has been extremely limited. In this study, we sequenced the chloroplast genomes of six taxa within the genus Catalpa. The purpose of this research was to 1) compare the chloroplast genomes of Catalpa to understand the evolution of their structure, 2) to identify a highly variable area for species identification, and 3) to clarify the phylogenetic relationships of Catalpa. The results provide genetic background information for hybridization breeding and drought resistance mechanism analysis of Catalpa species.
Fresh leaves of six taxa of the genus Catalpa, namely, C. fargesii f. duclouxii (Guiding County, Guizhou Province, China), C. fargesii (Tianshui City, Gansu Province, China), C. bungei (Luoyang, Henan Province, China), C. ovata (Tianshui, Gansu Province, China), C. bungei (Jinsiqiu) (Luoyang, Henan Province, China) and C. fargesii f. duclouxii (Huangxinzimu) (Fuquan, Guizhou Province, China), were collected. Six complete chloroplast genome sequences were deposited in GenBank with accession numbers OL628864 to OL628869 (Table 1). The samples were stored in silica gel and transported to the laboratory for low-temperature preservation (−40°C). Specimens of six taxa of the genus Catalpa preserved at the Institute of Forestry, Chinese Academy of Forestry, Beijing, were also examined (Table 1). Total DNA was extracted following the method of Li et al. (2013) and purified by a Wizard DNA cleanup system (Promega, Madison, WI, United States). DNA quality was assessed by spectrophotometry, and integrity was evaluated using a 1% (w/v) agarose gel (Promega, Madison, WI, United States).
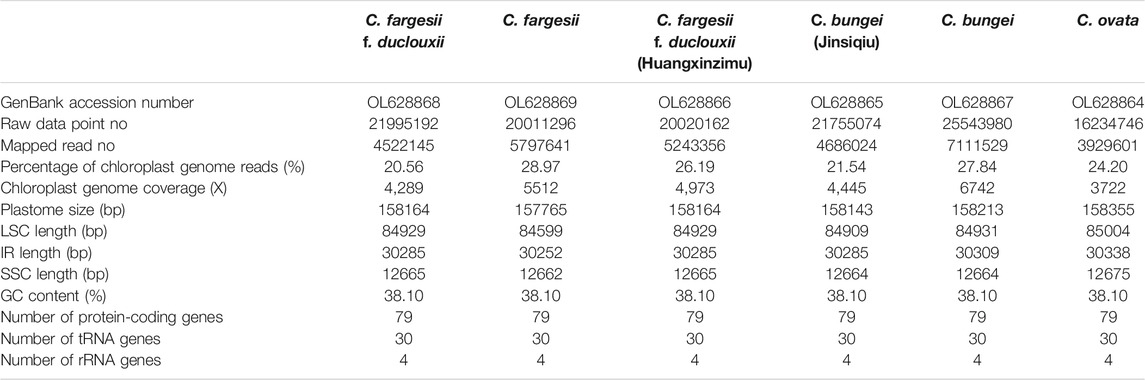
TABLE 1. Summaries of the complete chloroplast genomes of six taxa within the genus Catalpa.
Total DNA was fragmented into 350 bp fragments by ultrasound. A paired-end library was constructed by a NEBNext Ultra™ DNA library prep kit, and PE150 sequencing was performed on the Illumina HiSeq XTen platform. The NGS QC toolkit was used for quality control and to filter the low-quality reads. We used the obtained data for de novo assembly of the whole chloroplast genome with the GetOrganelle v1.7.5 pipeline using the following settings: F embplant_pt, -R 15, -K85 and 105. Using the published chloroplast genome of Tecomaria capensis (GenBank sequence acceptance number MG831880) of Bignoniaceae as a reference sequence, the Plann program (Huang and Cronk 2015) was used to annotate the chloroplast genes of Catalpa species. Some genes with unsuccessful or incorrect annotations were manually added in Sequin software. The structure map of the genome was first drawn using OrganellarGenomeDRAW-a (http://ogdraw.mpimp-golm.mpg.de/index.shtml) (Lohse et al., 2013) and then edited using Adobe Illustrator CS5. All chloroplast genome sequences were uploaded to the NCBI GenBank database for future reference.
GMATA (Wang and Wang 2016) software was used to analyze SSRs in the chloroplast genomes of the six taxa of the genus Catalpa with the parameters set as 1-10, 2-4, 3-4, 4-3, 5-3 and 6-3, that is, mononucleotide SSRs with a repeat unit of 1 and a repetition number ≥10, dinucleotide SSRs with a repeat unit of 2 and a repetition number ≥6, trinucleotide SSRs with a repeat unit of 3 and a repetition number ≥5, and tetranucleotide, pentanucleotide, and hexanucleotide SSRs with a repeat unit of 4, 5, and 6, respectively, and a repetition number ≥3 (Thiel et al., 2003). Two SSR markers separated by less than 100 bp were considered a composite microsatellite. The REPuter program (Kurtz et al., 2001) was used to find forward (F), palindromic (P), reverse (R) and complementary (C) oligonucleotide repeats with a minimum repeat size of 30 bp and a similarity of 90%. The REPuter program overestimated repeats, and redundant repeats were found in large repeats as well as in duplicated tRNAs.
The six assembled chloroplast genomes were compared with MAFFT (multiple alignments using fast Fourier transform) v7 software (Katoh and Standley 2013), and then the results were manually adjusted with MEGA7 software (Kumar et al., 2016; Abdullah et al., 2021b). MEGA7 was used to quantify the mutation sites and parsimony-informative sites in the chloroplast genomes of Catalpa. Taking the C. bungei sequence as the reference, the Shuffle-LAGAN model in the mVISTA program (http://genome.lbl.gov/vista/mvista/submit.shtml) was used to analyze the whole genome of Catalpa. First, we manually checked for small inversions and removed them from the alignment to avoid false results. The intergenic spacer regions and protein-coding regions were extracted from the alignment in Geneious R8.1 (Kearse et al., 2012) and visualized in DnaSP v.6 to determine the nucleotide diversity of each region (Rozas et al., 2017; Abdullah et al., 2021a).
All coding sequences (CDSs) were manually extracted from the chloroplast genomes. MEGA5 was used to analyze the codon usage frequency in each of the six Catalpa species (Kumar et al., 2008). Relative synonymous codon usage (RSCU) reflects whether a plastid gene is in a selected state, and codons with an RSCU value >1 are defined as high-frequency codons.
Thirty-three chloroplast genome sequences, including six from Catalpa and 17 from other species of Bignoniaceae, Lentibulariaceae and Lamiaceae from GenBank, were used for phylogenetic analysis. All chloroplast genome sequences were aligned using MAFFT v7, and regions with ambiguous alignment were trimmed by Gblocks 0.91b (Castresana 2000).
Phylogenetic analysis was carried out using the maximum likelihood (ML) and Bayesian inference (BI) methods. The optimal model was identified as TVM + F + I + G4 by ModelFinder based on the Bayesian information criterion (BIC) standard (recommended by the software) (Dong Zhang et al., 2020). ML calculations were performed using IQ-tree, with sampling repeated 1,000 times. BI of the phylogenies was implemented in MrBayes (Nguyen et al., 2015). Markov chain Monte Carlo (MCMC) analysis was run for 10,000,000 generations. Trees were sampled every 1,000 generations, and the initial 25% were discarded as burn-in. Finally, the average standard deviation of the split frequencies <0.01 was verified.
For the six taxa within the genus Catalpa, 2,435,211,900-3,831,597,000 bases of raw data with coverage ranging from 3722X-6742X were obtained (Table 1). The chloroplast genome of Catalpa has a typical structure, with a highly conserved, circular, double-stranded gene sequence mainly consisting of two IR regions separating two single-copy regions, namely, the LSC region and SSC region (Figure 1). The chloroplast genome length of the six taxa of the genus Catalpa ranged from 157,765 bp (C. fargesii) to 158,355 bp (C. ovata), the length of the LSC region ranged from 84,599 bp (C. fargesii) to 85,004 bp (C. ovata), and the length of the SSC region ranged from 12,662 bp (C. fargesii) to 12,675 bp (C. ovata) (Table 1). The length of the IR regions ranged from 30,252 bp (C. fargesii) to 30,338 bp (C. ovata). Therefore, the length variation of the LSC region was greater than that of the SSC and IR regions, and gene length variation was mainly caused by variation in the LSC region. The GC content of the genome is an important index for assessing the genetic relationships between species. The GC content of the chloroplast genomes of the six taxa within the genus Catalpa was 38.1%. There were almost no differences in the chloroplast genomes of the six taxa within the genus Catalpa.
