 Qing Xu1*†
Qing Xu1*† Shan-Ce Niu2,3†
Shan-Ce Niu2,3† Kang-Li Li1Pei-Ji Zheng1Xiao-Jing Zhang2Yin Jia2Yang Liu2
Kang-Li Li1Pei-Ji Zheng1Xiao-Jing Zhang2Yin Jia2Yang Liu2 Yun-Xia Niu4Li-Hong Yu1Duan-Fen Chen2*
Yun-Xia Niu4Li-Hong Yu1Duan-Fen Chen2* Guo-Qiang Zhang1,5*
Guo-Qiang Zhang1,5*- 1GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou, China
- 2College of Horticulture, Hebei Agricultural University, Baoding, China
- 3State Key Laboratory of North China Crop Improvement and Regulation, Hebei Agricultural University, Baoding, China
- 4School of Vocational Education, Tianjin University of Technology and Education, Tianjin, China
- 5Laboratory for Orchid Conservation and Utilization, The Orchid Conservation and Research Center of Shenzhen, The National Orchid Conservation Center of China, Shenzhen, China
Orchids constitute approximately 10% of flowering plant species. However, only about 10 orchid genomes have been published. Metabolites are the main way through which orchids respond to their environment. Dendrobium nobile, belonging to Dendrobium, the second largest genus in Orchidaceae, has high ornamental, medicinal, and ecological value. D. nobile is the source of many popular horticultural varieties. Among the Dendrobium species, D. nobile has the highest amount of dendrobine, which is regarded as one of the criteria for evaluating medicinal quality. Due to lack of data and analysis at the genomic level, the biosynthesis pathways of dendrobine and other related medicinal ingredients in D. nobile are unknown. In this paper, we report a chromosome-scale reference genome of D. nobile to facilitate the investigation of its genomic characteristics for comparison with other Dendrobium species. The assembled genome size of D. nobile was 1.19 Gb. Of the sequences, 99.45% were anchored to 19 chromosomes. Furthermore, we identified differences in gene number and gene expression patterns compared with two other Dendrobium species by integrating whole-genome sequencing and transcriptomic analysis [e.g., genes in the polysaccharide biosynthesis pathway and upstream of the alkaloid (dendrobine) biosynthesis pathway]. Differences in the TPS and CYP450 gene families were also found among orchid species. All the above differences might contribute to the species-specific medicinal ingredient biosynthesis pathways. The metabolic pathway-related analysis will provide further insight into orchid responses to the environment. Additionally, the reference genome will provide important insights for further molecular elucidation of the medicinal active ingredients of Dendrobium and enhance the understanding of orchid evolution.
Introduction
Jinchai Shihu (金钗石斛) Dendrobium nobile is a medicinal species belonging to Dendrobium, the second largest genus in the Orchidaceae. With high amounts of medicinal active ingredients, D. nobile is also one of the five Dendrobium species recorded in the Chinese Pharmacopoeia (2020 Edition). Polysaccharides and alkaloids are the main medicinal components of Dendrobium. They are mainly stored in the stem and have strong antioxidant, neuroprotective, antidiabetic, antihypertensive, and immunomodulatory activities (Wang et al., 2010; Li et al., 2011; Liu et al., 2011; Wang et al., 2020). Dendrobine, a sesquiterpenoid alkaloid, is the signature bioactive component and is regarded as one of the criteria for evaluating the quality of D. nobile. The biosynthesis of these medicinal ingredients varies with factors including the tissues sampled, species, and genetics (Li et al., 2019). Elucidating the biosynthesis pathway of polysaccharides and alkaloids in Dendrobium is an important research topic.
To date, many genes related to the biosynthesis of polysaccharides and alkaloids have been reported. Dendrobium contains active polysaccharides (Wang et al., 2014), with three main kinds in stems depending on the content; namely, non-starch mannan polysaccharides, glucose, and galactose (Ng et al., 2012). Thus far, many carbohydrate-related genes have been reported, such as genes encoding polysaccharide-metabolism-related enzymes involved in the biosynthesis of polysaccharides, monosaccharide-related genes, and the basic building blocks for polysaccharide-synthesis-related genes, as well as genes playing important roles in the regulatory mechanism of polysaccharide synthesis (Ren et al., 2020; Wang et al., 2020). The main kind of alkaloid in Dendrobium is dendrobine, a sesquiterpenoid alkaloid (Li et al., 2019). There are three main pathways involved in the upstream of alkaloid biosynthesis: the shikimate pathway, the methylerythritol phosphate (MEP) pathway, and the mevolonate (MVA) pathway; some genes involved in these three pathways have been reported, such as 3-dehydroquinate synthase, 1-deoxy-D-xylulose 5-phosphate synthase and 3-hydroxy-3-methyl-glutarylcoenzyme A reductase (Tzin et al., 2012; Wang et al., 2020). In addition, members of the terpene synthases (TPSs) and cytochrome P450 monooxygenases (CYP450s) have been reported to play an important role in dendrobine biosynthesis (Guo et al., 2013; Li et al., 2017; Yuan et al., 2018; Wang et al., 2020). Although some related genes or enzymes have been reported, the specific biosynthesis pathways of polysaccharides and alkaloids and even dendrobine biosynthesis in Dendrobium species remain unclear.
With omics technology development, many medicinal plant genomes have been sequenced, such as Dendrobium catenatum (Zhang et al., 2016), Salvia miltiorrhiza (Xu et al., 2016), Panax Notoginseng (Chen et al., 2017), Gelsemium elegans (Liu et al., 2020), and Platycodon grandifloras (Kim et al., 2020), suggesting that genomics is an effective method to mine the key genes of medicinal ingredients. In this study, with the use of PacBio sequencing and Hi-C technologies, the chromosome-level genome assembly of D. nobile was performed. Comparative genomic studies were conducted with D. catenatum (Zhang et al., 2016), Dendrobium chrysotoxum (Zhang et al., 2021), Apostasia shenzhenica (Zhang et al., 2017), and Phalaenopsis equestris (Cai et al., 2015). The genes involved in the biosynthesis pathway of polysaccharides and alkaloids (dendrobine) were identified in the present study, laying a foundation for further research on the gene functions of medicinal active ingredients and providing a reference for the breeding of new varieties. The metabolic pathway-related analysis in Dendrobium will provide further insight into how Dendrobium species respond to the environment.
Materials and Methods
Sample Preparation and Sequencing
The wild Dendrobium nobile (voucher specimen: China, Yunnan province, on rock in evergreen broad-leaf forest, alt. 1250 m, 15 April, 2019, GZMU001) plants were collected. The species was identified by comparison with the D. nobile specimen deposited in the herbarium of the National Orchid Conservation Center of China. Green young leaves were frozen for short reads sequencing and PacBio sequencing. Leaf buds from the plants were fixed as described in Wang et al., 2015a and Hi-C library construction is referred to Chen et al. (2020).
For short reads sequencing, genomic DNA was extracted from leaves of the D. nobile plants using a modified cetyltrimethylammonium bromide (CTAB) protocol, and then was shorn by Covaris ultrasonicator. DNA fragments between 300bp and 400bp were then selected by Agencourt AMPure XP-Medium kit for library construction and finally sequencing was performed on the MGISEQ-2000 platform. The raw data was generated from constructed paired-end libraries (PE150), and the clean data was obtained after data filtering, which was carried out by SOAPnuke v1.6.5 software (https://github.com/BGI-flexlab/SOAPnuke) (Chen et al., 2018), with the following parameters: -n 0.02; -l 20; -q 0.4; -Q 2; -i; -G; --seqType 0; –rmdup. The PacBio sequencing was performed on a PacBio Sequel II sequencer by BGI (Shenzhen, China). Furthermore, the data of Hi-C library sequenced by MGISEQ-2000 platform was used for Hi-C analysis.
For assisting gene annotation and gene expression analysis, total RNA was extracted from tender leaves, stems, and roots of three different individuals in the same growing stage of D. nobile using the RNAprep Pure Plant Kit and genomic DNA contamination was removed using RNase-Free DNase I (both from Tiangen), respectively. The integrity of RNA was evaluated on a 1.0% agarose gel stained with ethidium bromide (EB), and its quality and quantity were assessed using a Qubit2.0 Fluorometer and an Agilent 2,100 Bioanalyzer (Agilent Technologies). As the RNA integrity number (RIN) was greater than 7.0 for all samples, they were used in cDNA library construction and Illumina sequencing. The cDNA library was constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) and 3 μ g RNA per sample, following the manufacturer’s recommendations. The PCR products obtained were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2,100 system. Library preparations were sequenced on the Illumina Novaseq6000 sequencer, generating 150-bp paired-end reads.
Genome Assembly
Before genome assembly, genome size and heterozygosity were estimated by Jellyfish v.2.1.4 (Marcais and Kingsford, 2011) and GenomeScope (Vurture et al., 2017) based on a 17-K-mer distribution. Then, Canu v 2.2 (Koren et al., 2017) was used to correct the Pacbio raw data and assemble the genome with the following parameters: correctedErrorRate = 0.035 utgOvlErrorRate = 0.065 trimReadsCoverage = 2 trimReadsOverlap = 500 gridOptions = "--mem-per-cpu = 5 g”. Furthermore, pilon v1.22 (--fix bases--mindepth 10 --minqual 20 --diploid) (Walker et al., 2014) was used to correct the assembly using the data generated from the MGISEQ-2000 platform. For chromosome-level assembly, the Hi-C reads were filtered by SOAPnuke software (Chen et al., 2018) with the following parameters: -n 0.02; -l 20; -q 0.4; -Q 2; -i; -G; --seqType 1; –rmdup. Then, the obtained clean reads were compared with the preassembled contigs using Juicer software (Durand et al., 2016). After filtering the results and removing the misaligned reads, 3D-DNA (Dudchenko et al., 2017) software was used to preliminarily cluster, sequence, and direct the pseudochromosomes. Furthermore, Juicer-box was used to adjust, reset, and cluster the pseudochromosomes, and misassemblies and misjoins were manually corrected based on neighboring interactions. For the evaluation of Hi-C assembly results, the final pseudochromosome assemblies were divided into 150 kb bins with equal lengths, and the interaction signals generated by the valid mapped read pairs between each bin were visualized in a heat map. Finally, the completeness and quality of the final assembled genome were evaluated through Benchmarking Universal Single-Copy Ortholog (BUSCO v5.2.2) (Manni et al., 2021) tests.
