 Yingfeng Niu1†Guohua Li1Shubang Ni1Xiyong He1Cheng Zheng1Ziyan Liu1Lidan Gong1Guanghong Kong1
Yingfeng Niu1†Guohua Li1Shubang Ni1Xiyong He1Cheng Zheng1Ziyan Liu1Lidan Gong1Guanghong Kong1 Wei Li2*
Wei Li2* Jin Liu1*†
Jin Liu1*†- 1Yunnan Institute of Tropical Crops, Xishuangbanna, China
- 2School of Life Sciences, Institute of Life Sciences and Green Development, Hebei University, Baoding, China
Macadamia is an evergreen tree belonging to the Proteaceae family. The two commercial macadamia species, Macadamia integrifolia and M. tetraphylla, are highly prized for their edible kernels. The M. integrifolia genome was recently sequenced, but the genome of M. tetraphylla has to date not been published, which limits the study of biological research and breeding in this species. This study reports a high-quality genome sequence of M. tetraphylla based on the Oxford Nanopore Technologies technology and high-throughput chromosome conformation capture techniques (Hi-C). An assembly of 750.87 Mb with 51.11 Mb N50 length was generated, close to the 740 and 758 Mb size estimates by flow cytometry and k-mer analysis, respectively. Genome annotation indicated that 61.42% of the genome is composed of repetitive sequences and 34.95% is composed of long terminal repeat retrotransposons. Up to 31,571 protein-coding genes were predicted, of which 92.59% were functionally annotated. The average gene length was 6,055 bp. Comparative genome analysis revealed that the gene families associated with defense response, lipid transport, steroid biosynthesis, triglyceride lipase activity, and fatty acid metabolism are expanded in the M. tetraphylla genome. The distribution of fourfold synonymous third-codon transversion showed a recent whole-genome duplication event in M. tetraphylla. Genomic and transcriptomic analysis identified 187 genes encoding 33 crucial oil biosynthesis enzymes, depicting a comprehensive map of macadamia lipid biosynthesis. Besides, the 55 identified WRKY genes exhibited preferential expression in root as compared to that in other tissues. The genome sequence of M. tetraphylla provides novel insights for breeding novel varieties and genetic improvement of agronomic traits.
Introduction
Macadamia is an evergreen nut tree belonging to the Proteaceae family, genus Macadamia F. Muell, commercially grown for their high-value kernels (Toft et al., 2018). The genus Macadamia F. Muell. contains four different species, namely, M. integrifolia, M. tetraphylla, M. ternifolia, and M. jansenii (Akinsanmi et al., 2017), but only M. integrifolia, M. tetraphylla, and their hybrids (M. integrifolia × M. tetraphylla) are used for commercial nuts production (Hardner, 2016). Due to the large-scale commercial cultivation in Hawaii (United States) in 1948 (Ahmad Termizi et al., 2014), macadamia was also named as Hawaiian Nuts, which is native to the subtropical rainforest of Queensland, New South Wales, Australia (Neal et al., 2010; Shapcott and Powell, 2011).
Macadamia kernels are rich in unsaturated fatty acids, essential amino acids, trace elements, and vitamins, but monounsaturated fatty acids and palmitoleic acid contents are extremely high (Kaijser et al., 2000). Macadamia nuts are eaten directly or used as raw material for processing high-grade edible oil (Navarro and Rodrigues, 2018). Macadamia nuts are very nutritious and deeply subjected to a large consumer approbation fancy, to whom it is also known as the “Queen of nuts.” Long-term consumption of macadamia lowers blood cholesterol and viscosity of platelets, prevents arteriosclerosis, reduces heart disease, myocardial infarction, and other cardiovascular diseases (Garg et al., 2003; Garg et al., 2007), World consumption of macadamia nuts has rapidly grown in recent years with more than 400,000 tons in demand (FAO statistics, year), yet the current global supply is only ∼40,000 tons. Within the current and future periods, macadamia nuts production is still in short supply (Trueman and Turnbull, 1994). The commercial growing areas of macadamia are located in the tropical and subtropical belts, although the traditional cultivated area is in the United States and Australia (Trueman and Turnbull, 1994). The total macadamia planted area has rapidly grown over the past decade because the cultivation technology is simple, planting high income, the plants are more cold-resistant than rubber trees, bananas, and other traditional tropical crops, yet very suitable for the cool weather of tropical and subtropical countries. Globally, the largest macadamia orchard area is in China (300,000 ha), followed by South Africa, Australia, Kenya, Guatemala, and the United States in that order.
Macadamia is diploid (2n = 28) with genome size estimates of 652–780 Mb (Chagné, 2015). In recent years, the genome sequences of many important tropical crops have been reported, but reports on the genome of macadamia are very few. The chloroplast genomes of M. integrifolia, M. ternifolia, and M. tetraphylla were sequenced in 2014, 2017, and 2018 by Australian and Chinese researchers (Nock et al., 2014; Liu et al., 2017; Liu et al., 2018), and the draft genome and transcriptome of M. integrifolia cultivar 741 was sequenced in 2016. The total assembly length is 518 Mb, spanning ∼79% of the estimated genome size (Nock et al., 2016). However, there remains no report on the genome sequencing of M. tetraphylla to date. M. tetraphylla is an important parent species of commercially grown macadamia varieties (Pisanu et al., 2009), and the genome sequencing of M. tetraphylla will provide abundant genetic information and references for the screening of breeding materials.
