 Shan Tang1
Shan Tang1 Li Bai1
Li Bai1 Yuan Gao1Wei Hou1Wenyan Song1
Yuan Gao1Wei Hou1Wenyan Song1 Hui Liu1Zhongjie Hu1
Hui Liu1Zhongjie Hu1 Zhongping Duan1Liaoyun Zhang2*
Zhongping Duan1Liaoyun Zhang2* Sujun Zheng1*
Sujun Zheng1*- 1Beijing Youan Hospital, Capital Medical University, Beijing, China
- 2The First Hospital of Shanxi Medical University, Taiyuan, China
Type 3 hereditary hemochromatosis (HH) is a rare form of HH characterized by genetic mutation in the TFR2 gene. Clinical features reported in patients with type 3 HH include abnormal liver function, liver fibrosis, cirrhosis, diabetes, hypogonadism, cardiomyopathy, and skin pigmentation. Since its original description in 2000, 33 pathogenic TFR2 mutations associated with HH have been described until now. Here, we first reported a Chinese pedigree of TFR2-related hemochromatosis with a novel compound heterozygous mutation c.1288G > A (p.G430R)/c.960T > A (p.Y320X). Interestingly, different phenotypes were reported although the proband and his sister shared the same gene mutation. This inconsistency between genotypes and phenotypes indicates multifactorial etiology contributing to the development of HH. Our report broadens the mutation spectrum of the TFR2 gene associated with HH.
Introduction
Hereditary hemochromatosis (HH) is defined as an inherited iron overload disorder characterized by excessive absorption of iron (Kowdley et al., 2019). Over time, iron deposits in multiple organs including the liver, pancreas, heart, joints, and pituitary gland, further leading to liver cirrhosis, restrictive cardiomyopathy, heart failure, arthropathy, skin pigmentation, diabetes mellitus, and so on (Bacon et al., 2011). HH can be classified as types 1, 2, 3, and 4. Type 1 HH caused by the HFE gene mutation is the most prevalent form of HH in Caucasians (Brissot et al., 2018). Mutations in hemojuvelin BMP co-receptor (HJV) and hepcidin antimicrobial peptide (HAMP) genes (type 2a and 2b HH, respectively) result in juvenile hemochromatosis, the most severe form of HH (Papanikolaou et al., 2004). Type 4 HH, also known as ferroportin (FPN) disease, is the only autosomal dominant form of hemochromatosis due to the mutations in the solute carrier family 40-member 1(SLC40A1) gene (Joshi et al., 2015). Type 3 HH is a rare form of HH characterized by the genetic alterations in the transferrin receptor 2 (TFR2) gene, with an estimated allele prevalence between 0.0001 and 0.0004 among European populations (Wallace and Subramaniam, 2016). The human TFR2 gene is located on chromosome 7q22 and encodes transferrin receptor 2. TFR2 is a type II transmembrane glycoprotein, a member of the TFR family. It provides iron to the cell by internalization of the transferrin iron complex through receptor-mediated endocytosis (Silvestri et al., 2014). Hepcidin is the central regulator of systemic iron homeostasis, which can not only prevent absorption of iron from the gut but also prevent the release of iron from macrophages (Worthen and Enns, 2014). Hepcidin expression at an abnormally low levels leads to iron overload. Hemochromatosis, which is associated with mutations in TFR2, usually occurs at an earlier age than HFE-related hemochromatosis and usually has a more severe phenotype (Pietrangelo, 2010).
Here, we described a Chinese pedigree presented with typical features of HH. Also, we found a novel compound heterozygous mutation in the TFR2 gene, namely, c.1288G > A (p.G430R)/c.960T > A (p.Y320X). To the best of our knowledge, this is the first report on a Chinese pedigree of TFR2-related hemochromatosis.
Case Presentation
The patient (Figure 4, II-1), a 48-year-old male, was admitted to Beijing YouAn Hospital due to liver cirrhosis. Abdominal ultrasound suggested cirrhosis and splenomegaly 4 months ago even when the patient had no discomfort. In another hospital, he was diagnosed with diabetes and treated with insulin. He hardly ever drank and denied a family history of liver diseases. Physical examination revealed that there are no other abnormalities except for skin pigmentation.