Genome Annotation
Repetitive sequences are an important part of the genome and are divided into two types: tandem and interspersed repeats. RepeatMasker v4.0.7 and RepeatProteinMask v4.0.7 software (http://www.repeatmasker.org) were used to identify repetitive sequences based on the RepBase v21.12 database (http://www.girinst.org/repbase). For de novo prediction, a repetitive sequence database was constructed through RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) and LTR_FINDER v1.06 (http://tlife.fudan.edu.cn/ltr_finder/). RepeatMasker software was then used to predict the repeat sequences. Tandem Repeats Finder v4.09 (http://tandem.bu.edu/trf/trf.html) was used to find tandem repeats in the genome. The repeat-masked genome assembly was used for annotating high-quality protein-coding genes through an integration of homology-based, de novo, and transcriptome-based predictions. For homology-based prediction, protein sequences from seven species (Arabidopsis thaliana, Oryza sativa, Asparagus officinalis, A. shenzhenica, Gastrodia elata, P. equestris, and Vanilla planifolia) were used to align the D. nobile genome sequences through Genewise v2.4.1 (Birney et al., 2004) (https://www.ebi.ac.uk/Tools/psa/genewise/). Then, 5,000 complete genes from the homology-based prediction method were used to produce a training model using the Augustus (Stanke et al., 2006) (http://bioinf.uni-greifswald.de/augustus/) and SNAP (Johnson et al., 2008) (http://homepage.mac.com/iankorf/) software. The RNA sequencing data of D. nobile were mapped to the genome sequences through Hisat v2.1.0 and StringTie v1.3.4d (Kim et al., 2015; Pertea et al., 2015). Finally, Maker v2.31.8 (Holt and Yandell, 2011) was used to annotate and integrate the results produced by the three methods. BUSCO v5.2.2 (Manni et al., 2021) was used to evaluate the completeness and quality of the gene models.
Functional annotation of the predicted gene models was carried out by Blast v2.2.26 (Altschul, 1990) software, aligned against the Swissprot (Boeckmann et al., 2003) (http://www.uniprot.org/), TrEMBL (http://www.uniprot.org/), Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/), InterPro (Zdobnov and Apweiler, 2001) (https://www.ebi.ac.uk/interpro/), Nr (non-redundant database), KOG (Koonin et al., 2004) (clusters of euKaryotic Orthologous Groups), and Gene Ontology (GO) (Ashburner et al., 2000) databases. For non-coding RNA annotation, tRNAscan-SE 1.3.1 (Lowe and Eddy, 1997) (http://lowelab.ucsc.edu/tRNAscan-SE/) was used to annotate the tRNA sequences. BLASTN was used to search for rRNA. The miRNA and snRNA sequences were predicted by the INFERNAL (Griffiths-Jones, 2004) (http://infernal.janelia.org/) software.
Gene Family Identification
The protein sequences of D. nobile and 17 additional angiosperm species (D. catenatum, D. chrysotoxum, Cymbidium sinense, Amborella trichopoda, Spirodela polyrhiza, A. shenzhenica, A. officinalis, O. sativa, Ananas comosus, Musa acuminata, Phoenix dactylifera, Vitis vinifera, P. equestris, Sorghum bicolor, Populus trichocarpa, A. thaliana, and G. elata) were used for orthologous gene family identification and clustering. BLASTP was used to calculate the similarities between sequence pairs with a cut-off e value of 1e−5. Finally, OrthoMCL v.2.0.9 (Li et al., 2003) with default parameters was used to cluster the gene families.
Phylogenetic Tree Construction and Phylogenomic Dating
The protein sequences of each gene family of 313 identified single-copy gene families were aligned by MUSCLE (Edgar, 2004) (http://www.drive5.com/muscle/). Then, a super-alignment matrix generated from all the alignment results was used for the construction of the phylogenetic tree. Finally, RAxML (Stamatakis, 2014) was used for the construction of the phylogenetic tree of 18 angiosperm species with the GTRGAMMA model; the bootstrap value was 1,000.
PAML MCMCTREE (http://abacus.gene.ucl.ac.uk/software/paml.html) was used to estimate the divergence time. The following constraints were used for time calibrations: i) the B. distachyon and O. sativa divergence time [40.0–54.0 million years (Ma)] (International Brachypodium Initiative, 2010); ii) the divergence time of P. trichocarpa and A. thaliana (100.0–120.0 Ma) (Tuskan et al., 2006); iii) the monocot and eudicot divergence time (lower boundary of 130.0 Ma) (Jaillon et al., 2007); and iv) the time of the earliest-diverging angiosperms (<200.0 Ma) (Magallón et al., 2013).
Whole-Genome Duplication and Collinearity Analysis
The Ks distribution was used to infer whole-genome duplication (WGD) events in D. nobile based on paralogous gene pairs and the divergence between species based on orthologues. MCscanX v1.5.2 (Wang et al., 2012) was used to find the collinear regions. BLASTP was used to search for putative paralogous genes in each collinear region within D. nobile and orthologous genes between D. nobile and D. chrysotoxum, D. nobile and V. planifolia, and D. nobile and C. sinense. Furthermore, Codeml, in the PAML package (Yang, 1997) was used with the F3X4 model to calculate the Ks value of each gene pair.
For further analysis of the features of collinear regions, MCscan (https://zenodo.org/record/31631#.XpkUyTOeask) was used to find the collinear regions between D. nobile and D. chrysotoxum, D. nobile and V. planifolia, and D. nobile and C. sinense.
Expansion and Contraction of Gene Families
Based on the gene families clustered by OrthoMCL (Li et al., 2003), we filtered the gene families with a number higher than 200 in one species and lower than 2 in other species. CAFÉ software (De Bie et al., 2006) (http://sourceforge.net/projects/cafehahnlab/) was used to determine the expansion and contraction of orthologous gene families combined with divergence times.
Gene Family Analysis of the Biosynthesis of Bioactive Components
All the genes or gene families in the biosynthesis pathways of polysaccharides and alkaloids were identified by HMM or BLASTP searches. The HMM profiles (PF01128.20 for CMS genes, PF01264.22 for CS genes, PF13292.7 for DXS genes, PF02401.19 for HDR genes, PF04551.15 for HDS genes, PF08540.11 and PF01154.18 for HMGS genes, PF00288.27 for PMK genes, PF00275.21 for SHKG genes, PF01202.23 for SK genes, and PF03088.17 for STR genes) were downloaded from Pfam (pfam.xfam.org/), and the query sequences for the BLASTP methods were mostly from homologous genes of Arabidopsis, except for the SKDH genes from Pisum sativum (Weeden, 2018) and DHQS genes from tomatoes (Bischoff et al., 1996). After the candidate homologs were obtained, MAFFT (Katoh and Standley, 2013) and PhyML (Guindon et al., 2005) software were used for sequence alignment and phylogenetic tree construction, respectively.
TPS and CYP450 Gene Family Identification
The HMM profiles for PF01397 (Terpene_synth) and PF03936 (Terpene_synth_C) were downloaded from Pfam (pfam.xfam.org/), and two profiles were used to carry out HMM searches against the protein database for six species (D. nobile, D. chrysotoxum, D. catenatum, P. equestris, A. shenzhenica, and A. thaliana). These sequences were then manually checked, and the sequences with at least one of these domains were retained. The retained amino acid sequences were aligned using MAFFT (Katoh and Standley, 2013). Then, the aligned amino acids were used for phylogenetic tree construction by PhyML (Guindon et al., 2005). The tree was generated by the maximum likelihood method based on the Jones-Taylor-Thornton (JTT) matrix-based model (Jones et al., 1992) and the bootstrap method for phylogeny tests with 1,000 replications. The HMM profiles for PF00067.23 (CYP450) were downloaded from Pfam (pfam.xfam.org/). HMM searches were used to obtain the homologs in the protein database for seven species (D. nobile, D. chrysotoxum, D. catenatum, P. equestris, A. shenzhenica, A. thaliana, and O. sativa). The next steps were the same as those for TPS gene family identification, except for the rice genome. The identified CYP450 genes in A. thaliana and O. sativa in this study were further confirmed with the Arabidopsis Cytochrome P450 database (http://www.p450.kvl.dk/At_cyps/family.shtml) and rice (Wei and Chen, 2018). All gene expression analyses were carried out using Salmon v1.3.0 (Patro, et al., 2017) with the default settings.
Results
Genome Sequencing and Genomic Characteristics
D. nobile (Figure 1) has a karyotype of 2N = 2X = 38 (Zheng et al., 2018). For estimating the D. nobile genome size, a total of 130.62 Gb of raw data with 300-400 bp insert libraries were generated by MGISEQ-2000 sequencing and 122.52 Gb of clean data was obtained after data filtering (Supplementary Table S1). The estimated genome size was 1.16 Gb, with 1.35% heterozygosity based on K-mer analysis (Supplementary Figure S1). To obtain the assembly, 96.91 Gb (coverage of 83.54×) of PacBio sequencing data and 5.19 million subreads with an N50 read length of 21.4 kb was generated (Supplementary Table S1). Based on the PacBio and MGISEQ-2000 sequencing data, we then used 245.48 Gb of raw data from a Hi-C library (Supplementary Table S1) to reconstruct physical maps by recoding and clustering the assembled scaffolds into 19 pseudochromosomes, which represented the 19 chromosomes in the haploid genome of D. nobile (Figure 2; Supplementary Table S2). Finally, 211.34 Gb of clean data was produced using the data filtering. After the Hi-C analysis, 1,192,377,146 bp sequences were mapped to 19 pseudochromosomes, accounting for 99.45% of the raw assembly (1.19 Gb). The length of the 19 pseudochromosomes ranged from 37.78 to 95.36 Mb, with a scaffold N50 value of 64.46 Mb (Table 1; Supplementary Tables S2, S3). Furthermore, BUSCO estimation indicated that the completeness of the gene set of the assembled genome was 96.22%, while mapping short reads back to the assembly indicated that the completeness was 98.82% (Supplementary Tables S4, S5). The chromatin interaction data showed a high level of chromatin interaction between linked sequences and low chromatin interaction between non-linked sequences, suggesting the high quality of the Hi-C assembly (Figure 3).