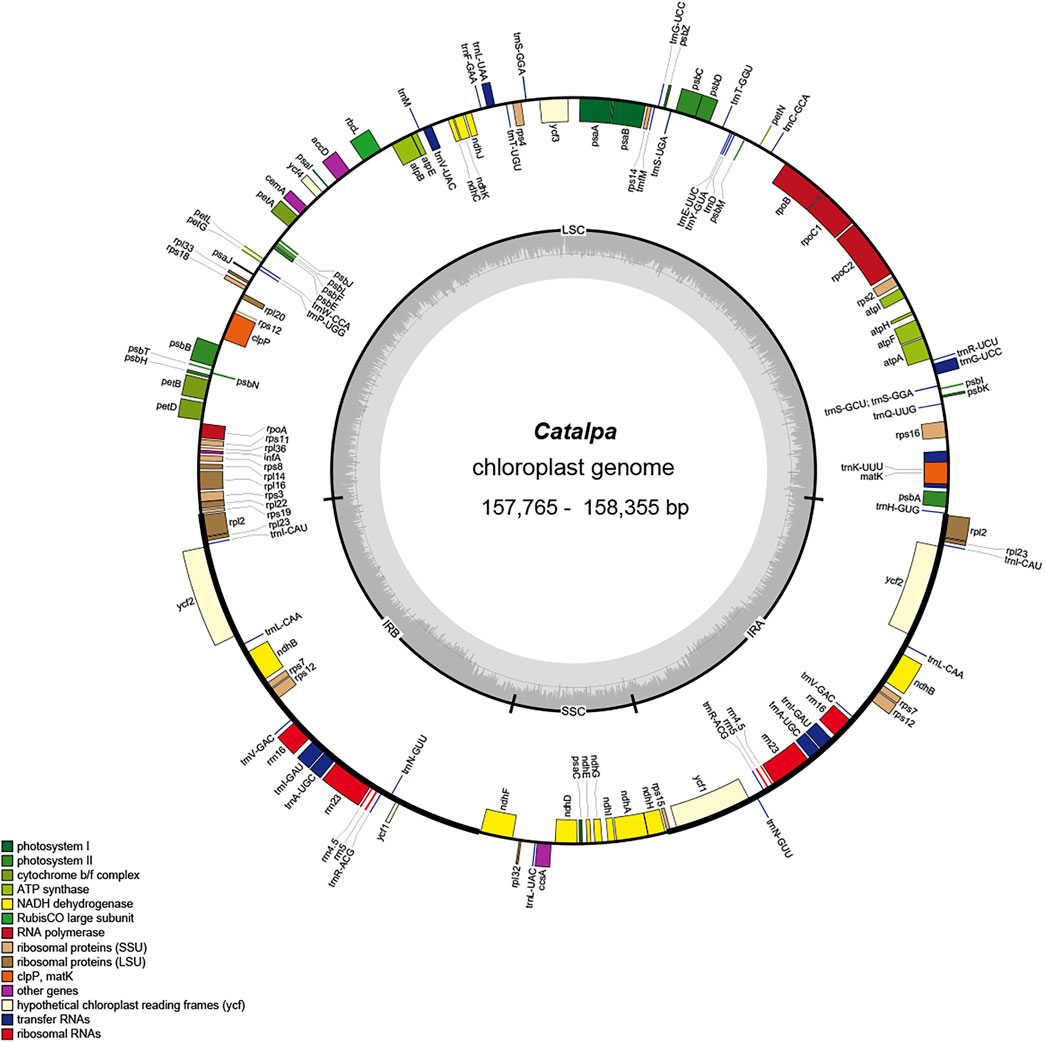
FIGURE 1. Gene map of the Catalpa chloroplast genome. Genes shown outside the outer circle are transcribed clockwise, and those insides are transcribed counterclockwise. Genes are color coded according to different functional groups. The darker gray in the inner circle indicates the GC content, and the lighter gray indicates the AT content. The inner circle also shows that the chloroplast genome contains two copies of inverted repeats (IRA and IRB), a large single-copy (LSC) region and a small single-copy (SSC) region. The map was constructed using OrganellarGenomeDRAW.
In the chloroplast genomes of the six taxa within the genus Catalpa, 113 genes were detected, 19 of which were located in the IR regions. The 113 genes included 79 protein-coding genes, 30 tRNA genes and four rRNA genes (rrn5, rrn4.5, rrn16, and rrn23), and all four rRNA genes were distributed in the IR regions, resulting in a much higher GC content in the IR regions than in the two single-copy regions (Figure 1; Table 2). The infA gene was a pseudogene in all species. According to their function, the detected genes can be divided into three categories. The first category included 47 genes related to photosynthesis, including Rubisco large subunit genes, genes for components in the photosynthetic electron transport chain and genes presumed to be NAD(P)H dehydrogenase subunits. The second category consisted of six genes involved in the biosynthesis of amino acids, fatty acids and other substances, as well as some genes with unknown functions. In the third group, most of the genes were tRNA genes, including RNA polymerase subunits, rRNA and ribosomal proteins and other products. These 60 genes were related to transcription and translation. Studies have shown that introns play an important role in gene expression regulation, and many introns can enhance the level of foreign gene expression at specific times and locations in plants, in turn controlling agronomic traits (Jiao et al., 2012). Fifteen of the 113 genes contained introns, 13 contained one intron, and ycf3 and clpP contained two introns. rps12 is a spliced gene with a 5′-terminal exon located in the LSC region and a 3′-terminal exon located in the IR regions.
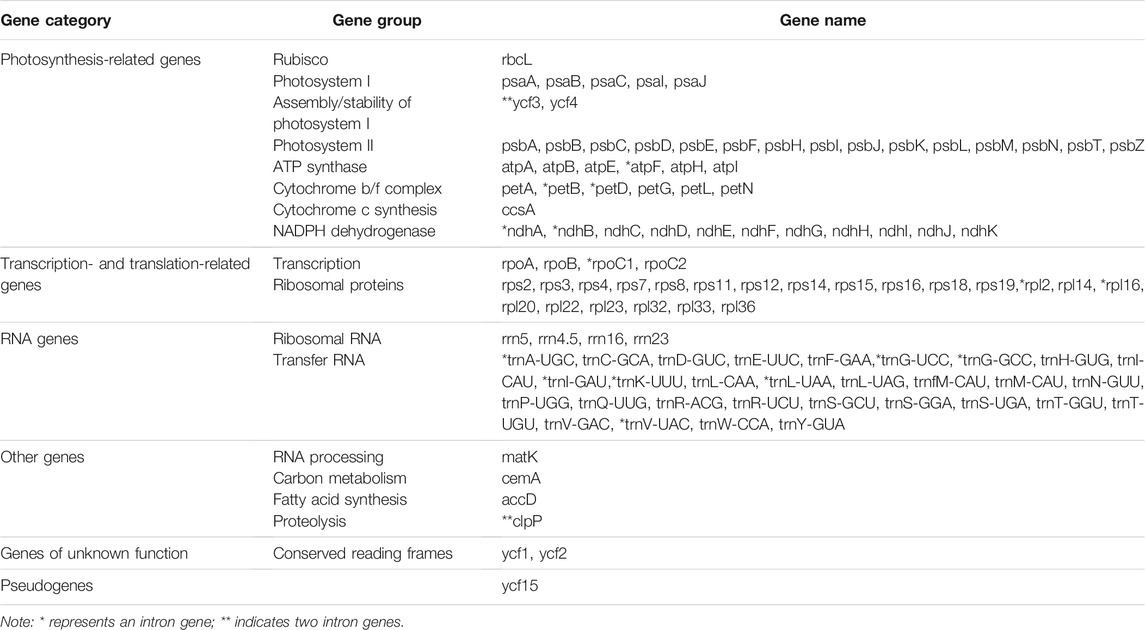
TABLE 2. Genes in the Catalpa chloroplast genomes.