Materials and Methods
Sample Collection, Library Construction, and Sequencing
A cultivated M. tetraphylla plant was collected from Xishuangbanna, Yunnan Province, China. The collected plant samples were immediately frozen in liquid nitrogen and stored at −80°C before DNA isolation. High-molecular-weight genomic DNA was extracted using a Qiagen plant genomic DNA extraction kit (QIAGEN, Hilden, Germany). Sequencing library was prepared with Nanopore Genomic Sequencing Kit SQK-MAP006 (ONT, United Kingdom) and a PCR-free “native barcoding” kit provided by ONT. Blunt/TA ligase Master Mix (M0367S, NEB) was used to ligate native barcode adapters for 10 min at room temperature. A 1:1 volume of AMPure XP beads was used to purify the barcoded DNA, and the DNA was eluted in 26 μL nuclease free water. Hairpin adapters were ligated and added to the pooled library DNA to give a final reaction volume of 100 μL. The reaction mixture was incubated for 10 min at room temperature. The final reaction was cleaned using prewashed Dynabeads MyOne Streptavidin C1 beads (65001; Thermo Fisher Scientific). The library was loaded on a single R9.4 flow cell and sequenced on a GridION X5 platform (Oxford Nanopore Technology, OX4 4DQ, Oxford, United Kingdom). Reads were trimmed according to a minimum read quality of Q15. Reads with length shorter than 30 bp were excluded and sequencing adapters were removed. MinION (TM) 2D reads were filtered into passed and failed reads using Metrichore basecaller.
To construct the Hi-C library, ∼4–6 g young leaves were harvested and frozen in liquid nitrogen. The nuclear DNA was cross-linked in situ in 2% formaldehyde at room temperature before extraction. The nuclei extracted were then restricted with HindIII restriction enzyme. Free ends were then biotinylated, diluted and ligated randomly. The generated libraries were sequenced on Illumina Hiseq4000 platform.
Total RNA was isolated from five tissues (young leaves, young inflorescences, flowering inflorescences, proteoid roots, and barks) using the Column Plant RNAout kit (TIANDZ, Beijing, China). A 300-cycle kit was used for a 2 × 150 bp paired-end run and the generated library was sequenced on the Illumina HiSeq2500 platform.
Estimation of Genome Size and Heterozygosity
The M. tetraphylla genome size was estimated by flow cytometry following the protocol described by Dolezel (Doležel et al., 2007) and k-mer frequency analysis. In brief, Jellyfish v2.1.0 (Marçais and Kingsford, 2011) was used to generate the 17-mer frequency distribution of paired-end reads. The genome size was estimated according to the formula: G = K_num/peak depth (G: genome size; K_num: total number of k-mers; peak depth: depth of the major peak). The GenomeScope (Vurture et al., 2017) estimated heterozygosity of the M. tetraphylla genome is 1.03%.
Genome Assembly
De novo assembly pipeline was used to alleviate the dual effects of the highly heterozygous genome and highly repetitive DNA sequences. The Nanopore raw reads were corrected and trimmed using the Canu v1.8 (Koren et al., 2017) software. The corrected reads were uploaded to the WTDBG v2.2 (Ruan and Li, 2020) for genome assembly using the following parameters: S 2 --edge-min 2 --rescue-low-cov-edges -x ccs -g 800 m. Iterative polishing was performed using the Pilon v1.23 (Walker et al., 2014) software with the Illumina genomic data to fix bases, fill gaps, and correct local misassemblies. The polished contigs were subsequently processed by the 3d-DNA pipeline (version 170123) (Dudchenko et al., 2017) using default parameters. Juicebox (https://github.com/aidenlab/Juicebox) was used to visualize the resulting Hi-C contact matrix, which was manually corrected based on the neighboring interactions. A total of 14 high-confidence clusters were identified in the M. tetraphylla genome. A visualization of the assembly contiguity was generated using assembly-stats (https://github.com/rjchallis/assembly-stats).
The Illumina sequencing reads were mapped to the genome using bowtie2 v2.2.6 (Langmead and Salzberg, 2012) to assess the completeness and accuracy of the genome assembly. Additionally, the assembly was evaluated by BUSCO (Benchmarking Universal SingleCopy Orthologs) (Simão et al., 2015). To further evaluate the genome assembly, the RNA reads were mapped to the genome using HISAT2 (Daehwan et al., 2015). The GC content was calculated with a 2 kb non-overlapping sliding window. To assess the accuracy of the genome assembly, we also mapped the ONT long reads to the genome using minimap2 v2.17-r941 (Li, 2018) with the “map-ont” option.