TABLE 1. Genome assembly statistics.

FIGURE 1. The plant of Dendrobium nobile.
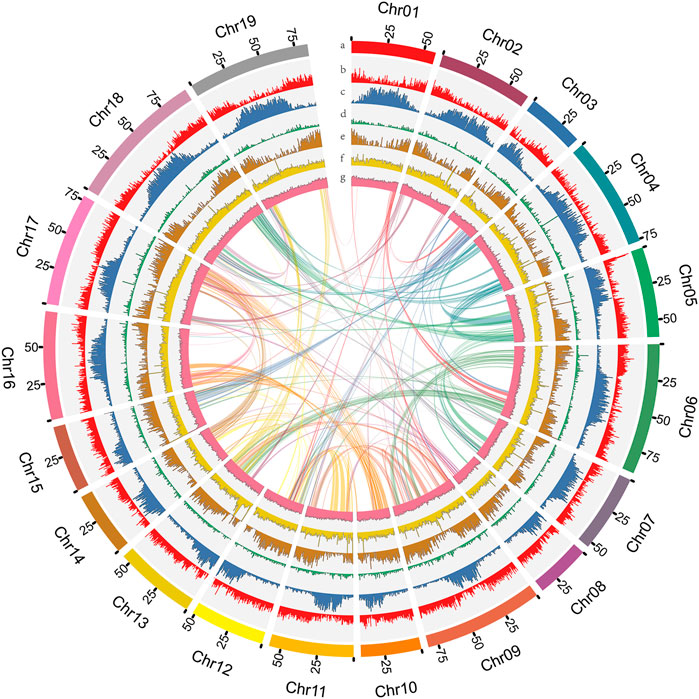
FIGURE 2. The genome features of Dendrobium nobile. (A) Chromosomes. (B) LTR Gypsy density. (C) LTR Copia density. (D) DNA transposon density. (E) Gene density. (F) Coverage of second-generation data. (G) GC content within sliding windows of 500 kb.
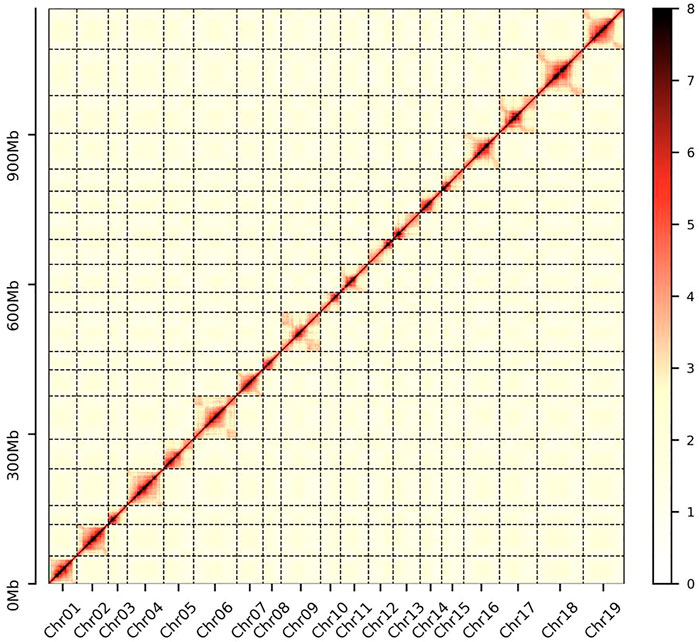
FIGURE 3. Intensity signal heat map of the Hi-C chromosome. A higher value on the scale bar indicates a higher contact frequency.
Gene Prediction and Annotation
A total of 29,476 protein-coding genes were annotated in D. nobile (Supplementary Table S6). Among them, 98.28% of ≥50% CD regions overlapped based on homolog or de novo data (Supplementary Table S7), with 93.43% exhibiting high completeness (Supplementary Table S8).
The average lengths of genes and introns in D. nobile were similar to those in other orchid species, except for A. shenzhenica (Zhang et al., 2017), and much higher than those in most other angiosperms (Zhang et al., 2017; Supplementary Figure S2). This suggests that the gene length is positively correlated with the intron length, and that genes and introns in orchids except for A. shenzhenica have a longer average length, which may be a unique characteristic. Furthermore, 76 microRNAs, 386 transfer RNAs, 958 ribosomal RNAs, and 457 small nuclear RNAs were identified in the D. nobile genome (Supplementary Table S9).
In this study, it was estimated that 61.07% of the D. nobile genome consisted of repetitive sequences (Supplementary Table S10), similar to the 62% in P. equestris and 62.81% in D. chrysotoxum, and lower than the 78.1% in D. catenatum (Cai et al., 2015; Zhang et al., 2016). Furthermore, the notably higher percentage of repeats predicted by the de novo method compared to the homology-based search method indicates that D. nobile has many characteristic repeats (Supplementary Table S10). Among these elements, long terminal repeats (LTRs) accounted for 51.31% of the genome (Supplementary Table S11), similar to the 53.15% in D. chrysotoxum and higher than the 46% in D. catenatum, 46.47% in P. equestris, and 22.06% in A. shenzhenica (Zhang et al., 2016; Zhang et al., 2017).
In addition, 27,765 (94.20%) predicted genes were functionally annotated (Supplementary Table S12). Among them, 27.601 (93.64%) and 25,870 (87.77%) genes were annotated to the Nr and TrEMBL databases, respectively, (Supplementary Table S14). The number of annotated genes was 24,044 (81.57%), 20,215 (68.58%), and 19,855 (67.36%) in the Interpro, KEGG, and Swissprot databases, respectively (Supplementary Table S12).
Evolution of Gene Families
For the phylogenetic relationship and divergence times among different plant species, a high-confidence phylogenetic tree and the estimated divergence times of 18 different plant species based on genes extracted from a total of 313 single-copy families were constructed (Supplementary Figures S3, S4; Supplementary Table S13). As expected, D. nobile, D. catenatum, and D. chrysotoxum were sisters to P. equestris, forming an Epidenroideae clade. G. elata and A. shenzhenica were located at the base of the Orchidaceae branches (Supplementary Figure S4). The estimated Orchidaceae divergence time was 113.3 (104.3–121.0) Mya, the divergence time of the subfamily Apostasioideae was 76.4 (61.1–90.8) Mya, and the divergence time between D. nobile and P. equestris was 41.7 (29.2–54.9) Mya (Figure 4). Finally, D. chrysotoxum appeared at 11.9 (6.5–18.1) Mya, and the divergence time between D. nobile and D. catenatum was 5.6 (2.9–8.6) Mya. Then, the expansion and contraction of orthologous gene families were determined. According to the results (Figure 5), 110 gene families were expanded in the lineage leading to the Orchidaceae, whereas 1,566 gene families were contracted. Furthermore, 595 gene families expanded and 321 gene families contracted, leading to the Dendrobium genus. In D. nobile, 859 gene families were expanded compared with 417 in D. catenatum, 721 in D. chrysotoxum, and 872 in P. equestris. For the contracted gene families, 325 gene families were contracted in D. nobile compared with 1,010 in D. catenatum, 1,510 in D. chrysotoxum, and 900 in P. equestris. In the D. nobile clade, 859 gene families were expanded, including 3,134 genes, and 325 gene families were contracted, including 213 genes. To further investigate the functions of the expanded gene families and their species specificity, KEGG enrichment analysis was conducted for the expanded gene families. The KEGG term “Biosynthesis of other secondary metabolites” was found to be the most significantly enriched, and contained the most genes (Supplementary Table S14). This may be related to the synthesis of specific medicinal ingredients in D. nobile.
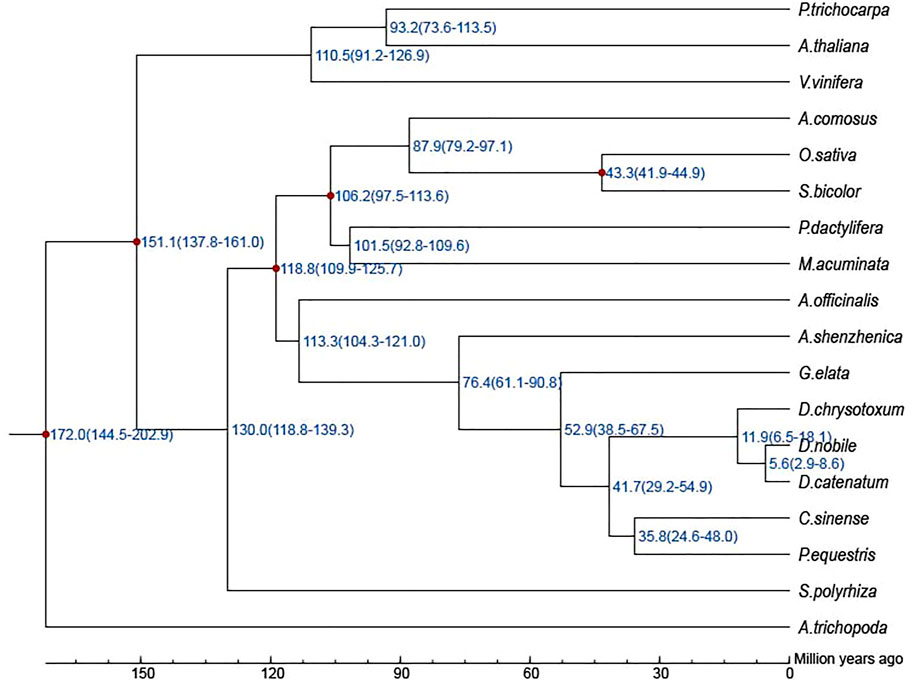
FIGURE 4. Divergence time among 19 species.