SSRs, also known as microsatellites, are composed of repeating units with a length of 1-6 bp. In this study, a total of 248 SSRs were detected in the 6 chloroplast genomes of Catalpa. In terms of distribution, 197 SSRs were located in the LSC region (79.44%), 21 SSRs were located in the SSC region (8.47%), and 30 SSRs were located in the IR regions (12.1%). Therefore, the distribution of SSRs in the chloroplast genome of Catalpa is uneven (Figure 2A). The largest number of SSRs observed among the taxa within the genus Catalpa was 51, and the smallest was 43. The remaining four species (C. fargesii f. duclouxii, C. fargesii f. duclouxii (Huangxinzimu), C. bungei (Jinsiqiu), and C. bungei) had 46 SSR loci (Figure 2C). The chloroplast genomes of the six taxa within the genus Catalpa included mono-, di-, tetra-, and pentanucleotide SSRs (Figure 2D). Trinucleotide SSRs were observed in only one Catalpa species, and none of the six species contained hexanucleotide SSRs. Among the 248 SSR sites in the chloroplast genomes of Catalpa (Figure 2B), 219 sites (78.78%) were composed of A/T, and only seven sites (2.52%) contained G/C, indicating an SSR base composition preference for A/T. These findings are consistent with previous reports that SSRs are typically composed of polyadenine (PolyA) and polythymine (PolyT) repeats (Cheng et al., 2015; Shen et al., 2016). Tetranucleotides accounted for the largest percentage of SSRs (21.94%), followed by dinucleotides and pentanucleotides (both 5.04%). There were differences in the number and distribution of SSRs among the six species within the Genus Catalpa, which may be due to the deletion and mutation of gene sequences during the evolution of Catalpa. We also analyzed oligonucleotide repeats by REPuter and found four categories: palindromic (P), forward (F), reverse (R), and complementary (C). The abundance of the repeats varied among species based on the type of repeat. In the chloroplast genomes of C. ovata, C. bungei, and C. fargesii, REPuter revealed 49 repeats (F = 23, R = 26, P = 0, and C = 0), whereas in those of C. bungei (Jinsiqju), C. fargesii f. duclouxi (Huangxinzimu), and C. fargesii f. duclouxi, 49 repeats (F = 24, R = 25, P = 0, and C = 0) were detected (Figure 2E). Most of the repeats were between 35 bp to 39 bp and 40 bp to 44 bp long (Figure 2F).
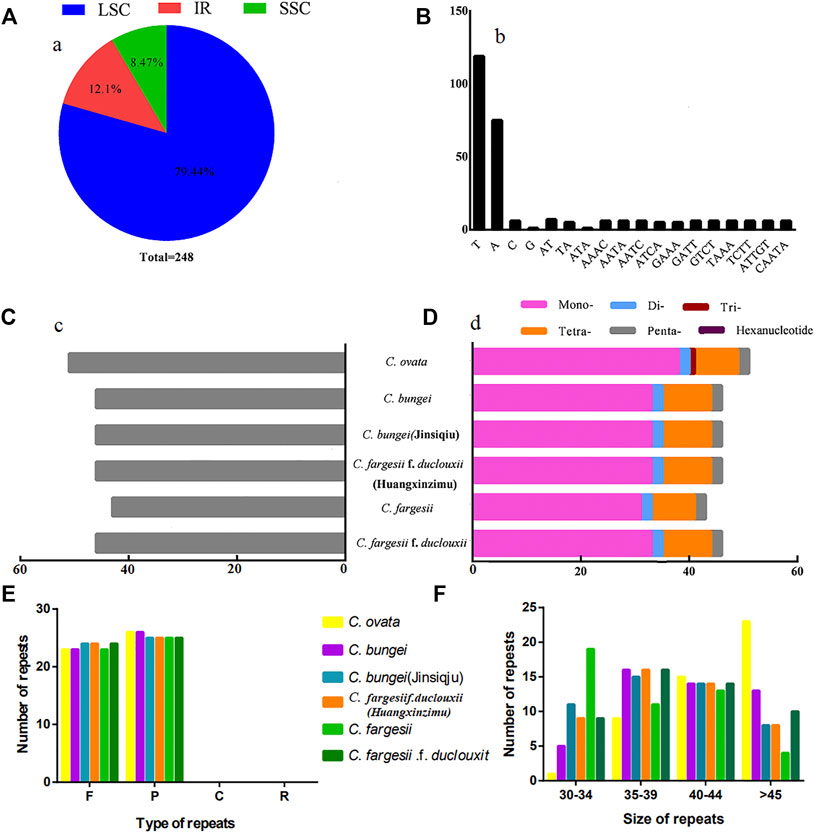
FIGURE 2. Microsatellite and oligonucleotide repeat analyses. (A) Frequency of identified SSRs in LSC, IR, and SSC regions. (B) Frequency of identified SSR motifs in different repeat class types. (C) Number of SSRs detected in six chloroplast genomes. (D) Number of SSR types detected in six chloroplast genomes. (E) Comparison of the various types of oligonucleotide repeats. (F) Comparison of repeats based on size.
The expansion and contraction of IR regions in the chloroplast genome are important evolutionary events in plants and relatively common phenomena, ultimately causing changes in the size and gene content of the chloroplast genome (Huang et al., 2014). To explore the potential expansion and contraction of IRs, the distributions of IR and SC border regions in the chloroplast genomes of six taxa within the genus Catalpa were compared. Genes with a boundary distribution of JLB, JSB, JSA and JLA included rps19, rpl2, rps15, ndHF, ndhH and trnH (Figure 3). The JLB, JSB, JSA and JLA boundaries showed very similar gene distributions. In C. bungei, C. fargesii, C. fargesii f. duclouxii, C. fargesii f. duclouxii (Huangxinzimu) and C. bungei (Jinsiqiu), rps19 was located in the LSC region, 3 bp from JLB, while rps19 was located 31 bp from JLB. The distributions of genes at the JLB, JSB, JSA, and JLA borders were very similar among the six taxa of the genus Catalpa. The length of the rps15 gene was 228 bp in the IR region of C. bungei, C. fargesii, C. fargesii f. duclouxii, C. fargesii Bur f. duclouxii (Huangxinzimu) and C. bungei (Jinsiqiu) but 231 bp in the IR region of C. ovata. Only the trnH gene of C. ovata was 8 bp away from JLA, and the trnH gene was located in the LSC region and 13 bp away from JLA.
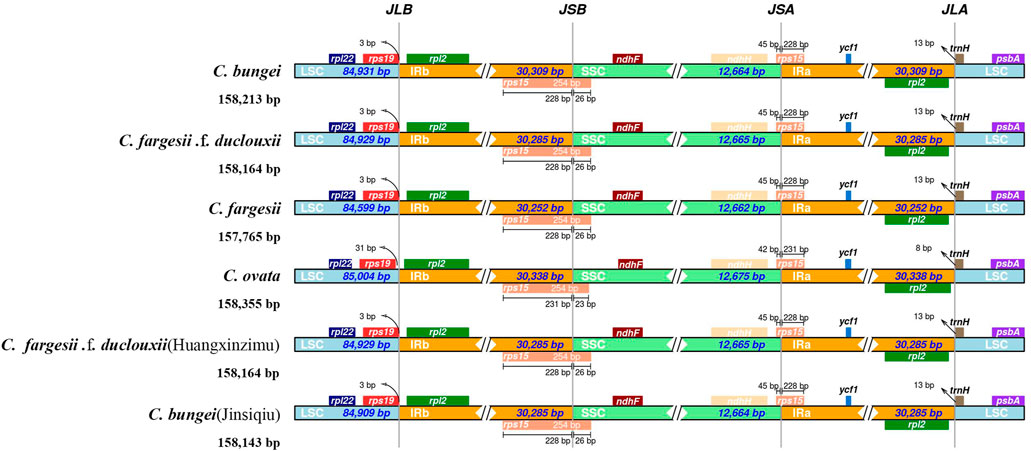
FIGURE 3. Comparisons of LSC, SSC and IR junctions among six taxa within the genus Catalpa.
Using the chloroplast genome of C. bungei as a reference, the mVISTA tool was applied to perform multiple sequence alignment, and the sequence similarity results were visualized to determine the degree of differentiation. The chloroplast genome sequences of the six taxa within the genus Catalpa were highly similar and conserved. The variation in the LSC and SSC regions was significantly greater than that in the IR regions, the rRNA gene was highly conserved with almost no variation, and the sequence variation in coding regions was lower than that in noncoding regions. The gene regions with large variations were accD, psaI-ycf4 and ycf1-trnN (Figure 4). The conservation degree of other genes was very high, with most of the genes being more than 90% conserved.