A thorough examination was carried out to find out the cause of cirrhosis. The liver function test showed that the levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were elevated (Table 1). Serum markers for viral hepatitis and autoimmune liver diseases were all negative. Notably, the patient had abnormal plasma iron indices: iron, 47 μmol/L; transferrin saturation, 97.9%; and ferritin>2000 ng/ml. HbA1c was significantly elevated. Testosterone was normal (18.26 nmol/L).
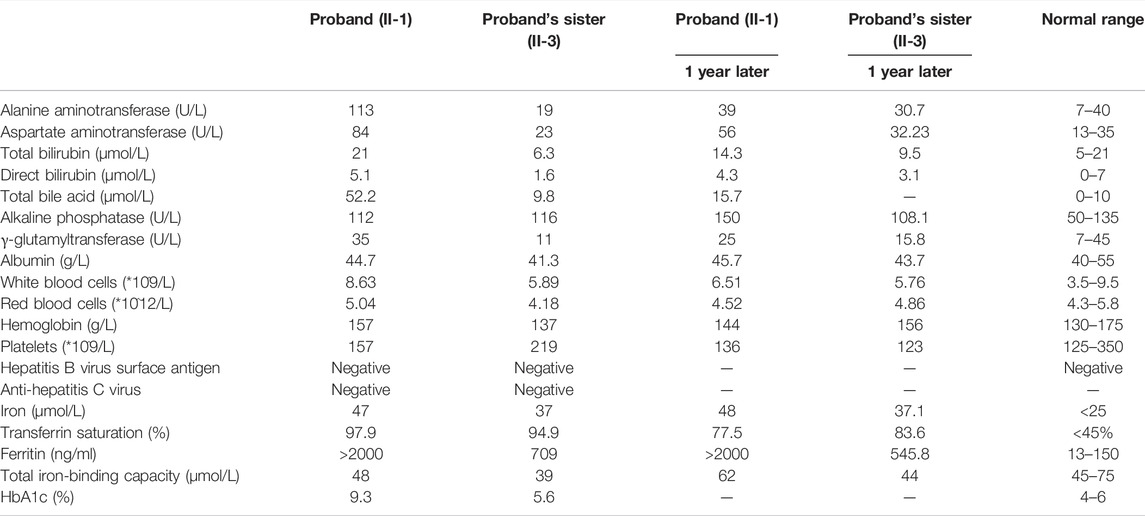
TABLE 1. Laboratory test results of the proband and the proband’s family.
Pre-contrast computed tomography (CT) revealed hepatomegaly with an increased attenuation over the liver and spleen, splenomegaly, and collateral circulation formation (Figure 1A). Magnetic resonance imaging (MRI) showed a typical decrease in T2-weighted signal intensity over the liver (Figure 1B).
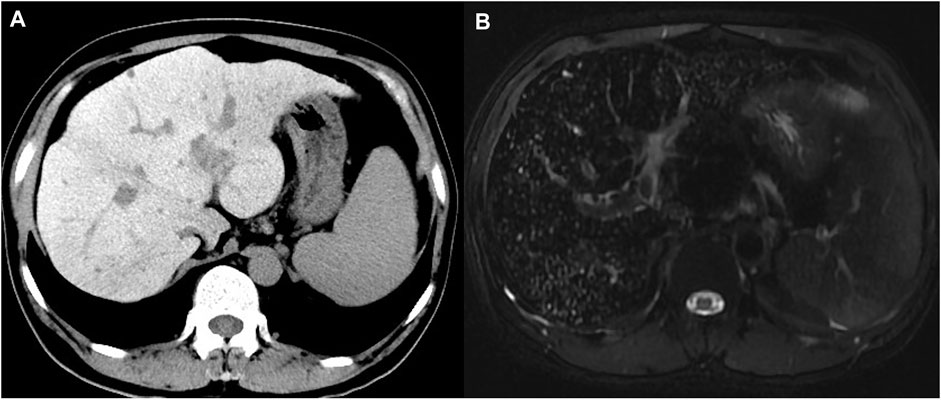
FIGURE 1. Computed tomography and magnetic resonance imaging of the proband. (A) Precontrast CT showed hyper-attenuation of liver parenchyma; (B) MRI T2-weighted image showed diffuse low signal intensity of liver parenchyma obviously.