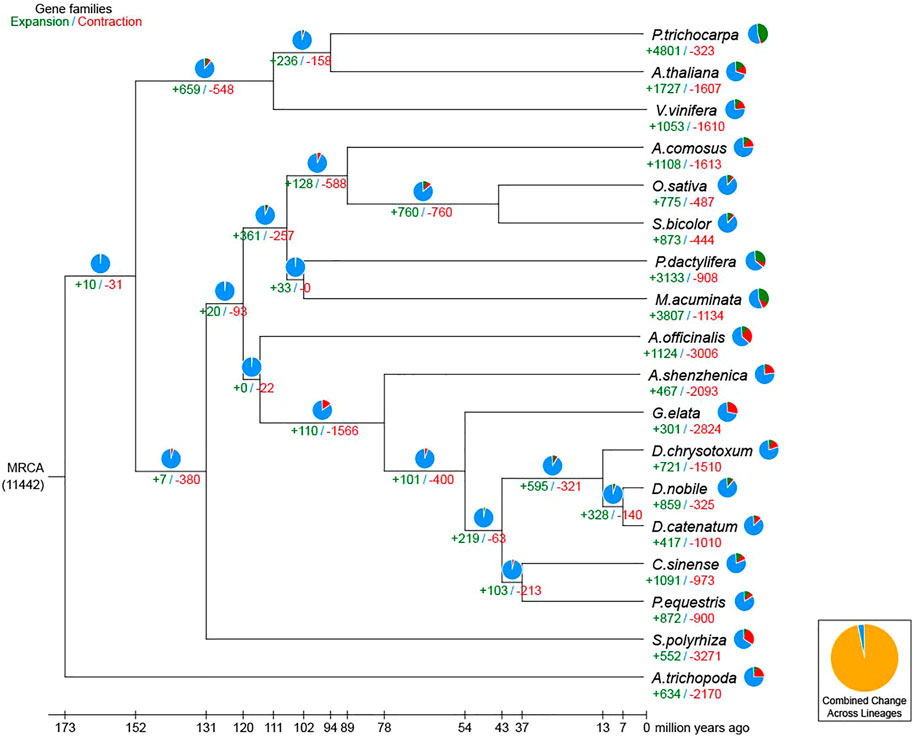
FIGURE 5. Expansion and contraction of gene families. The green and red numbers are the numbers of expanded and contracted gene families, respectively.
Whole-Genome Duplication and Synteny Analysis
To detect the occurrence of WGDs in D. nobile, the Ks distribution pattern of paralogous genes in collinear regions was analyzed. There were two peaks, and the Ks values were 0.8 and 1.5 (Figure 6), suggesting the occurrence of two recent WGD events in D. nobile. To determine the time of the two WGD events, the Ks distribution of the homologous genes between D. nobile and P. equestris, A. shenzhenica, D. chrysotoxum, and G. elata was further analyzed. The most recent WGD event in D. nobile (Ks = 0.8) occurred before the divergence between D. nobile and A. shenzhenica, suggesting that the WGD events were shared among all extant orchids (Zhang et al., 2017). The second recent WGD event in D. nobile (Ks = 1.5) was shared with most monocots (Zhang et al., 2017). For the collinear regions, the syntenic relationships among D. nobile, V. planifolia, C. sinense, and D. chrysotoxum were further analyzed. There were 25,166 collinear gene pairs located in 1,404 collinear regions between D. nobile and V. planifolia. 31,498 collinear gene pairs located in 120 collinear regions between D. nobile and D. chrysotoxum, and 30,650 collinear gene pairs located in 430 collinear regions between D. nobile and C. sinense (Supplementary Figures S5–S7; Supplementary Table S15).
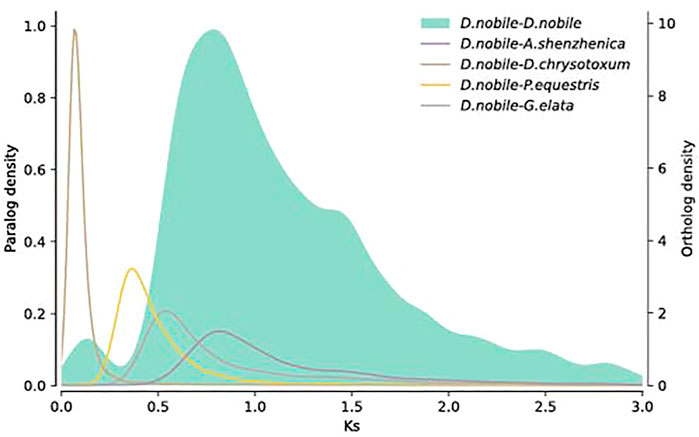
FIGURE 6. Distribution of Ks of the whole paranome of Dendrobium nobile. The Ks distribution of paralogues in the D. nobile genome is shown in the gray histogram and gray density curve. The other density curves show the Ks distribution of orthologous genes between D. nobile and Phalaenopsis equestris, D. nobile and Apostasia shenzhenica, D. nobile and Dendrobium chrysotoxum, and D. nobile, and Gastrodia elata.
Genes in the Polysaccharide Biosynthesis Pathway
The key genes encoding enzymes in the potential polysaccharide synthesis pathway were identified in D. nobile, D. catenatum, D. chrysotoxum, A. shenzhenica, P. equestris, and A. thaliana (Figure 7; Supplementary Table S16). All six gene families were multigene families. There were seven alkaline/neutral invertase (NI) genes, two PGM genes, three SPS genes, four SUS genes, two UGP genes, and five UGE genes in D. nobile. Based on the phylogenetic analysis, these gene families could be further divided into several branches. The number of genes in different branches among these species varied greatly, especially those in the NI, PGM, and UGE gene families (Figure 7, in red font). The number of NI genes in the cytosolic branch, mitochondria branch, and chloroplastic branch of D. nobile was five, two, and one, respectively, compared with seven, one, and one in D. chrysotoxum, respectively, and three, three, and one in D. catenatum, respectively (Supplementary Table S17). For PGM genes, one gene in D. nobile was clustered into the pPGM branch, and one gene were divided into the cPGM branch (Supplementary Table S16). Interestingly, there were five PGM genes of D. chrysotoxum on the cPGM branch and one gene on the pPGM branch (Supplementary Table S16). There were five and zero members divided into group 1 and group 2 in the UGE gene family, respectively, while there were one to two different values among other species (Supplementary Table S16). The gene number variations in different branches of each gene family may be related to the different amounts and constituent polysaccharides among Dendrobium species. Furthermore, for the SPS4F branch in the SPS gene family, three homologous genes were identified in three Dendrobium species, and no genes were found in other orchid species (A. shenzhenica and P. equestris), suggesting a Dendrobium-specific gene (Figure 7, in green font; Supplementary Table S16). The gene expression patterns in different tissues of the three Dendrobium species were also analyzed, but most were not significantly different among tissues.
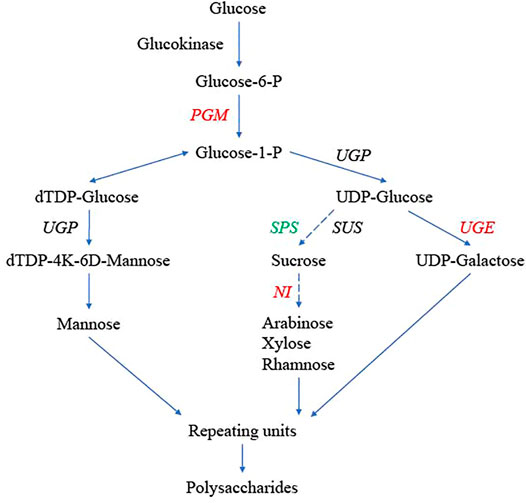
FIGURE 7. Genes in the polysaccharide synthesis pathway in Dendrobium nobile, adapted from Wang (Wang et al., 2020). Hydrolysis or hydrolysis-derived reactions could generate monosaccharides, such as mannose, glucose, and galactose. These monosaccharides are also the basic building blocks and repeating units for synthesizing polysaccharides. Genes in red indicate gene copy number variation among D. nobile, Dendrobium catenatum, Dendrobium chrysotoxum, Phalaenopsis equestris, Apostasia shenzhenica, and Arabidopsis thaliana. Genes in green are Dendrobium-specific. The dashed lines indicate multiple steps.
Genes Upstream of the Alkaloid Biosynthesis Pathway
To determine the genetic variation of the alkaloid biosynthesis pathway, all key genes (20 genes or gene families) upstream of the alkaloid biosynthesis pathway were identified among three Dendrobium species (D. nobile, D. catenatum, and D. chrysotoxum) and three other species (A. shenzhenica, P. equestris, and A. thaliana) (Supplementary Table S17)). Most were single-copy genes, except for the DHS, DXS, and HMGR gene families. Interestingly, no SKDH, HMGR, or MVD genes were found in D. chrysotoxum, which hinted at a significant variation in genes or the possibility of alternative pathways playing a dominant role in this process. The gene expression patterns in different tissues among the three Dendrobium species were also determined. Three genes or gene families in the shikimate pathway exhibited higher expression in stems than in leaves: Dnobile10G00647.1 (DHS), Dnobile17G01430.1 (DHS), Dnobile08G00887.1 (DHQS), and Dnobile02G00904.1 (DHD-SKDH) (Figure 8, red five-pointed star; Supplementary Figure S8). The Dnobile08G00649.1 (HMGS) and Dnobile01G00682.1 (MVD) genes in the MVA pathway also had higher expression in stems (Figure 8, red five-pointed star). All genes in the MEP pathway were expressed equally in stems and leaves. For D. chrysotoxum, most genes had higher expression in leaves than in stems (Supplementary Figure S9), such as Guchui-Maker62728 (DHS), Guchui-Maker69398 (DXR), Guchui-Maker111118 (MCS), Guchui-Maker68853 (HDS), and Guchui-Maker109159 (HDR). For STR genes, Dnobile14G00298.1 (STR12/13) was mainly expressed in stems (Supplementary Figure S10), while other members mainly low or equally expression between the two tissues. Interestingly, Guchui-Maker98640 (STR9) was mainly expressed in stems, and Guchui-Maker96976 (STR10) was mainly expressed in leaves (Supplementary Figure S10). Dendrobine is mainly produced in the stems of D. nobile (Li et al., 2019) and is rare in D. chrysotoxum. These opposite gene expression patterns may be related to the difference between D. nobile and D. chrysotoxum in terms of alkaloid (dendrobine) biosynthesis.