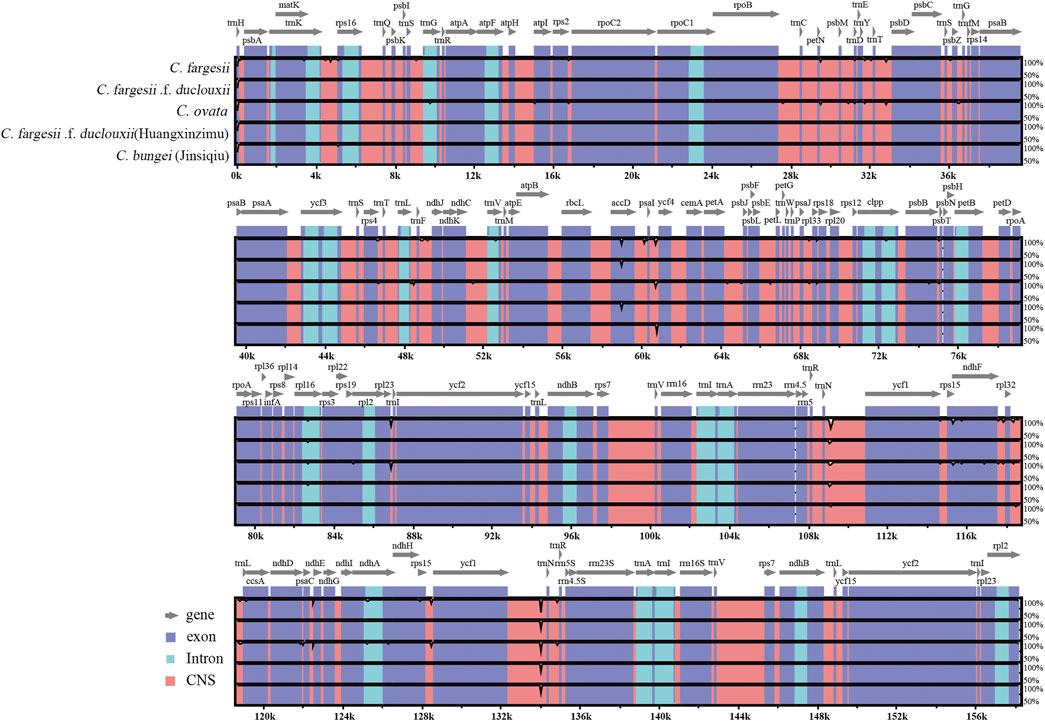
FIGURE 4. Visualization of the chloroplast genome alignments of six taxa within the genus Catalpa using C. bungei as a reference in mVISTA. The x-axis represents the position in the chloroplast genome. The sequence similarity of the aligned regions is shown as horizontal bars indicating the average percent identity within 50–100%.
After the chloroplast genomes were compared by software, three base mutations and nondimorphic mutations were excluded from subsequent analysis. The sequence length was 159,629 bp, with a total of 301 polymorphic sites (polymorphic, S), 36 parsimony-informative sites, and six haplotypes. The nucleotide diversity (Pi) of the sequences was 0.00059 (Table 3). The IR segment had the fewest mutation sites, with 18 polymorphic sites and one parsimony-informative site. The sequence had four haplotypes, and the sequence polymorphism of this region was only 0.00018. The patterns of SNPs, 60 transitions (Ts) and 149 transversions (Tv) were determined, and the overall Ts:Tv ratio was 0.403, indicating a preference for transversions (Figure 5). The high-frequency SNPs were C to T and G to A, and mutations from A to T and from T to A exhibited the lowest frequency.

TABLE 3. DNA polymorphisms identified in the six Catalpa plastomes.
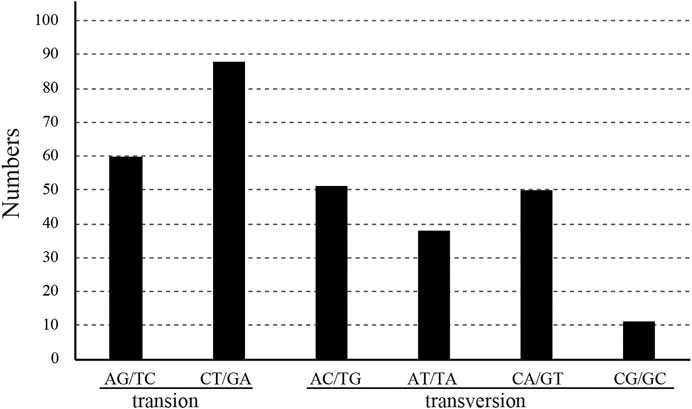
FIGURE 5. Pattern of single-nucleotide substitutions in the six Catalpa chloroplast genomes.
We recorded higher average polymorphism for intergenic spacer regions (0.0021) than for protein-coding sequences (0.0011). The polymorphisms of all regions are shown in Figure 6. We ignored loci <200 bp and selected 5 polymorphic regions with nucleotide diversity >0.003, of which three belonged to intergenic spacer regions and one to a protein-coding region (trnH-psbA, rps2-rpoC2, rpl22, ycf15-trnl-CAA, and rps15). rpl22 showed a nucleotide diversity of 0.00378 and contained four substitutions with 459 missing data points. A similar approach was used for ycf15-trnl-CAA, selecting a 364 bp region, which had a nucleotide diversity of 0.00458 and contained three substitutions. The selected regions may act as suitable and cost-effective markers (Table 4). These polymorphic loci might be helpful for phylogenetic inference and population genetic studies of Catalpa species.
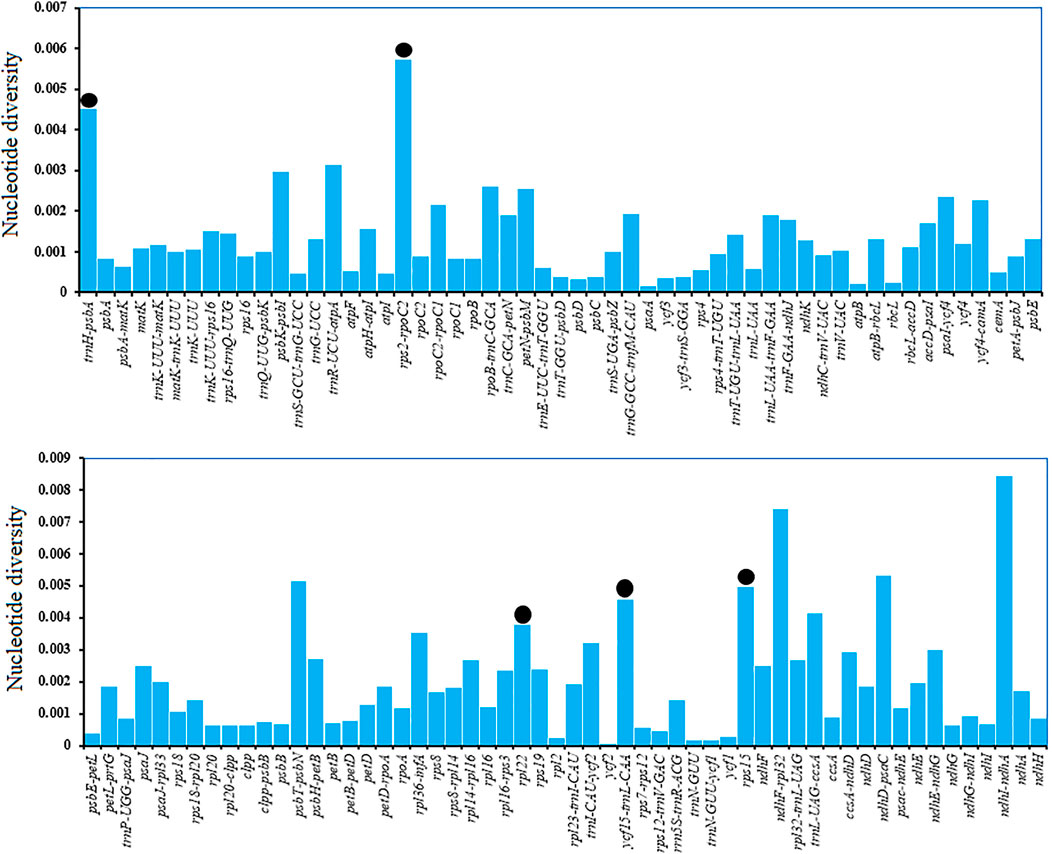
FIGURE 6. Extent of polymorphism in all plastid regions. Regions with no nucleotide diversity were excluded and are not shown here. The black circle indicates the five suitable polymorphic loci with a length >200 bp. The x-axis shows plastid regions, and the y-axis shows nucleotide diversity.

TABLE 4. Identified suitable polymorphic loci based on comparative plastome analysis of taxa within the genus Catalpa.
The base compositions and AT/GC contents of the six genomes of Catalpa were identical. Using CDSs of the chloroplast genome, we estimated the codon usage frequency of the six taxa of the genus Catalpa. The total number of codons detected in C. bungei and C. fargesii was 27,012 and 26,746, respectively, while the number in the other four species was 26,750 (Figure 7). The chloroplast genome of Catalpa encodes 20 amino acids at all codons. Leucine (Leu) was the most frequently used in the six taxa of the genus Catalpa, with a frequency ranging from 10.4% (2808) - 10.46% (2825), while cysteine (Cys) was the lowest in the six taxa of the genus, with a frequency reaching only 1.17% 315) - 1.18% (318). The RSCU values of the CDSs of Catalpa were calculated. Synonymous codon usage (RSCU value) refers to the relative probability of synonymous codons encoding corresponding amino acids for a specific codon, which eliminates the influence of amino acid composition on codon use. If there is no preference for the use of a codon, the RSCU value of the codon is equal to 1. When the RSCU value of a codon is greater than 1, it means that the codon is used more often than another, and vice versa. The results showed that the RSCU values of the six taxa included in the present study were similar. There were 30 codons with an RSCU value > 1, only one of which ended with G (UUG); the remaining 29 codons ended with A and T. The codons with an RSCU value < 1, except for UGA (stop codon) and CUA ending in A, ended in C or G. Therefore, the codon pairs ending with C and G in the Catalpa chloroplast genome have low bias, and they are nonpreferred codons. Due to usage frequency variation, the RSCU values of the chloroplast genome are a valuable form of evolutionary information resulting from mutation and selection that are essential in studying organismal evolution (Morton 2003).