A liver biopsy was performed, and the liver histology was evaluated by an experienced pathologist. The findings confirmed established cirrhosis with a marked deposition of iron in the hepatocytes and biliary epithelia, which is consistent with hemochromatosis (Figures 2A,B).
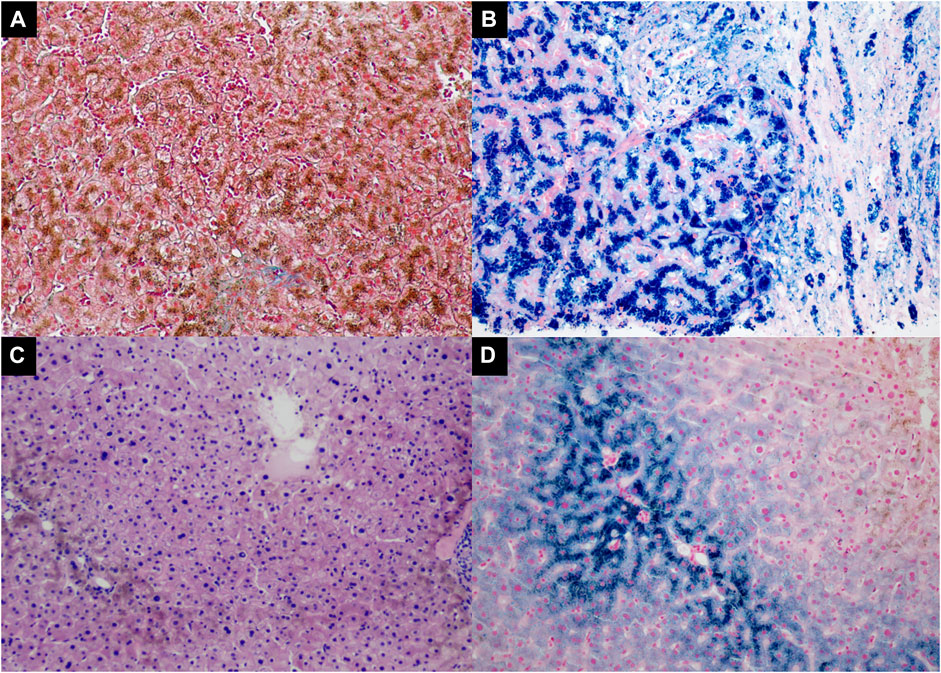
FIGURE 2. The liver histopathology of the proband [Panel (A, B), II-1] and the proband’s sister [Panel (C, D), II-3]. (A) Increased collagen fibers stained green by Masson Trichrome stain in the expanded fibrous portal tract with cirrhosis (Masson trichrome, 100×); (B) Marked iron deposition in the hepatocytes and biliary epithelium (Prussian blue, 200×); (C) Marked deposition of hemosiderin in the hepatocytes (H&E, 100×); (D) The proband’s sister had iron deposited in the periportal hepatocytes (Prussian blue, 200×).
We also investigated one of the patient’s sisters (Figure 4, II-3), and the results showed that she had abnormal plasma iron indices as well: iron, 37 μmol/L; transferrin saturation, 94.9%; and ferritin>709 ng/ml (Table 1), while her HbA1c and sex hormones levels were normal (HbA1c 5.6%, estradiol <37 pmol/L, follicle stimulating hormone 58.02 IU/L, luteinizing hormone 25.86 IU/L, progesterone 0.5 nmol/L, prolactin 464.13 mIU/L, and testosterone 0.94 nmol/L). Also, a fine needle aspiration biopsy was performed, which showed iron deposition in the periportal hepatocytes (Figures 2C,D).