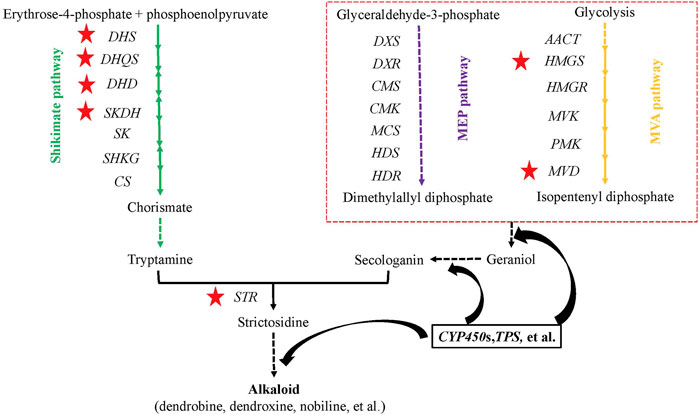
FIGURE 8. Genes upstream of the alkaloid biosynthesis pathway in Dendrobium nobile, adapted from Pu (Pu et al., 2021) and Wang (Wang et al., 2020). In the methylerythritol phosphate (MEP) pathway, there are seven consecutive steps catalyzed by the following enzymes: DXS, 1-deoxy-D-xylulose 5-phosphate (DXP) synthase; DXR, DXP-reductoisomerase; MCT, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CMK, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; MDS, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; HDS, 4-hydroxy-3-methylbut-2-enyldiphosphate (HMBPP) synthase; and HDR, HMBPP reductase. The mevolonate (MVA) pathway contains ACCT, acetyl-coenzyme A (CoA) C-acetyltransferase; HMGS, HMG-CoA synthase; HMGR, HMG-CoA reductase; MVK, mevalonic acid kinase; PMK, phosphomevalonate kinase; and MVD, mevalonate-5-diphosphate decarboxylase. The shikimate pathway contains DHS, 3-dexoy-7-phosphoheptulonate synthase; DHQS, 3-dehydroquinate synthase; DHD, 3-dehydroquinate dehydratase; SKDH, shikimate dehydrogenase; SK, shikimate kinase; SHKG, 3-phosphoshikimate 1-carboxyvinyltransferase; and CS, chorismate synthase. The red five-pointed stars show higher gene expression in stems than in leaves. The dashed lines indicate multiple steps.
TPS Gene Family and Dendrobine Biosynthesis
Dendrobine, a sesquiterpenoid alkaloid, is the dominant type of alkaloid in D. nobile. TPSs catalyze the biosynthesis of sesquiterpenes (C15) using farnesyl diphosphate (FPP) as a substrate (McGarvey and Croteau, 1995). TPSs may have originated from isoprenyl diphosphate synthase genes, which are involved in dendrobine biosynthesis (Jiang et al., 2019; Wang et al., 2020). CrGES (a gene encoding geraniol synthase that is present in Catharanthus roseus, and is the homologue of TPS02) and TPS21 genes are involved in dendrobine biosynthesis (Kuang et al., 2016; Wang et al., 2020).
In this study, 51 TPS genes were identified in D. nobile, which was more than the 48 identified in D. chrysotoxum and the 42 identified in D. catenatum (Figure 9). The number of TPS genes in Dendrobium was much higher than that in A. shenzhenica and P. equestris, which indicated a relationship with dendrobine biosynthesis. The TPS gene family was divided into five subfamilies, TPS-a, TPS-b, TPS-c, TPS-e/f, and TPS-g, and the TPS-b subfamily was further clustered into TPS-b-I, TPS-b-II, and TPS-b-III. The TPS-a and TPS-b subfamilies showed different evolutionary patterns between monocots and dicots. Interestingly, there were 21, 16, and 17 TPS-a genes in D. nobile, D. chrysotoxum, and D. catenatum, respectively. As the TPS-a subfamily encodes only sesqui-TPSs (Jiang et al., 2019), a higher number of TPS-a genes in D. nobile may be an important factor in the higher production of dendrobine compared with other Dendrobium species (Li et al., 2019). Based on gene expression pattern analysis, genes of the TPS-a subfamily were mainly expressed in the stem (Supplementary Figure S11), which further confirmed that these genes contributed to the production of more dendrobine in D. nobile through the existence of more genes with higher expression in the stems. Furthermore, the gene numbers of TPS-b-II and TPS-b-III were three and 13 in D. nobile, respectively, compared with four and 10 in D. catenatum, respectively, while the numbers were 14 and seven in D. chrysotoxum, respectively. There were several TPS genes with higher expression in the leaves than in the stem (Supplementary Figure S11), such as Dnobile18G01782.1, Dnobile18G01781.1, Dnobile18G01775.1, and Dnobile18G01780.1. As the TPS-b subfamily encoding monoTPSs (Jiang et al., 2019), the opposite distribution and gene expression pattern may have contributed to the differences in the amounts and constitution of alkaloids between D. nobile and D. chrysotoxum in different tissues.
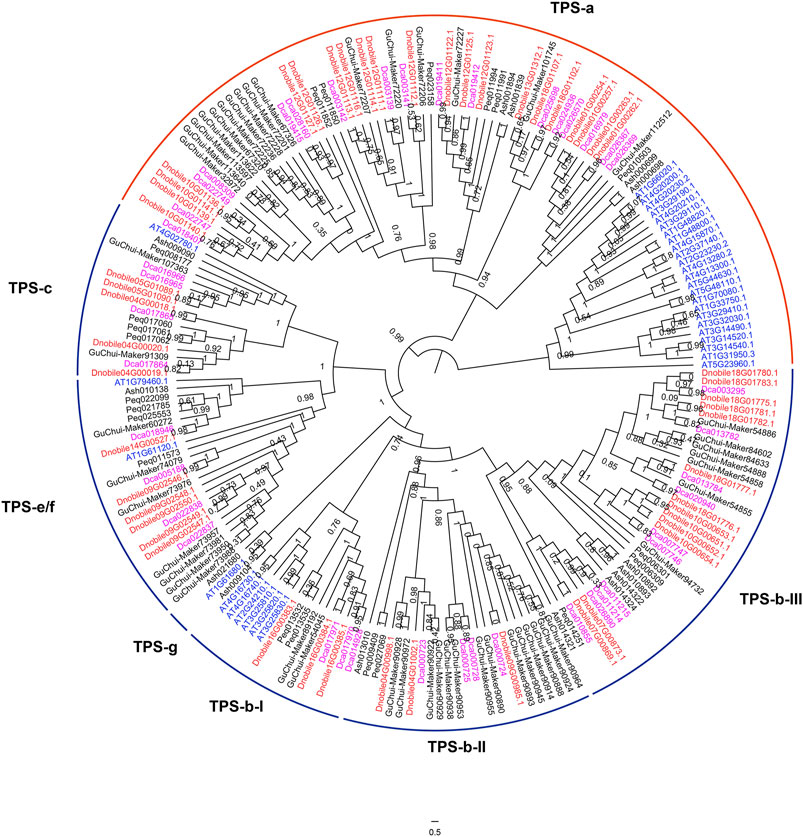
FIGURE 9. Analysis of terpene synthase (TPS) genes in Dendrobium nobile. Phylogenetic analysis of TPS genes in D. nobile, Dendrobium chrysotoxum, Dendrobium catenatum, Apostasia shenzhenica, Arabidopsis thaliana, and Phalaenopsis equestris. Dnobile, D. nobile; Ash, A. shenzhenica; Guchui-Maker, D. chrysotoxum; Dca, D. catenatum; Peq, P. equestris; AT, A. thaliana.
CYP450 Gene Family and Dendrobine Biosynthesis
Cytochrome P450 monooxygenases (CYP450s) are integral components in terpenoid and alkaloid pathways (Schuler, 2015). Biosynthetic CYP450s in these pathways can be considered organism-specific (Schuler, 2015). In this study, all the CYP450 genes were identified in three Dendrobium species (D. nobile, D. chrysotoxum, and D. catenatum), two other orchid species (P. equestris and A. shenzhenica), A. thaliana, and rice. There were 123, 170, 228, 257, and 210 CYP450 gene members in A. shenzhenica, P. equestris, D. nobile, D. chrysotoxum, and D. catenatum, respectively. The number of genes in Dendrobium species was much higher than that in P. equestris and A. shenzhenica, mainly because of tandem gene duplication (Supplementary Figure S12).
CYP450 families are grouped into the 10 plant CYP450 clans (Nelson et al., 2004), and many duplications and divergence events, such as chemical defense pathways in particular plants or groups of plants, occur mainly in the expanded multiple-family clans. The species-specific synthesis and catabolism pathways of terpenoids are mainly caused by gene neofunctionalizations within the CYP85 clan (Schuler, 2015). In this study, the differences in CYP450 genes among species were also mainly detected in the CYP85 clan. The CYP720 subfamily, which mediates the oxygenation of monoterpenes (myrcene and pinenes), sesquiterpenes (farnesene), and diterpenes (abietadienol and abietic acid) (Ro et al., 2005; Zulak and Bohlmann, 2010; Hamberger et al., 2011), was only identified in dicots in a previous study (Wei and Chen, 2018). In this study, the orthologous genes of CYP720 in orchids were identified (Supplementary Figure S12, red). Differences among CYP85 clans may involve the dendrobine signaling pathway in D. nobile.