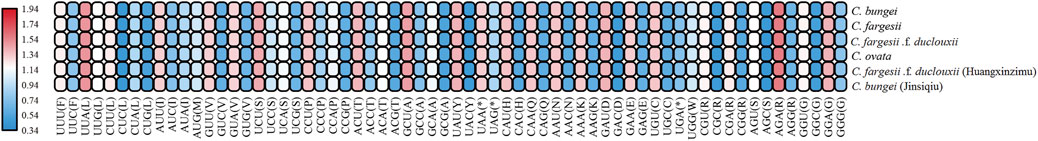
FIGURE 7. Analysis of codon bias in the chloroplast genome of Catalpa.
The phylogenetic trees constructed using the ML and BI methods for the whole genome sequences of Catalpa chloroplasts had highly similar topologies (Figure 8). Strong bootstrap support and high posterior probabilities were recorded at all branch nodes. The seven species of Catalpa formed a monophyletic clade, with C. ovata diverging before the other six taxa included in the present study, with a high support rate. All taxa of the Bignoniaceae family were grouped together. In terms of the interspecific relationships of Catalpa, in the phylogenetic tree constructed based on the whole chloroplast genome sequences, C. ovata was located at the base and formed sister branches with the other six species, and C. speciosa formed sister branches with the other five species (C. fargesii f. duclouxii, C. fargesii, C. fargesii f. duclouxii (Huangxinzimu), C. bungei (Jinsiqiu) and C. bungei). C. fargesii f. duclouxii (Huangxinzimu) and C. fargesii Bur f. duclouxii formed a subbranch and were sister to C. fargesii.
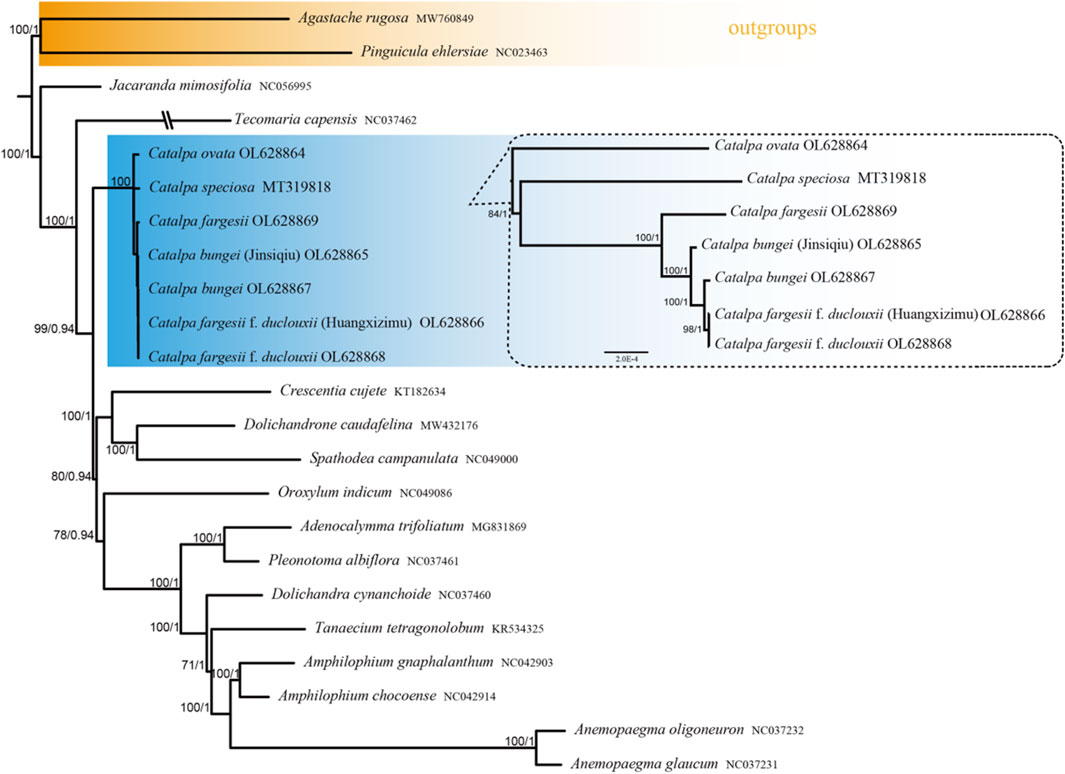
FIGURE 8. Phylogenetic tree constructed using the maximum likelihood (ML) and Bayesian inference (BI) methods based on the whole chloroplast genomes from 23 different species. The numbers above the branches represent the MI bootstrap values and Bl posterior probabilities.
In this study, the chloroplast genomes of some taxa of the genus Catalpa (C. fargesii f. duclouxii (Huangxinzimu) and C. bungei (Jinsiqiu)) were sequenced for the first time. The chloroplast genome size ranged from 157,765 bp (C. fargesii) to 158,355 bp (C. ovata), displaying six haplotypes. There were 113 genes in the chloroplast genome of all species, including 79 protein-coding genes, 30 tRNA genes and four rRNA genes. The two genes ycf15 and ycf68 were not annotated in this study, possibly because they are pseudogenes (Lu et al., 2016; Wang et al., 2020), consistent with results reported for other Catalpa species (Ma et al., 2020a; Ma et al., 2020b; Li et al., 2020; Wang et al., 2020). The accD, rpl32 and ycf2 genes are lost from the chloroplast genome in some cases (Jansen et al., 2008; Oliver et al., 2010; Dong et al., 2018), but these genes were present in the Catalpa chloroplast genome. The overall structure of the chloroplast genome of Catalpa is relatively conserved, and no major gene deletions or genome rearrangements were found. The total GC content was highly consistent among species, while genome size differed slightly but not significantly. The mVISTA results and nucleotide diversity tests revealed high degrees of similarity between the chloroplast genomes, implying that the chloroplast genomes of Catalpa are less diverged than those of other species (Li et al., 2018).
Variation in genome structure is another form of information that helps reveal the genetic diversity of species or aspects of their population biology or evolution. The most common SSRs in the chloroplast genome of C. ovata were mononucleotides mainly composed of A or T and rarely G or C. Microsatellites are very important for the study of population genetics. There were significantly fewer di-, tetra-, tri-, and pentanucleotide motif repeats and no hexanucleotide repeats in the six studied species, similar to the results of Rono et al. (Rono et al., 2020). The codon is crucial to the correct expression of genetic information. In general, the start codon sequences of chloroplast genomic DNA are ATG, ATT and ATA. There were two unique patterns in RSCU and usage frequency values based on six haplotypes of protein-coding genes. First, in addition to the stop codons, all codons showed a preference for ending in A or T, but the low-frequency codons were biased toward ending in C or G. Second, the two stop codons (UAA and UGG) showed no bias, consistent with the findings of previous studies (Rono et al., 2020; Wen et al., 2021). Overall, apart from codon usage, the SNPs and SSRs of the Catalpa chloroplast genomes were different and can be used as excellent resources for evaluating population genetic diversity. The chloroplast genomes of the six taxa of the genus Catalpa showed high genetic diversity.
There are 10 species of Catalpa worldwide, with several varieties. However, due to the low genetic differentiation of Catalpa, the systematic relationships among these taxa are not clear (Jia et al., 2012). The pollen morphology of C. bungei showed that some morphological characteristics of C. fargesii f. duclouxii are the same as those of C. fargesii but different from those of C. bungei, which is differentiated by its morphology. The use of several chloroplast markers, such as ndhF and nuclear ribosomal DNA, for phylogenetic reconstruction is sufficient to draw firm conclusions about the interspecies relationships within Catalpa (Li 2008). Therefore, sampling of additional genetic features is expected to improve phylogenetic resolution. The large-scale application of Illumina HiSeq technology has improved the ability to sequence entire chloroplast genomes so that these genome sequences can be used to analyze the close relationships between species (Sheng et al., 2021; Tian et al., 2021).