Genetic Testing Results
Genetic analysis was performed for the patient and his family to explore the genetic factor for hemochromatosis. Panel sequencing was performed for major genes involved in the liver metabolism and carbohydrate metabolism including the common HH-related geness, namely, HFE, HJV, HAMP, TFR2, and SLC40A1. The SureSelect Human All ExonV6 custom chip was used to capture exons, and the Illumina sequencing platform was used for high-throughput sequencing. BWA (0.7.12-r1039) (Li and Durbin, 2009) was used to compare next-generation sequencing data with the human genome; ANNOVAR ($ Date: 2015-06-17) (Park and Park, 2021) was used to annotate mutation sites according to dbSNP, Clinvar, ExAC, 1,000 Genomes, and other databases. Possible pathogenicity of gene mutation was graded by the ACMG (American Society of Medical Genetics and Genomics) genetic mutation grading system. Then, the target sequence of the suspected pathogenic mutation was amplified by PCR and sequenced via an ABI3730xL sequencer (Applied Biosystems, Foster City, CA, United States). Finally, the comparisons and analysis for Sanger sequencing data were performed by SeqMan (DNASTAR, Madison, Wisconsin, United States). Potential functional effects of detected missense mutations were predicted by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), SIFT (http://sift.jcvi.org/), PROVEAN (http://provean.jcvi.org/index.php), MutationTaster (http://mutationtaster.org/), and FATHMM (http://fathmm.biocompute.org.uk/).
The results showed that the patient (II-1) had new heterozygous mutations c.1288G > A (p.G430R)/c.960T > A (p.Y320X) in the TFR2 gene. These mutations were further confirmed by Sanger sequencing (Supplementary Figure S1). As a result, the novel c.960T > A mutation led to a premature stop codon at amino acid 320 (p.Y320X). According to the ACMG guideline, the mutation c.960T > A (p.Y320X) was identified as pathogenic (PVS1: null variant + PM2: absent from controls + PP4: the phenotype of the patient is highly consistent with HH type 3). The mutation c.1288G > A (p.G430R) is a pathogenic mutation that has been reported earlier (Majore et al., 2013). Here, we also predicted its pathogenicity by utilizing six kinds of prediction software. All software programs except for FATHMM showed that the p.G430R mutation was damaging (Table 2). The mutation was identified as likely pathogenic according to the ACMG guideline (PM1: the variant is in the domain + PM3: there is a literature suggesting this variant + PM2: absent from controls + PP3: bioinformatics protein function comprehensive prediction software predicts outcomes as harmful + PP4: the phenotype of the patient is highly consistent with HH type 3). The PhastCons scores of these two mutations were both 1, and the corresponding PhyloP values were 4.594 and 4.844, respectively, suggesting the high conservation of these two amino acids. Moreover, amino acid sequence alignments for the corresponding parts in the TFR2 protein in multiple species were studied (Supplementary Figure S3). The results showed that the two amino acids at positions 430 and 320 were absolutely conserved.
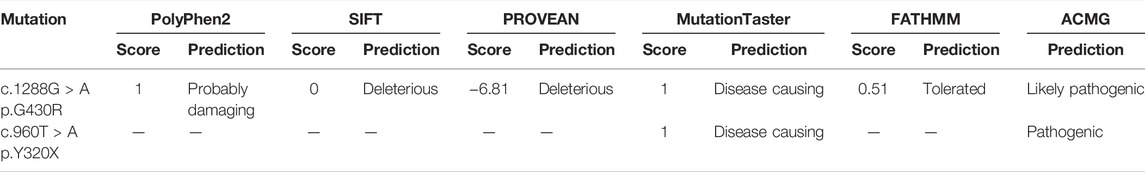
TABLE 2. Bioinformatics analysis of the TFR2 mutations.
The molecular structure of TFR2 was constructed by SWISS-MODEL (https://swissmodel.expasy.org/interactive). Also, molecular graphics were analyzed by PyMOL software. As shown in Figure 3, the secondary structure of the predicted mutation was not affected by the mutation c.1288G > A (p.G430R) but affected by the mutation c.960T > A (p.Y320X). There is no electricity in the blank space in the model evaluation graph (Supplementary Figure S2), indicating that the model quality is acceptable.