Furthermore, other differences in orchid-specific related traits in CYP450 genes were also identified. CYP78 is involved in regulating organ size and cell proliferation (Nagasawa et al., 2013; Wang X. et al., 2015). More genes expanded by tandem duplication were found in Dendrobium species (Supplementary Figure S12, pink), such as Dnobile03G00500.1, Dnobile03G00501.1, and Dnobile03G00502.1 in D. nobile, GuChui-Maker69281, GuChui-Maker69269, GuChui-Maker69278, GuChui-Maker23937, and GuChui-Maker23933 in D. chrysotoxum, which may be involved in the formation of large capsules and swollen stems. CYP715, as a single gene family, plays a role as a key regulator in flower maturation, synchronizing petal expansion, and volatile emission in Arabidopsis (Liu et al., 2015). Interestingly, there were two to three CYP715 homologous genes in three Dendrobium species and P. equestris (Supplementary Figure S12, blue), indicating that gene expansion occurred in the Epidendroideae subfamily, potentially as a result of floral development.
Discussion
To date, three Dendrobium species recorded in the Chinese Pharmacopoeia (2020 Edition) have had their complete genome sequenced. The chromosome-level genome assembly of D. nobile should be helpful for further functional research as the model of dendrobine biosynthesis in Dendrobium. As one of the most important medicinal plants in traditional Chinese medicine, D. nobile is mainly composed of dendrobine and polysaccharides with pharmacological activity. This study contributed to improving the understanding of the metabolism of these medicinal components.
The heterozygosity of the D. nobile genome is 1.35%, meaning that it is difficult to assemble (Xin et al., 2019). In this study, Pacbio Sequel II sequencing technology with longer read lengths was used. Indeed, the long sequencing reads led to a BUSCO completeness estimate of 96.22%, which was a huge improvement. Polyploidization occurs frequently in angiosperms, which contributes to plant adaption to the environment and plant genome evolution (Van de Peer et al., 2017). One polyploidization event occurred in the most recent common ancestor of orchids (Cai et al., 2015; Zhang et al., 2016; Zhang et al., 2017). In this study, D. nobile was estimated to have undergone a WGD event at the same time, which further confirmed that the WGD event occurred at the most recent common ancestor of orchids. Furthermore, the average length of genes and introns in orchids, with the exception of A. shenzhenica, was much higher than that in most other angiosperms (Zhang et al., 2017). There are regulatory elements that are frequently contained within introns, and alternative splicing events often occur among different introns and exons, diversifying the protein coding of the genome. All of these factors may contribute to the genome structure evolution, genome size, gene function diversification, and gene expression pattern of a species (Castillo-Davis et al., 2002; De La Torre et al., 2014; Sena et al., 2014; Keane and Seoighe, 2016). For example, the long intron transcriptional delay in Drosophila is particularly important for the proper development of the embryo (Swinburne and Silver, 2008; Artieri and Fraser, 2014). This characteristic of orchids requires further analysis and research.
The content and composition of alkaloids and polysaccharides vary with factors including the tissues sampled, species, and genetics. For the genes in the polysaccharide biosynthesis pathway, NI genes are localized in the mitochondria, chloroplasts, and cytosol (Roitsch and Gonzalez, 2004). The diversity of subcellular localization suggests that NIs have a variety of physiological functions. Sucrose unloaded in the sink cell can be cleaved in the cytosol by NIs, which are involved in cellulose biosynthesis (Roitsch and Gonzalez, 2004; Rende et al., 2017), suggesting that the cytosolic NIs contribute to the supply of substrate for cellulose biosynthesis (Rende et al., 2017). In this study, the number of cytosolic NIs was found to be three, four, and seven in D. catenatum, D. nobile, and D. chrysotoxum, respectively. These large differences in quantity may contribute to the differences in the biosynthesis of polysaccharides among Dendrobium species. Cytosolic phosphoglucomutase (cPGM) interconverts glucose-6-phosphate and glucose-1-phosphate and is a key enzyme in central metabolism (Egli et al., 2010). Interestingly, five cPGM members were detected in D. chrysotoxum, one in D. nobile, and one in D. catenatum, suggesting that different molecular mechanisms may occur among Dendrobium species. Sucrose-phosphate synthase (SPS; E.C. 2.4.1.14) is a plant enzyme that plays vital roles in sucrose production across various plant species and is involved in photosynthesis (Huber and Huber, 1996; Ma et al., 2020). The SPS genes were only found in Dendrobium in this study, indicating a Dendrobium-specific sucrose biosynthesis mechanism.
Compared with multi-gene families in the polysaccharide pathway, most of the gene families in the alkaloid pathway were single-copy, except for DHS, DXS, and HMGR gene families. Therefore, the main difference in genes in the alkaloid pathway among Dendrobium species was the gene expression pattern in stems and leaves, with the exception of the loss of several genes. The expression pattern of the upstream genes in the alkaloid biosynthesis pathway may be the main factor contributing to the differences in the content and composition of alkaloids among Dendrobium species.
TPSs are a diverse class of enzymes that catalyze the biosynthesis of all kinds of terpenes (McGarvey and Croteau, 1995). TPSs may have originated from isoprenyl diphosphate synthase genes, which are involved in dendrobine biosynthesis (Jiang et al., 2019; Wang et al., 2020). The CrGES (encoding geraniol synthase in C. roseus, the homologue of TPS02) and TPS21 genes are involved in dendrobine biosynthesis (Simkin et al., 2013; Kuang et al., 2016; Wang et al., 2020). In this study, the number of TPS genes in Dendrobium was much higher than that in A. shenzhenica and P. equestris, indicating a relationship between TPS genes and dendrobine biosynthesis. Interestingly, the phylogenetic analysis revealed that there were 21, 16, and 17 TPS-a genes in D. nobile, D. chrysotoxum, and D. catenatum, respectively. As the TPS-a subfamily encodes only sesqui-TPSs (Jiang et al., 2019), the higher number of TPS-a genes in D. nobile and their expression mainly in the stem may be important factors involved in the production of more dendrobine compared to other Dendrobium species (Li et al., 2019). Furthermore, the opposite gene distribution in TPS-b-II and TPS-b-III among the three Dendrobium species may contribute to the differences in the amounts and constitution of alkaloids between D. nobile and D. chrysotoxum in various tissues, as the TPS-b subfamily encodes monoTPSs (Jiang et al., 2019).
For the CYP450 gene family, the main difference was found in the CYP85 clan, which was mainly involved in the species-specific synthesis and catabolism pathway of terpenoids (Schuler, 2015). There were twice as many CYP87B-C genes in D. nobile compared to other Dendrobium species. A previous study found that the CYP720 subfamily only occurred in dicots (Wei and Chen, 2018), mediating oxygenation of monoterpenes, sesquiterpenes, and diterpenes (Ro et al., 2005; Zulak and Bohlmann, 2010; Hamberger et al., 2011). This subfamily was also identified in this study, suggesting a contribution to the biosynthesis of orchid-specific components. All of the differences in CYP85 clans may be involved in the dendrobine signaling pathway in D. nobile.
Conclusion
Although D. nobile has high medicinal and ornamental value, the lack of omics data has hampered molecular mechanism research and the development of medicinal ingredients in this species. In this study, a chromosome-level reference genome of D. nobile was obtained, with an assembled genome size of 1.19 Gb and 29,476 annotated protein-coding genes. Two polyploidization events occurred in D. nobile based on Ks analysis: the recent WGD shared with other orchid species and the ancient polyploidization event shared with most monocots (the tau event). Phylogenetic analysis of the D. nobile gene family involved in the polysaccharide synthesis pathway showed that the gene number variation and Dendrobium-specific genes may be related to the fleshy stems with abundant polysaccharides. The analysis results of the TPS and CYP450 gene families suggest that there are more TPS-a genes in D. nobile, and the opposite distribution pattern in TPS-b-II and TPS-b-III among Dendrobium species may contribute to the species-specific alkaloid biosynthesis pathways. The differences in CYP85 clans among Dendrobium species may also play important roles in alkaloid biosynthesis and floral development. The analysis of D. nobile revealed the mechanism through which the fleshy stem produces abundant polysaccharides and alkaloids, as well as the floral development regulation, which is critical for industrial development. This was the first study to fully analyze the characteristics of genes in the biosynthesis pathways of polysaccharides and alkaloids in Dendrobium. The results of this study provide a high-quality genome of Dendrobium and important insights into the molecular elucidation of medicinal active ingredients, molecular breeding, and orchid responses to the environment, enhancing the understanding of orchid evolution.
Accession Codes and Genome Links
All data from this study were submitted to the NCBI database under Bioproject ID: PRJNA725550. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAGYWB000000000. The version described in this paper is version JAGYWB010000000.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
QX, G-QZ, S-CN, and D-FC designed the project. S-CN, QX, G-QZ, and D-FC wrote the draft manuscript. K-LL and P-JZ collected the materials and conducted the experiments. G-QZ, S-CN, and QX performed the genome analysis. S-CN contributed to the gene family and phylogenetic analyses. X-JZ, YJ, Y-XN, YL, K-LL, P-JZ, and L-HY took part in data analysis. The final manuscript has been read and approved by all authors. QX and S-CN contributed equally to this work.