In this study, we used plastome sequences to assess the phylogenetic relationships within Catalpa. The results revealed deep phylogenetic relationships in this genus. Dode (Dode 1907) described samples of C. fargesii f. duclouxii collected from Yunnan, China, and found that the collected samples differed from C. fargesii in that the undersurface of leaves and petioles were hairless. Rehder (Rehder 1913) considered C. fargesii f. duclouxii to be a variety of C. fargesii. Gilmour (Gilmour 1936) further elaborated on this view: C. fargesii f. duclouxii is a hairless variety of C. fargesii, with C. fargesii being more closely related than C. bungei. Chloroplast genome ndhF and ribosomal DNA internal transcribed spacer ITS (nrDNA ITS) sequences were used to study the interspecific relationships of Catalpa, and phylogenetic trees constructed with ITS and chloroplast sequences showed that C. fargesii f. duclouxii formed its own branch and formed a sister branch with C. bungei and C. fargesii (Li 2008). The results of this study show that C. fargesii f. duclouxii and C. bungei are more closely related, C. fargesii f. duclouxii and C. fargesii f. duclouxii (Huangxinzimu) form a branch, and the branches formed with C. bungei and C. fargesii are sister branches. The results of this study do not support the conclusion that C. fargesii Bur f. duclouxii is a variant of C. fargesii as proposed by Rehder (1913) and Gilmour (1936). It is suggested that C. fargesii f. duclouxii be treated as a species independent of C. bungei and C. fargesii. This study also showed that all the taxa of Bignoniaceae clustered into one group, and similar family groups formed sister branches. Catalpa has sufficient genetic information, and Tecomaria capensis (NC 037462) is closely related to Catalpa, consistent with the results of previous studies (Gilmour 1936; Li et al., 2020). The results of this study provide strong evidence for elucidating the evolutionary history of these species and deeply analyzing the evolutionary events of Catalpa and even Bignoniaceae. The further development of sequencing technology will help fully reveal the general characteristics and patterns of variation of the chloroplast genome and provide a foundation for resolving the differences between morphological and genetic classification and for obtaining an in-depth understanding of plant evolution (Xu et al., 2021). However, due to the limited number of published chloroplast genomes of taxa within the genus Catalpa, there are still many difficulties in phylogenetic studies of this group. In the future, more data will be needed to explore their phylogenetic relationships.
Not all genes are phylogenetically useful in resolving taxonomic discrepancies. Oligonucleotide repeats exist widely in the plastome (Abdullah et al., 2021a). Mono-nucleotide, Palindromic, and forward repeats were the most common repeated sequences (Meng et al., 2019). Oligonucleotide repeats are also reported among the mutational events in chloroplast genomes (Abdullah, et al., 2020a). They consist of small repeats that exist in duplicate form (Kurtz et al., 2001) and mostly reported in size from 14 bp to 50 bp in chloroplast genome, unlike simple sequence repeats, which are one to six nucleotide tandem repeat units (Henriquez et al., 2014; Menezes et al., 2018; Abdullah et al., 2020a; Shahzadi et al., 2020). The results of Oligonucleotide repeats in this study are completely consistent with those mentioned above. In the taxonomy of the Catalpa, ndhF and the nrDNA ITS region can be discussed lower level relationships of plant groups (Baldwin et al., 1995; Soltis et al., 1998; Li 2008). However, the discriminatory power of these markers in Catalpa molecular phylogenetic investigations or DNA barcoding is deficient (Li 2008). Therefore, Chloroplast genome sequences provide an opportunity to elucidate patterns of genome evolution and provide valuable genetic resources for further research. Mutation events are not generally randomly distributed in the chloroplast genome but are concentrated in certain areas, forming “hotspots” (Dong et al., 2012; Wang et al., 2021). Comparing chloroplast genome sequences is an effective strategy for identifying mutation hotspots, and these highly variable regions can be used as DNA barcodes to distinguish species within specific taxa (Kuang et al., 2011; Abdullah et al., 2020a) and germplasm resources (Zhou et al., 2018; Ge et al., 2019). On the basis of the current study results, specifically, the results on nucleotide diversity among six Catalpa species or varieties, we suggest using a set of five divergent regions (≥200 bp) to solve taxonomic discrepancies and provide barcodes for the genus Catalpa. Regions of the plastome showed different polymorphisms, and certain regions were more predisposed to mutations. In this study, we identified five hypervariable regions, namely, trnH-psbA, rps2-rpoC2, rpl22, ycf15-trnl-CAA and rps15. These five regions had nucleotide diversity values of 0.00574 to 0.00378 from highest to lowest. The chloroplast genome sequences of the six taxa within the genus Catalpa were highly similar and conserved, and the noncoding regions had more variation than the coding regions, consistent with the results of previous studies (Perry and Wolfe 2002; Xiao-Feng Zhang et al., 2020). These variable regions can also be used to evaluate the phylogenetic relationships and interspecific differences of Catalpa (Yildirim et al., 2013). In this study, chloroplast genome data provided effective markers for inferring the phylogenetic relationships within Catalpa.
In this study, the chloroplast genomes of six taxa within the genus Catalpa were sequenced and assembled, providing valuable genetic resources for taxa within the genus Catalpa. Through phylogenetic analysis of the whole chloroplast genome, the relationships within this genus were clarified for the first time. Moreover, comparative analysis of the chloroplast genomes revealed variable regions that can be used as specific DNA barcodes. The genetic resources obtained herein will contribute to studies on the population genetics, species identification, phylogenetics and conservation biology of catalpa. In the future, we will expand genome sampling, including nuclear genomes, and comprehensively assess and discuss the phylogeny and evolutionary relationships of taxa within the genus Catalpa.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: In this study has been submitted to National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) and obtained the GenBank accession number (OL628864, OL628865,OL628866, OL628867, OL628868, OL628869).
WM and FL conceived and designed the experiments; FL analyzed the data; FL performed the experiments; WM, YL, JW, PX, JZ, KZ, MZ, and HY summarized the data; FL wrote the manuscript; PX, JW, WM, and FL revised the manuscript.
This study was financially supported by the National Key R&D Program of China (2021YFD2200301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank AJE (https://www.aje.cn/) for its linguistic assistance during the preparation of this manuscript. We also thank Yiheng Wang for helping upload data and collate pictures.
Abdullah, Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., et al. (2020a). Chloroplast Genome of Hibiscus Rosa-Sinensis (Malvaceae): Comparative Analyses and Identification of Mutational Hotspots. Genomics 112, 581–591. doi:10.1016/j.ygeno.2019.04.010
Abdullah, Mehmood, F., Shahzadi, I., Ali, Z., Islam, M., Naeem, M., et al. (2020b). Correlations Among Oligonucleotide Repeats, Nucleotide Substitutions, and Insertion-Deletion Mutations in Chloroplast Genomes of Plant Family Malvaceae. J. Syst. Evol. 59, 388–402. doi:10.1111/jse.12585
Abdullah, Mehmood, F., Rahim, A., Heidari, P., Ahmed, I., and Poczai., P. (2021a). Comparative Plastome Analysis of Blumea , with Implications for Genome Evolution and Phylogeny of Asteroideae. Ecol. Evol. 11, 7810–7826. doi:10.1002/ece3.7614
Abdullah, Henriquez, C. L., Mehmood, F., Hayat, A., Sammad, A., Waseem, S., et al. (2021b). Chloroplast Genome Evolution in the Dracunculus Clade (Aroideae, Araceae). Genomics 113, 183–192. doi:10.1016/j.ygeno.2020.12.016
Baldwin, B. G., Sanderson, M. J., Porter, J. M., Wojciechowski, M. F., Campbell, C. S., and Donoghue, M. J. (1995). The ITS Region of Nuclear Ribosomal DNA: a Valuable Source of Evidence on Angiosperm Phylogeny. Ann. Mo. Bot. Garden 82, 247–277. doi:10.2307/2399880
Bi, S. F., Wen, X., Pan, Y. H., Cai, H. J., Zhong, H., and Wang, A. K. (2020). Application and Research Progress of Chloroplast DNA Barcoding in Forest Trees. Mol. Plant Breed. 18, 5444–5452.