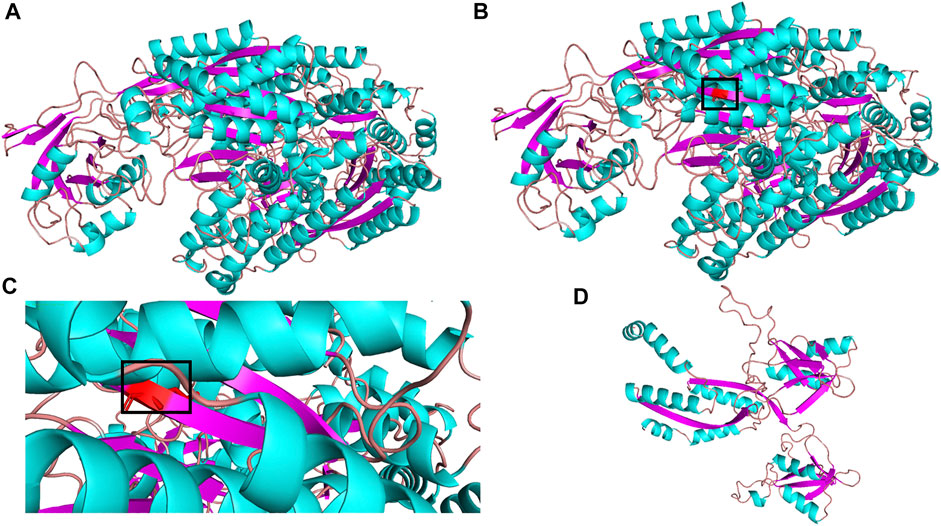
FIGURE 3. 3D structure of TFR2 protein. (A) wild-type; (B) mutant type c.G1288A (p.G430R); (C) partial mutation graph c.G1288A (p.G430R); (D) mutant type c.T960A (p.Y320X).
Moreover, we tested the mutations in the TFR2 gene for his family members (Figure 4). The same heterozygous mutations were found in one of his sisters (II-3). In addition, one of the patient’s sisters (II-4) was found to carry a heterozygous mutation c.1288G > A (p.G430R). However, TFR2 gene mutation was not found in the other sister (II-2). The patient’s parents had passed away, so we cannot perform their genetic sequencing. According to the genetic sequencing results of his sisters, we speculated the mutations in the proband were compound heterozygous. In addition, the nephew (III-1) of the proband was also confirmed to have a heterozygous mutation c.1288G > A (p.G430R).
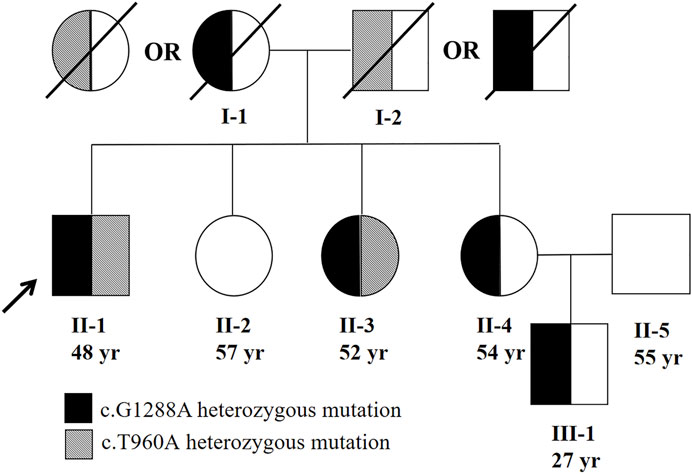
FIGURE 4. Family pedigree of patient. The parents of the proband should each had a heterozygous mutation (c.960T > A or c.G1288A). The proband’s parents were not consanguineous.
Ethical Statement
All procedures involving human participants were in accordance with the ethical standards of the Institute Ethical Committee of Beijing YouAn Hospital, Capital Medical University, Beijing, China, and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient and his family members.