Funding
This project was supported by the Talent Project of Guangzhou Medical University High-level University Construction (B195002005025, 06-410-2106132, 02-412-B205002-1005025), the Young Talent Project of the Hebei Agricultural University Foundation (YJ201848), and the Youth Fund of the Hebei Province Natural Science Foundation (C2019204295). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.844622/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi:10.1016/s0022-2836(05)80360-2
Artieri, C. G., and Fraser, H. B. (2014). Transcript Length Mediates Developmental Timing of Gene Expression across Drosophila. Mol. Biol. Evol. 31, 2879–2889. doi:10.1093/molbev/msu226
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 25, 25–29. doi:10.1038/75556
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14 (5), 988–995. doi:10.1101/gr.1865504
Bischoff, M., Rosler, J., Raesecke, H. R., Gorlach, J. r., Amrhein, N., and Schmid, J. r. (1996). Cloning of a cDNA Encoding a 3-dehydroquinate Synthase from a Higher Plant, and Analysis of the Organ-specific and Elicitor-Induced Expression of the Corresponding Gene. Plant Mol. Biol. 31 (1), 69–76. doi:10.1007/bf00020607
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT Protein Knowledgebase and its Supplement TrEMBL in 2003. Nucleic Acids Res. 31 (1), 365–370. doi:10.1093/nar/gkg095
Cai, J., Liu, X., Vanneste, K., Proost, S., Tsai, W.-C., Liu, K.-W., et al. (2015). The Genome Sequence of the Orchid Phalaenopsis Equestris. Nat. Genet. 47 (1), 65–72. doi:10.1038/ng.3149
Castillo-Davis, C. I., Mekhedov, S. L., Hartl, D. L., Koonin, E. V., and Kondrashov, F. A. (2002). Selection for Short Introns in Highly Expressed Genes. Nat. Genet. 31, 415–418. doi:10.1038/ng940
Chen, M. S., Niu, L., Zhao, M. L., Xu, C., Pan, B. Z., Fu, Q., et al. (2020). De Novo genome Assembly and Hi-C Analysis Reveal an Association between Chromatin Architecture Alterations and Sex Differentiation in the Woody Plant Jatropha Curcas. Gigascience 9, 1–12. doi:10.1093/gigascience/giaa009
Chen, W., Kui, L., Zhang, G., Zhu, S., Zhang, J., Wang, X., et al. (2017). Whole-Genome Sequencing and Analysis of the Chinese Herbal Plant Panax Notoginseng. Mol. Plant 10 (6), 899–902. doi:10.1016/j.molp.2017.02.010
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 7 (1), 1–6. doi:10.1093/gigascience/gix120
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a Computational Tool for the Study of Gene Family Evolution. Bioinformatics 22 (10), 1269–1271. doi:10.1093/bioinformatics/btl097
De La Torre, A. R., Birol, I., Bousquet, J., Ingvarsson, P. K., Jansson, S., Jones, S. J., et al. (2014). Insights into Conifer Giga-Genomes. Plant Physiol. 166, 1724–1732. doi:10.1104/pp.114.248708
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De Novo assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 356, 92–95. doi:10.1126/science.aal3327
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S. P., Huntley, M. H., Lander, E. S., et al. (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cel Syst. 3 (1), 95–98. doi:10.1016/j.cels.2016.07.002
Edgar, R. C. (2004). MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi:10.1093/nar/gkh340
Egli, B., Kölling, K., Köhler, C., Zeeman, S. C., and Streb, S. (2010). Loss of Cytosolic Phosphoglucomutase Compromises Gametophyte Development in Arabidopsis. Plant Physiol. 154 (4), 1659–1671. doi:10.1104/pp.110.165027
Griffiths-Jones, S. (2004). Rfam: Annotating Non-coding RNAs in Complete Genomes. Nucleic Acids Res. 33 (Database issue), D121–D124. doi:10.1093/nar/gki081
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML Online-Aa Web Server for Fast Maximum Likelihood-Based Phylogenetic Inference. Nucleic Acids Res. 33, W557–W559. doi:10.1093/nar/gki352
Guo, X., Li, Y., Li, C., Luo, H., Wang, L., Qian, J., et al. (2013). Analysis of the Dendrobium Officinale Transcriptome Reveals Putative Alkaloid Biosynthetic Genes and Genetic Markers. Gene 527 (1), 131–138. doi:10.1016/j.gene.2013.05.073
Hamberger, B., Ohnishi, T., Hamberger, B., Séguin, A., and Bohlmann, J. (2011). Evolution of Diterpene Metabolism: Sitka Spruce CYP720B4 Catalyzes Multiple Oxidations in Resin Acid Biosynthesis of Conifer Defense against Insects. Plant Physiol. 157 (4), 1677–1695. doi:10.1104/pp.111.185843
Holt, C., and Yandell, M. (2011). MAKER2: an Annotation Pipeline and Genome-Database Management Tool for Second-Generation Genome Projects. BMC Bioinformatics 12, 491. doi:10.1186/1471-2105-12-491
Huber, S. C., and Huber, J. L. (1996). Role and Regulation of Sucrose-Phosphate Synthase in Higher Plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 431–444. doi:10.1146/annurev.arplant.47.1.431
International Brachypodium Initiative (2010). Genome Sequencing and Analysis of the Model Grass Brachypodium Distachyon. Nature 463 (7282), 763–768. doi:10.1038/nature08747
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 449 (7161), 463–467. doi:10.1038/nature06148
Jiang, S.-Y., Jin, J., Sarojam, R., Ramachandran, S., and Alba, M. (2019). A Comprehensive Survey on the Terpene Synthase Gene Family Provides New Insight into its Evolutionary Patterns. Genome Biol. Evol. 11 (8), 2078–2098. doi:10.1093/gbe/evz142
Johnson, A. D., Handsaker, R. E., Pulit, S. L., Nizzari, M. M., O'Donnell, C. J., and de Bakker, P. I. W. (2008). SNAP: a Web-Based Tool for Identification and Annotation of Proxy SNPs Using HapMap. Bioinformatics 24 (24), 2938–2939. doi:10.1093/bioinformatics/btn564
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The Rapid Generation of Mutation Data Matrices from Protein Sequences. Bioinformatics 8 (3), 275–282. doi:10.1093/bioinformatics/8.3.275
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30 (4), 772–780. doi:10.1093/molbev/mst010
Keane, P. A., and Seoighe, C. (2016). Intron Length Coevolution across Mammalian Genomes. Mol. Biol. Evol. 33, 2682–2691. doi:10.1093/molbev/msw151
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Kim, J., Kang, S.-H., Park, S.-G., Yang, T.-J., Lee, Y., Kim, O. T., et al. (2020). Whole-genome, Transcriptome, and Methylome Analyses Provide Insights into the Evolution of Platycoside Biosynthesis in Platycodon Grandiflorus, a Medicinal Plant. Hortic. Res. 7 (1), 112. doi:10.1038/s41438-020-0329-x
Koonin, E. V., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Krylov, D. M., Makarova, K. S., et al. (2004). A Comprehensive Evolutionary Classification of Proteins Encoded in Complete Eukaryotic Genomes. Genome Biol. 5 (2), R7. doi:10.1186/gb-2004-5-2-r7
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi:10.1101/gr.215087.116
Kuang, X. J., Wang, C. X., Zou, L. Q., Zhu, X. X., and Sun, C. (2016). Advance in Biosynthesis of Terpenoid Indole Alkaloids and its Regulation in Catharanthus Roseus. Zhongguo Zhong Yao Za Zhi 41 (22), 4129–4137. doi:10.4268/cjcmm20162208
Li, L., Stoeckert, C. J., Roos, D. S., and Roos, D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13 (9), 2178–2189. doi:10.1101/gr.1224503
Li, Q., Ding, G., Li, B., and Guo, S.-X. (2017). Transcriptome Analysis of Genes Involved in Dendrobine Biosynthesis in Dendrobium Nobile Lindl. Infected with Mycorrhizal Fungus MF23 (Mycena sp.). Sci. Rep. 7 (1), 316. doi:10.1038/s41598-017-00445-9
Li, Y., Li, F., Gong, Q., Wu, Q., and Shi, J. (2011). Inhibitory Effects of Dendrobium Alkaloids on Memory Impairment Induced by Lipopolysaccharide in Rats. Planta Med. 77 (2), 117–121. doi:10.1055/s-0030-1250235
Li, Z., Wang, Y., Han, B., Yang, Y., Wang, Z., and Sun, Z. (2019). Research Progress on Constituents of Alkaloids in Plants from Dendrobium Sw. Chin. Traditional Herbal Drugs 50 (13), 3246–3254.