Castresana, J. (2000). Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 17, 540–552. doi:10.1093/oxfordjournals.molbev.a026334
Cheng, B.-b., Zheng, Y.-q., and Sun, Q.-w. (2015). Genetic Diversity and Population Structure of Taxus Cuspidata in the Changbai Mountains Assessed by Chloroplast DNA Sequences and Microsatellite Markers. Biochem. Syst. Ecol. 63, 157–164. doi:10.1016/j.bse.2015.10.009
Cheng, H., Li, J., Zhang, H., Cai, B., Gao, Z., Qiao, Y., et al. (2017). The Complete Chloroplast Genome Sequence of Strawberry (Fragaria × ananassaDuch.) and Comparison with Related Species of Rosaceae. Peerj 5, e3919. doi:10.7717/peerj.3919
Cros, J., Combes, M. C., Trouslot, P., Anthony, F., Hamon, S., Charrier, A., et al. (1998). Phylogenetic Analysis of Chloroplast DNA Variation inCoffeaL. Mol. Phylogenet. Evol. 9, 109–117. doi:10.1006/mpev.1997.0453
Cui, Y., Chen, X., Nie, L., Sun, W., Hu, H., Lin, Y., et al. (2019). Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species. Ijms 20, 4040. doi:10.3390/ijms20164040
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast Genomes: Diversity, Evolution, and Applications in Genetic Engineering. Genome Biol. 17, 134–163. doi:10.1186/s13059-016-1004-2
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly Variable Chloroplast Markers for Evaluating Plant Phylogeny at Low Taxonomic Levels and for DNA Barcoding. Plos One 7, e35071–9. doi:10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., and Zhou, S. (2013). Sequencing Angiosperm Plastid Genomes Made Easy: a Complete Set of Universal Primers and a Case Study on the Phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi:10.1093/gbe/evt063
Dong, W.-L., Wang, R.-N., Zhang, N.-Y., Fan, W.-B., Fang, M.-F., and Li, Z.-H. (2018). Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Ijms 19, 716. doi:10.3390/ijms19030716
Ge, Y., Dong, X., Wu, B., Wang, N., Chen, D., Chen, H., et al. (2019). Evolutionary Analysis of Six Chloroplast Genomes from Three Persea Americana Ecological Races: Insights into Sequence Divergences and Phylogenetic Relationships. Plos One 14, e0221827. doi:10.1371/journal.pone.0221827
Gu, C., Tembrock, L. R., Zhang, D., and Wu, Z. (2017). Characterize the Complete Chloroplast Genome of Lagerstroemia Floribunda (Lythraceae), a Narrow Endemic Crape myrtle Native to Southeast Asia. Conservation Genet. Resour. 9, 91–94. doi:10.1007/s12686-016-0628-6
Henriquez, C. L., Arias, T., Pires, J. C., Croat, T. B., and Schaal, B. A. (2014). Phylogenomics of the Plant Family Araceae. Mol. Phylogenet. Evol. 75, 91–102. doi:10.1016/j.ympev.2014.02.017
Huang, D. I., and Cronk, Q. C. B. (2015). Plann: A Command-Line Application for Annotating Plastome Sequences. Appl. Plant Sci. 3, 1500026. doi:10.3732/apps.1500026
Huang, H., Shi, C., Liu, Y., Mao, S.-Y., and Gao, L.-Z. (2014). Thirteen Camelliachloroplast Genome Sequences Determined by High-Throughput Sequencing: Genome Structure and Phylogenetic Relationships. BMC Evol. Biol. 14, 151. doi:10.1186/1471-2148-14-151
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2008). Analysis of 81 Genes from 64 Plastid Genomes Resolves Relationships in Angiosperms and Identifies Genome-Scale Evolutionary Patterns. Proc. Natl. Acad. Sci. U S A. 104, 19369–19374. doi:10.1073/pnas.0709121104
Jia, J. W., Ma, W. J., Wang, J. H., Zhang, J. F., Zhang, S. G., Zhang, J. G., et al. (2012). Pollen Morphology of Several Species in Catalpa and its Taxonomic Significance. Entia Silvae Sin. 48, 182–185. doi:10.1007/s11783-011-0280-z
Jiao, Y., Jia, H.-m., Li, X.-w., Chai, M.-l., Jia, H.-j., Chen, Z., et al. (2012). Development of Simple Sequence Repeat (SSR) Markers from a Genome Survey of Chinese Bayberry (Myrica Rubra). BMC Genomics 13, 201. doi:10.1186/1471-2164-13-201
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Kuang, D.-Y., Wu, H., Wang, Y.-L., Gao, L.-M., Zhang, S.-Z., and Lu, L. (2011). Complete Chloroplast Genome Sequence of Magnolia Kwangsiensis (Magnoliaceae): Implication for DNA Barcoding and Population Genetics. Genome 54, 663–673. doi:10.1139/g11-026
Kumar, S., Nei, M., Dudley, J., and Tamura, K. (2008). MEGA: a Biologist-Centric Software for Evolutionary Analysis of DNA and Protein Sequences. Brief. Bioinform. 9, 299–306. doi:10.1093/bib/bbn017
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 29, 4633–4642. doi:10.1093/nar/29.22.4633
Lee, C., Choi, I.-S., Cardoso, D., de Lima, H. C., de Queiroz, L. P., Wojciechowski, M. F., et al. (2021). The Chicken or the Egg? Plastome Evolution and a Novel Loss of the Inverted Repeat in Papilionoid Legumes. Plant J.. doi:10.1101/2021.02.04.429812
Li, X. W., Hu, Z. G., Lin, X. H., Li, Q., Gao, H. H., Luo, G. A., et al. (2012). High-throughput Pyrosequencing of the Complete Chloroplast Genome of Magnolia Officinalis and its Application in Species Identification. Yao Xue Xue Bao 47, 124–130.
Li, J. L., Wang, S., Yu, J., Wang, L., and Zhou, S. L. (2013). A Modified CTAB Protocol for Plant DNA Extraction. Chin. Bull. Bot. 48, 72–78. doi:10.3724/SP.J.1259.2013.00072
Li, W., Liu, Y., Yang, Y., Xie, X., Lu, Y., Yang, Z., et al. (2018). Interspecific Chloroplast Genome Sequence Diversity and Genomic Resources in Diospyros. BMC Plant Biol. 18, 210. doi:10.1186/s12870-018-1421-3
Li, W.-Q., Lu, Y.-Z., Xie, X.-M., Han, Y., Wang, N., Sun, T., et al. (2020). The Complete Chloroplast Genome of Catalpa 'Bairihua', a Hybrid Variety with Multi Season Flowering. Mitochondrial DNA B 5, 2760–2762. doi:10.1080/23802359.2020.1788445
Li, H.-L. (1952). Floristic Relationships between Eastern Asia and Eastern North America. Trans. Am. Philos. Soc. 42, 371–429. doi:10.2307/1005654
Li, J. H. (2008). Phylogeny of Catalpa (Bignoniaceae) Inferred from Sequences of Chloroplast ndhF and Nuclear Ribosomal DNA. J. Syst. Evol. 46, 341–348. doi:10.1016/j.ympev.2007.10.009
Liu, Q., Li, X., Li, M., Xu, W., Schwarzacher, T., and Heslop-Harrison, J. S. (2020). Comparative Chloroplast Genome Analyses of Avena: Insights into Evolutionary Dynamics and Phylogeny. BMC Plant Biol. 20, 1–20. doi:10.1186/s12870-020-02621-y
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW-a Suite of Tools for Generating Physical Maps of Plastid and Mitochondrial Genomes and Visualizing Expression Data Sets. Nucleic Acids Res. 41, W575–W581. doi:10.1093/nar/gkt289
Lu, R.-S., Li, P., and Qiu, Y.-X. (2016). The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 7, 2054. doi:10.3389/fpls.2016.02054
Ma, Q.-g., Zhang, J.-g., and Zhang, J.-p. (2020a). The Complete Chloroplast Genome of Catalpa Ovata G. Don. (Bignoniaceae). Mitochondrial DNA Part B 5, 1800–1801. doi:10.1080/23802359.2020.1750979
Ma, Q.-g., Zhang, J.-g., and Zhang, J.-p. (2020b). The Complete Chloroplast Genome of Catalpa Speciosa (Warder) Engelmann (Bignoniaceae). Mitochondrial DNA Part B 5, 2089–2090. doi:10.1080/23802359.2020.1765213
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast Genomes of Byrsonima Species (Malpighiaceae): Comparative Analysis and Screening of High Divergence Sequences. Sci. Rep. 8, 2210–2212. doi:10.1038/s41598-018-20189-4
Meng, D., Xiaomei, Z., Wenzhen, K., Xu, Z., and Chiang, T. Y. (2019). Detecting Useful Genetic Markers and Reconstructing the Phylogeny of an Important Medicinal Resource Plant, Artemisia Selengensis, Based on Chloroplast Genomics. Plos One 14, e0211340. doi:10.1371/journal.pone.0211340
Morton, B. R. (2003). The Role of Context-dependent Mutations in Generating Compositional and Codon Usage Bias in Grass Chloroplast DNA. J. Mol. Evol. 56, 616–629. doi:10.1007/s00239-002-2430-1
Nazareno, A. G., Carlsen, M., and Lohmann, L. G. (2015). Complete Chloroplast Genome of Tanaecium Tetragonolobum: The First Bignoniaceae Plastome. Plos One 10, e0129930. doi:10.1371/journal.pone.0129930
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., et al. (1986). Chloroplast Gene Organization Deduced from Complete Sequence of Liverwort Marchantia Polymorpha Chloroplast DNA. Nature 322, 572–574. doi:10.1038/322572a0
Oliver, M. J., Murdock, A. G., Mishler, B. D., Kuehl, J. V., Boore, J. L., Mandoli, D. F., et al. (2010). Chloroplast Genome Sequence of the moss Tortula Ruralis: Gene Content, Polymorphism, and Structural Arrangement Relative to Other green Plant Chloroplast Genomes. BMC Genomics 11, 143. doi:10.1186/1471-2164-11-143
Olsen, R. T., and Kirkbride, J. H. (2017). Taxonomic Revision of the Genus Catalpa (Bignoniaceae). Brittonia 69, 387–421. doi:10.1007/s12228-017-9471-7
Perry, A. S., and Wolfe, K. H. (2002). Nucleotide Substitution Rates in Legume Chloroplast DNA Depend on the Presence of the Inverted Repeat. J. Mol. Evol. 55, 501–508. doi:10.1007/s00239-002-2333-y
Rehder, A. (1913). “Bignoniaceae,” in Sargente Sed, Plantae Wilsonianae I (Cambridge: Harvard University Press).