Discussion
In recent years, increasing mutations in the TFR2 gene which are related to type 3 HH have been documented (Hernández et al., 2021; Joshi et al., 2015). As shown in Supplementary Table S1, a total of 33 pathogenic TFR2 mutations associated with HH have been described in the Human Gene Mutation Database (Stenson et al., 2020). Most patients were Caucasian (27/33, 81.8%), males (17/25, 68.0%). The average age of the patients was 31.4 years, and most of them had liver cirrhosis. The data on the mutations in the TFR2 gene in Asian patients are relatively rare with only five articles reported.
In the present study, we described a Chinese Han pedigree suffering from type 3 HH with a novel compound heterozygous mutation c.1288G > A (p.G430R)/c.960T > A (p.Y320X) in the TFR2 gene. The proband exhibits very high plasma transferrin saturation and deposition of iron in the hepatocytes and biliary epithelia, and this phenotype highly suggested hemochromatosis. The gene sequencing results confirm the diagnosis of type 3 hemochromatosis. One of his sisters shares the same TFR2 gene mutation; however, her liver pathology is milder than that in the proband. Except for the hepatic manifestations, other clinical features including diabetes, hypogonadism, and skin pigmentation have been reported in patients with type 3 hemochromatosis. Given this, we evaluated these symptoms in the proband and his family members. As a result, the proband developed skin pigmentation and diabetes, while his sister showed no other extrahepatic manifestations. We considered that the difference in the phenotype between the proband and his sister can be ascribed to other factors including sex (menstruation), environment (iron intake), or possibly other unknown genetic factors. These factors apart from TFR2 gene mutation probably contribute to the development of HH. At present, both the proband and his sister have received phlebotomy therapy and resulted in improvement in the laboratory test (Table 1).
This case report highlights three important clinical issues. First, our team discovered the TFR2 gene c.960T > A (p.Y320X) mutation, which led to a premature stop codon at amino acid 320 (p.Y320X). According to the ACMG genetic mutation grading system, this mutation is pathogenic. We also analyzed the predictive mutation structure and found that the Y320X mutation affects the secondary structure of the TFR2 protein and may destroy its function. Our report expands the mutation spectrum of the TFR2 gene associated with HH. Second, this is the first time to report a novel compound heterozygous mutation c. G1288A (p.G430R)/c.T960A (p.Y320X) in the TFR2 gene in a Chinese pedigree. We analyzed the minor allele frequency (MAF) of the c. G1288A (p.G430R) mutation by utilizing different public databases in different populations (Supplementary Table S2). The MAF of c. G1288A (p.G430R) is between 0.000003978 and 0.000008 in different databases, which indicates that the mutation is extremely rare. Third, early diagnosis and treatment for patients with HH have been documented to improve the prognosis as well as decrease mortality. It is helpful to remind even physicians in Asia that where the incidence of HH is low, it is important to keep the diagnosis of HH in mind, when a patient is presented with symptoms such as iron overload, liver fibrosis, diabetes, and so on.