Liu, X. F., Zhu, J., Ge, S. Y., Xia, L. J., Yang, H. Y., Qian, Y. T., et al. (2011). Orally Administered Dendrobium Officinale and its Polysaccharides Enhance Immune Functions in BALB/c Mice. Nat. Prod. Commun. 6 (6), 867–870. doi:10.1177/1934578x1100600627
Liu, Y., Tang, Q., Cheng, P., Zhu, M., Zhang, H., Liu, J., et al. (2020). Whole-genome Sequencing and Analysis of the Chinese Herbal Plant Gelsemium Elegans. Acta Pharmaceutica Sinica B 10 (2), 374–382. doi:10.1016/j.apsb.2019.08.004
Liu, Z., Boachon, B., Lugan, R., Tavares, R., Erhardt, M., Mutterer, J., et al. (2015). A Conserved Cytochrome P450 Evolved in Seed Plants Regulates Flower Maturation. Mol. Plant 8 (12), 1751–1765. doi:10.1016/j.molp.2015.09.002
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 25 (5), 955–964. doi:10.1093/nar/25.5.955
Ma, P., Zhang, X., Chen, L., Zhao, Q., Zhang, Q., Hua, X., et al. (2020). Comparative Analysis of Sucrose Phosphate Synthase (SPS) Gene Family between Saccharum Officinarum and Saccharum Spontaneum. BMC Plant Biol. 20 (1), 422. doi:10.1186/s12870-020-02599-7
Magallón, S., Hilu, K. W., and Quandt, D. (2013). Land Plant Evolutionary Timeline: Gene Effects Are Secondary to Fossil Constraints in Relaxed Clock Estimation of Age and Substitution Rates. Am. J. Bot. 100 (3), 556–573. doi:10.3732/ajb.1200416
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654. doi:10.1093/molbev/msab199
Marçais, G., and Kingsford, C. (2011). A Fast, Lock-free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27 (6), 764–770. doi:10.1093/bioinformatics/btr011
McGarvey, D. J., and Croteau, R. (1995). Terpenoid Metabolism. Plant Cell 7 (7), 1015–1026. doi:10.1105/tpc.7.7.1015
Nagasawa, N., Hibara, K.-I., Heppard, E. P., Vander Velden, K. A., Luck, S., Beatty, M., et al. (2013). GIANTEMBRYOencodes CYP78A13, Required for Proper Size Balance between Embryo and Endosperm in rice. Plant J. 75 (4), 592–605. doi:10.1111/tpj.12223
Nelson, D. R., Schuler, M. A., Paquette, S. M., Werck-Reichhart, D., and Bak, S. (2004). Comparative Genomics of Rice and Arabidopsis. Analysis of 727 Cytochrome P450 Genes and Pseudogenes from a Monocot and a Dicot. Plant Physiol. 135 (2), 756–772. doi:10.1104/pp.104.039826
Ng, T. B., Liu, J., Wong, J. H., Ye, X., Wing Sze, S. C., Tong, Y., et al. (2012). Review of Research on Dendrobium, a Prized Folk Medicine. Appl. Microbiol. Biotechnol. 93 (5), 1795–1803. doi:10.1007/s00253-011-3829-7
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 14, 417–419. doi:10.1038/nmeth.4197
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Pu, X., Dong, X., Li, Q., Chen, Z., and Liu, L. (2021). An Update on the Function and Regulation of Methylerythritol Phosphate and Mevalonate Pathways and Their Evolutionary Dynamics. J. Integr. Plant Biol. 63 (7), 1211–1226. doi:10.1111/jipb.13076
Ren, Z., Ji, X., Jiao, Z., Luo, Y., Zhang, G.-Q., Tao, S., et al. (2020). Functional Analysis of a Novel C-Glycosyltransferase in the Orchid Dendrobium Catenatum. Hortic. Res. 7, 111. doi:10.1038/s41438-020-0330-4
Rende, U., Wang, W., Gandla, M. L., Jönsson, L. J., and Niittylä, T. (2017). Cytosolic Invertase Contributes to the Supply of Substrate for Cellulose Biosynthesis in Developing wood. New Phytol. 214 (2), 796–807. doi:10.1111/nph.14392
Ro, D.-K., Arimura, G.-I., Lau, S. Y. W., Piers, E., and Bohlmann, J. (2005). Loblolly pine Abietadienol/abietadienal Oxidase PtAO (CYP720B1) Is a Multifunctional, Multisubstrate Cytochrome P450 Monooxygenase. Proc. Natl. Acad. Sci. 102 (22), 8060–8065. doi:10.1073/pnas.0500825102
Roitsch, T., and González, M.-C. (2004). Function and Regulation of Plant Invertases: Sweet Sensations. Trends Plant Sci. 9 (12), 606–613. doi:10.1016/j.tplants.2004.10.009
Schuler, M. A. (2015). P450s in Plants, Insects, and Their Fungal Pathogens. Cytochrome P450, 409–449. doi:10.1007/978-3-319-12108-6_7
Simkin, A. J., Miettinen, K., Claudel, P., Burlat, V., Guirimand, G., Courdavault, V., et al. (2013). Characterization of the Plastidial Geraniol Synthase from Madagascar Periwinkle Which Initiates the Monoterpenoid branch of the Alkaloid Pathway in Internal Phloem Associated Parenchyma. Phytochemistry 85, 36–43. doi:10.1016/j.phytochem.2012.09.014
Stamatakis, A. (2014). RAxML Version 8: A Tool for Phylogenetic Analysis and post-analysis of Large Phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 34, W435–W439. doi:10.1093/nar/gkl200
Stival Sena, J., Giguère, I., Boyle, B., Rigault, P., Birol, I., Zuccolo, A., et al. (2014). Evolution of Gene Structure in the conifer Picea glauca: a Comparative Analysis of the Impact of Intron Size. BMC Plant Biol. 14, 95. doi:10.1186/1471-2229-14-95
Swinburne, I. A., and Silver, P. A. (2008). Intron Delays and Transcriptional Timing during Development. Develop. Cel 14, 324–330. doi:10.1016/j.devcel.2008.02.002
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The Genome of Black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313 (5793), 1596–1604. doi:10.1126/science.1128691
Tzin, V., Malitsky, S., Zvi, M. M. B., Bedair, M., Sumner, L., Aharoni, A., et al. (2012). Expression of a Bacterial Feedback‐insensitive 3‐deoxy‐ D ‐arabino‐heptulosonate 7‐phosphate Synthase of the Shikimate Pathway in Arabidopsis Elucidates Potential Metabolic Bottlenecks between Primary and Secondary Metabolism. New Phytol. 194 (2), 430–439. doi:10.1111/j.1469-8137.2012.04052.x
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 18 (7), 411–424. doi:10.1038/nrg.2017.26
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). Genomescope Fast Reference-free Genome Profiling from Short Reads. Bioinformatics 33, 2202. doi:10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9 (11), e112963. doi:10.1371/journal.pone.0112963
Wang, C., Liu, C., Roqueiro, D., Grimm, D., Schwab, R., Becker, C., et al. (2015a). Genome-wide Analysis of Local Chromatin Packing in Arabidopsis thaliana. Genome Res. 25 (2), 246–256. doi:10.1101/gr.170332.113
Wang, J.-H., Zha, X.-Q., Luo, J.-P., and Yang, X.-F. (2010). An Acetylated Galactomannoglucan from the Stems of Dendrobium Nobile Lindl. Carbohydr. Res. 345 (8), 1023–1027. doi:10.1016/j.carres.2010.03.005
Wang, X., Li, Y., Zhang, H., Sun, G., Zhang, W., and Qiu, L. (2015b). Evolution and Association Analysis of GmCYP78A10 Gene with Seed Size/weight and Pod Number in Soybean. Mol. Biol. Rep. 42 (2), 489–496. doi:10.1007/s11033-014-3792-3
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Wang, Z., Wang, C., Su, T., and Zhang, J. (2014). Antioxidant and Immunological Activities of Polysaccharides from Gentiana Scabra Bunge Roots. Carbohydr. Polym. 112, 114–118. doi:10.1016/j.carbpol.2014.05.077
Wang, Z., Zhao, M., Cui, H., Li, J., and Wang, M. (2020). Transcriptomic Landscape of Medicinal Dendrobium Reveals Genes Associated with the Biosynthesis of Bioactive Components. Front. Plant Sci. 11, 391. doi:10.3389/fpls.2020.00391
Weeden, N. F. (2018). Domestication of Pea (Pisum Sativum L.): The Case of the Abyssinian Pea. Front. Plant Sci. 9, 515. doi:10.3389/fpls.2018.00515
Wei, K., and Chen, H. (2018). Global Identification, Structural Analysis and Expression Characterization of Cytochrome P450 Monooxygenase Superfamily in rice. BMC Genomics 19 (1), 35. doi:10.1186/s12864-017-4425-8
Xin, T., Zhang, Y., Pu, X., Gao, R., Xu, Z., and Song, J. (2019). Trends in Herbgenomics. Sci. China Life Sci. 62 (3), 288–308. doi:10.1007/s11427-018-9352-7
Xu, H., Song, J., Luo, H., Zhang, Y., Li, Q., Zhu, Y., et al. (2016). Analysis of the Genome Sequence of the Medicinal Plant Salvia Miltiorrhiza. Mol. Plant 9 (6), 949–952. doi:10.1016/j.molp.2016.03.010
Yang, Z. (1997). PAML: a Program Package for Phylogenetic Analysis by Maximum Likelihood. Bioinformatics 13 (5), 555–556. doi:10.1093/bioinformatics/13.5.555
Yuan, Y., Yu, M., Jia, Z., Song, X., Liang, Y., and Zhang, J. (2018). Analysis of Dendrobium Huoshanense Transcriptome Unveils Putative Genes Associated with Active Ingredients Synthesis. BMC Genomics 19 (1), 978. doi:10.1186/s12864-018-5305-6
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan - an Integration Platform for the Signature-Recognition Methods in InterPro. Bioinformatics 17, 847–848. doi:10.1093/bioinformatics/17.9.847
Zhang, G.-Q., Liu, K.-W., Li, Z., Lohaus, R., Hsiao, Y.-Y., Niu, S.-C., et al. (2017). The Apostasia Genome and the Evolution of Orchids. Nature 549 (7672), 379–383. doi:10.1038/nature23897
Zhang, G.-Q., Xu, Q., Bian, C., Tsai, W.-C., Yeh, C.-M., Liu, K.-W., et al. (2016). The Dendrobium Catenatum Lindl. Genome Sequence Provides Insights into Polysaccharide Synthase, floral Development and Adaptive Evolution. Sci. Rep. 6, 19029. doi:10.1038/srep19029
Zhang, Y., Zhang, G.-Q., Zhang, D., Liu, X.-D., Xu, X.-Y., Sun, W.-H., et al. (2021). Chromosome-scale Assembly of the Dendrobium Chrysotoxum Genome Enhances the Understanding of Orchid Evolution. Hortic. Res. 8, 183. doi:10.1038/s41438-021-00621-z
Zheng, S.-g., Hu, Y.-d., Zhao, R.-x., Yan, S., Zhang, X.-q., Zhao, T.-m., et al. (2018). Genome-wide Researches and Applications on Dendrobium. Planta 248 (4), 769–784. doi:10.1007/s00425-018-2960-4
Keywords: Dendrobium nobile, chromosome-level assembly, polysaccharide and alkaloid (dendrobine), gene family, transcriptome
Citation: Xu Q, Niu S-C, Li K-L, Zheng P-J, Zhang X-J, Jia Y, Liu Y, Niu Y-X, Yu L-H, Chen D-F and Zhang G-Q (2022) Chromosome-Scale Assembly of the Dendrobium nobile Genome Provides Insights Into the Molecular Mechanism of the Biosynthesis of the Medicinal Active Ingredient of Dendrobium. Front. Genet. 13:844622. doi: 10.3389/fgene.2022.844622
Received: 28 December 2021; Accepted: 11 February 2022;
Published: 01 March 2022.
Edited by:
Zefeng Yang, Yangzhou University, ChinaReviewed by:
Yunqing Cheng, Jilin Normal University, ChinaFatma Aydinoglu, Gebze Technical University, Turkey
Xiaojun Zhou, Luoyang Normal University, China
Copyright © 2022 Xu, Niu, Li, Zheng, Zhang, Jia, Liu, Niu, Yu, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Xu, eHFAZ3pobXUuZWR1LmNu; Duan-Fen Chen, Y2hlbmR1YW5mZW5AMTYzLmNvbQ==; Guo-Qiang Zhang, Z3VvcWlhbmd6aGFuZ2NuQDE2My5jb20=
†These authors have contributed equally to this work