Rono, P. C., Dong, X., Yang, J.-X., Mutie, F. M., Oulo, M. A., Malombe, I., et al. (2020). Initial Complete Chloroplast Genomes of Alchemilla (Rosaceae): Comparative Analysis and Phylogenetic Relationships. Front. Genet. 11, 560368. doi:10.3389/fgene.2020.560368
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 34, 3299–3302. doi:10.1093/molbev/msx248
Shahzadi, I., AbdullahMehmood, F., Mehmood, F., Ali, Z., Ahmed, I., and Mirza, B. (2020). Chloroplast Genome Sequences of Artemisia Maritima and Artemisia Absinthium: Comparative Analyses, Mutational Hotspots in Genus Artemisia and Phylogeny in Family Asteraceae. Genomics 112, 1454–1463. doi:10.1016/j.ygeno.2019.08.016
Shen, L., Guan, Q., Amin, A., Zhu, W., Li, M., Li, X., et al. (2016). Complete Plastid Genome of Eriobotrya Japonica (Thunb.) Lindl and Comparative Analysis in Rosaceae. Springerplus 5, 2036. doi:10.1186/s40064-016-3702-3
Sheng, J., Yan, M., Wang, J., Zhao, L., Zhou, F., Hu, Z., et al. (2021). The Complete Chloroplast Genome Sequences of Five Miscanthus Species, and Comparative Analyses with Other Grass Plastomes. Ind. Crops Prod. 162, 113248. doi:10.1016/j.indcrop.2021.113248
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The Complete Nucleotide Sequence of the Tobacco Chloroplast Genome: its Gene Organization and Expression. EMBO J. 5, 2043–2049. doi:10.1002/j.1460-2075.1986.tb04464.x
Soltis, D. E., Soltis, P. S., and Doyle, J. J. (1998). Molecular Systematics of Plants II: DNA Sequencing. Norwell, MA: Kluwer Academic Publishers. doi:10.1007/978-1-4615-5419-611
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum Vulgare L.). Theor. Appl. Genet. 106, 411–422. doi:10.1007/s00122-002-1031-0
Tian, S., Lu, P., Zhang, Z., Wu, J. Q., Zhang, H., and Shen, H. (2021). Chloroplast Genome Sequence of Chongming lima Bean (Phaseolus Lunatus l.) and Comparative Analyses with Other Legume Chloroplast Genomes. BMC Genomics 22, 194. doi:10.1186/s12864-021-07467-8
Tong, W., Kim, T.-S., and Park, Y.-J. (2016). Rice Chloroplast Genome Variation Architecture and Phylogenetic Dissection in Diverse Oryza Species Assessed by Whole-Genome Resequencing. Rice 9, 57. doi:10.1186/s12284-016-0129-y
Twyford, A. D., and Ness, R. W. (2017). Strategies for Complete Plastid Genome Sequencing. Mol. Ecol. Resour. 17, 858–868. doi:10.1111/1755-0998.12626
Wang, X., and Wang, L. (2016). GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front. Plant Sci. 7, 1350. doi:10.3389/fpls.2016.01350
Wang, Y., Zhao, Y., Wang, K., Wang, L., Feng, Y., Qi, L., et al. (2020). The Complete Chloroplast Genome of Catalpa Ovata (Bignoniaceae): an Important Ornamental and Medicinal Plant. Mitochondrial DNA Part B 5, 1675–1676. doi:10.1080/23802359.2020.1742226
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., and Guo, L. (2021). Chloroplast Genome Variation and Phylogenetic Relationships of Atractylodes Species. BMC Genomics 22, 103. doi:10.1186/s12864-021-07394-8
Wen, F., Wu, X., Li, T., Jia, M., Liu, X., and Liao, L. (2021). The Complete Chloroplast Genome of Stauntonia Chinensis and Compared Analysis Revealed Adaptive Evolution of Subfamily Lardizabaloideae Species in China. BMC Genomics 22, 161. doi:10.1186/s12864-021-07484-7
Wu, C.-S., Wang, Y.-N., Hsu, C.-Y., Lin, C.-P., and Chaw, S.-M. (2011). Loss of Different Inverted Repeat Copies from the Chloroplast Genomes of Pinaceae and Cupressophytes and Influence of Heterotachy on the Evaluation of Gymnosperm Phylogeny. Genome Biol. Evol. 3, 1284–1295. doi:10.1093/gbe/evr095
Wu, Y., Zhou, H., and Li, Y. (2012). Evolution Analysis of Sugarcane Chloroplast DNA Microsatellites in Poaceae. Genomics Appl. Biol. 31, 624–633. doi:10.1089/dna.2012.1642
Xin, Y. X., Dong, Z. H., Qu, S. H., Liu, C., Ye, P., and Xin, P. Y. (2020). Analysis on Codon Usage Bias of Chloroplast Genome in Pyrus Betulifolia Bge. J. Hebei Agric. Univ. 43, 51–59. doi:10.13320/j.cnki.jauh.2020.0112
Xu, J., Liu, C., Song, Y., and Li, M. (2021). Comparative Analysis of the Chloroplast Genome for Four Pennisetum Species: Molecular Structure and Phylogenetic Relationships. Front. Genet. 12, 687844. doi:10.3389/fgene.2021.687844
Yang, Z., Zhao, T., Ma, Q., Liang, L., and Wang, G. (2018). Comparative Genomics and Phylogenetic Analysis Revealed the Chloroplast Genome Variation and Interspecific Relationships of Corylus (Betulaceae) Species. Front. Plant Sci. 9, 927. doi:10.3389/fpls.2018.00927
Yang, J. P., Zhu, Z. L., Fan, Y. J., Zhu, F., Chen, Y. J., Niu, Z. T., et al. (2020). Comparative Plastomic Analysis of Three Bulboplyllum Medicinal Plants and its Significance in Species Identification. Acta Pharm. Sin. 55, 2736–2745.
Yildirim, A., Inci, A., Duzlu, O., Onder, Z., and Ciloglu, A. (2013). Detection and Molecular Characterization of the Wolbachia Endobacteria in the culex Pipiens (Diptera: Culicidae) Specimens Collected from Kayseri Province of turkey. Ankara Univ. Veteriner Fakultesi Dergisi 60, 189–194. doi:10.1501/Vetfak_0000002577
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 20, 348–355. doi:10.1111/1755-0998.13096
Zhang, X.-F., Landis, J. B., Wang, H.-X., Zhu, Z.-X., and Wang, H.-F. (2020). Comparative Analysis of Chloroplast Genome Structure and Molecular Dating in Myrtales. BMC Plant Biol. 21, 219. doi:10.1186/s12870-021-02985-9
Zhao, B., Li, J. J., Mao, S. Z., and Tang, W. (2016). Application of DNA Barcoding Based on the Chloroplast ndhA Intron Region in Classification of Begonia (Begoniaceae). North. Hortic. (16), 103–107. doi:10.11937/bfyy.2016.16027
Keywords: catalpa, chloroplast genome, chloroplast structure, codon bias, simple sequence repeat, phylogenetic
Citation: Li F, Liu Y, Wang J, Xin P, Zhang J, Zhao K, Zhang M, Yun H and Ma W (2022) Comparative Analysis of Chloroplast Genome Structure and Phylogenetic Relationships Among Six Taxa Within the Genus Catalpa (Bignoniaceae). Front. Genet. 13:845619. doi: 10.3389/fgene.2022.845619
Received: 30 December 2021; Accepted: 01 March 2022;
Published: 16 March 2022.
Edited by:
Madhav P. Nepal, South Dakota State University, United StatesReviewed by:
Jia-Yu Xue, Nanjing Agricultural University, ChinaCopyright © 2022 Li, Liu, Wang, Xin, Zhang, Zhao, Zhang, Yun and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenjun Ma, bXdqbHhAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.