CONCLUSION
In conclusion, we first reported a Chinese pedigree of TFR2-related hemochromatosis with a novel compound heterozygous mutation c.1288G > A (p.G430R)/c.960T > A (p.Y320X). Our report broadens the mutation spectrum of the TFR2 gene associated with HH. On the other hand, our results reveal inconsistency between genotypes and phenotypes, which indicates multifactorial etiology contributing to the development of HH.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Beijing You'An Hospital of Capital Medical University and the Declaration of Helsinki (revised in 2013). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by grants from the Key Medical Professional Development Plan (Sailing plan) of Beijing Hospital Management Center (ZYLX202125), the Beijing Advanced Innovation Center for Big Data-Based Precision Medicine (1212040205), the Beijing Municipal Natural Science Foundation (7202068, 72222093, and 7222094), and the Digestive Medical Coordinated Development Center of Beijing Municipal Administration of Hospitals (XXZ0503). Capital Health Development Research Project (2022-1-2182).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Jing Zhao and LB for their help in polishing the language of our article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.836431/full#supplementary-material
References
Bacon, B. R., Adams, P. C., Kowdley, K. V., Powell, L. W., and Tavill, A. S. (2011). Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology 54, 328–343. doi:10.1002/hep.24330
Brissot, P., Pietrangelo, A., Adams, P. C., De Graaff, B., Mclaren, C. E., and Loréal, O. (2018). Haemochromatosis. Nat. Rev. Dis. Primers 4, 18016. doi:10.1038/nrdp.2018.16
Hernández, G., Ferrer-Cortès, X., Venturi, V., Musri, M., Pilquil, M. F., Torres, P. M. M., et al. (2021). New Mutations in HFE2 and TFR2 Genes Causing Non HFE-Related Hereditary Hemochromatosis. Genes 12, 1980. doi:10.3390/genes12121980
Joshi, R., Shvartsman, M., Morán, E., Lois, S., Aranda, J., Barqué, A., et al. (2015). Functional Consequences of Transferrin Receptor‐2 Mutations Causing Hereditary Hemochromatosis Type 3. Mol. Genet. Genomic Med. 3, 221–232. doi:10.1002/mgg3.136
Kowdley, K. V., Brown, K. E., Ahn, J., and Sundaram, V. (2019). ACG Clinical Guideline: Hereditary Hemochromatosis. Am. J. Gastroenterol. 114, 1202–1218. doi:10.14309/ajg.0000000000000315
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Majore, S., Ricerca, B. M., Radio, F. C., Binni, F., Cosentino, I., and Gallusi, G. (2013). Type 3 Hereditary Hemochromatosis in a Patient from Sub-Saharan Africa: Is There a Link between African Iron Overload and TFR2 Dysfunction?. Blood Cells Mol. Dis. 50 (1), 31–32. doi:10.1016/j.bcmd.2012.08.007
Papanikolaou, G., Samuels, M. E., Ludwig, E. H., Macdonald, M. L. E., Franchini, P. L., Dubé, M.-P., et al. (2004). Mutations in HFE2 Cause Iron Overload in Chromosome 1q-Linked Juvenile Hemochromatosis. Nat. Genet. 36, 77–82. doi:10.1038/ng1274
Park, K.-J., and Park, J.-H. (2021). Variations in Nomenclature of Clinical Variants between Annotation Tools. Lab. Med. lmab074. doi:10.1093/labmed/lmab074
Pietrangelo, A. (2010). Hereditary Hemochromatosis: Pathogenesis, Diagnosis, and Treatment. Gastroenterology 139, 393–408. doi:10.1053/j.gastro.2010.06.013
Silvestri, L., Nai, A., Pagani, A., and Camaschella, C. (2014). The Extrahepatic Role of TFR2 in Iron Homeostasis. Front. Pharmacol. 5, 93. doi:10.3389/fphar.2014.00093
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The Human Gene Mutation Database (HGMD): Optimizing its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 139, 1197–1207. doi:10.1007/s00439-020-02199-3
Wallace, D. F., and Subramaniam, V. N. (2016). The Global Prevalence of HFE and Non-HFE Hemochromatosis Estimated from Analysis of Next-Generation Sequencing Data. Genet. Med. 18, 618–626. doi:10.1038/gim.2015.140
Keywords: case report, TFR2 gene, compound heterozygous mutation, hereditary hemochromatosis, Chinese
Citation: Tang S, Bai L, Gao Y, Hou W, Song W, Liu H, Hu Z, Duan Z, Zhang L and Zheng S (2022) A Novel Mutation of Transferrin Receptor 2 in a Chinese Pedigree With Type 3 Hemochromatosis: A Case Report. Front. Genet. 13:836431. doi: 10.3389/fgene.2022.836431
Received: 17 December 2021; Accepted: 11 March 2022;
Published: 08 April 2022.
Edited by:
María L. Couce, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Jinchen Li, Central South University, ChinaEduardo Arroyo-Pardo, Complutense University of Madrid, Spain
Copyright © 2022 Tang, Bai, Gao, Hou, Song, Liu, Hu, Duan, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sujun Zheng, emhlbmdzdWp1bkBjY211LmVkdS5jbg==; Liaoyun Zhang, emx5c2d6eUAxNjMuY29t