Repeat Annotation
Two complementary methods were used to identify repetitive sequences in the M. tetraphylla genome. First, the Tandem Repeats Finder v4.09 (Benson, 1999) was employed to identify the tandem repeats. Second, a combined strategy was selected to predict transposable elements (TEs). For the homology-based annotation of TEs, RepeatMasker v1.332 (http://www.repeatmasker.org) was employed to search the RepBase database (v18.07) (Bao et al., 2015) for repetitive DNA, and RepeatProteinMasker (Tarailo-Graovac and Chen, 2009) to search the protein database for TE-related proteins. A de novo library was constructed using three software, including RepeatModeler v1.05 (http://www.repeatmasker.org/RepeatModeler.html), RepeatScout v1.05 (Price et al., 2005), and Piler v1.06 (Edgar and Myers, 2005). The RepeatMasker was then applied to identify TEs comprehensively.
Simple sequence repeats (SSRs) in the M. tetraphylla genome were identified using the MISA program (Thiel et al., 2003) with the following parameters: at least twelve repeats for monomer, six repeats for the dimer, four repeats for trimer, three repeats for tetramer, pentamer, and hexamer.
Gene Prediction and Functional Annotation
The Augustus v2.7 (Mario et al., 2004) and SNAP v2006-07-28 (Korf, 2004) software were used to perform de novo prediction. Genes were predicted from the combination of de novo, homology, and EST predictions. The assembled transcripts were used for iterative self-training, and the optimized parameters were applied for further annotation by the Augustus and SNAP software. For homology prediction, protein sequences from Arabidopsis thaliana (Initiative, 2000), Malus domestica (Velasco et al., 2010), Nelumbo nucifera (Ming et al., 2013), and Rosa chinensis (Saint-Oyant et al., 2018) were aligned to the genome using the genblastA v1.0.1 (She et al., 2009) software. The homologous genomic regions of the target genes were extended in both 3′ and 5′ directions and then loaded to the GeneWise v2.2.0 (Birney et al., 2004) software to obtain accurate spliced alignments. The transcripts were also mapped to the genome to generate spliced alignments using the Program to Assemble Spliced Alignments (PASA) pipeline (version 2.0.2) (Haas et al., 2003). Finally, all these predictions were consolidated into a consensus gene set using EVidenceModeler (r2012-06-25) (Haas et al., 2008).
Functional assignment was performed using BLASTP (with 1e-5 e-value) to search against the SwissProt database (Bairoch and Apweiler, 2000). The KAAS server (Yuki et al., 2007) was used to map the predicted genes onto KEGG metabolic pathways. InterProScan v5.10-50.0 (Jones et al., 2014) software determined the motifs and functional domains. The GO term and Pfam domains were directly obtained from the InterProScan results.
The Arabidopsis gene (TAIR10) was used to search for genes related to the oil biosynthesis pathway (Supplementary Table S1). Target genes with ≥50% sequence coverage in length and functional annotations were classified into corresponding gene families. The identified genes were further refined by searching the Pfam database.
Noncoding RNA Annotation
Five types of noncoding RNA genes, tRNA, rRNA, snRNA, snoRNA, and miRNA, were identified in the M. tetraphylla genome. The tRNA genes were identified using the tRNAscan-SE v2.0 (Schattner et al., 2005) software with default parameters. The RNAmmer v1.2 (Lagesen et al., 2007) software was performed to predict rRNAs and their subunits with default parameters. The snRNA genes were predicted using the INFERNAL software (v1.1.2) (Nawrocki et al., 2009) with cm models from the Rfam database (Griffiths-Jones et al., 2005). The snoRNA genes were identified by the snoscan v0.9.1 (Lowe and Eddy, 1999) software.
Comparative Genome Analyses
Orthologous gene clusters were computed using the OrthoMCL pipeline (Li et al., 2003) with the following finished genomes: Actinidia chinensis, Coffea canephora, Populus trichocarpa, Oryza sativa, A. thaliana, R. chinensis, N. nucifera, and M. tetraphylla. An all-against-all comparison was performed using the BLASTP search tool with a 1e-5 e-value. For each single-copy gene family, protein sequences were aligned by the MUSCLE software (Edgar, 2004) and subsequently joined into one supergene for each species. The phylogenetic tree was inferred using the RAxML software (Stamatakis, 2014) with PROTGAMMAWAG model and 1000 bootstraps. The CAFE (Computational Analysis of Gene Family Evolution) tool (De Bie et al., 2006) was used to detect gene family expansion and contraction with a probabilistic graphical model. The GSEA (Gene Set Enrichment Analysis) was performed with the Fisher’s exact test (p-value < 0.05) on InterPro domains.
To further analyze the major evolutionary events in M. tetraphylla, the 4DTv (fourfold synonymous third-codon transversion) distribution in the M. tetraphylla genome was calculated. The two proteomes were compared using BLASTP (1e-5 e-value). Syntenic blocks were identified using the MCscanX toolkit (Wang et al., 2012) with <5 intervening genes between hits. The 4DTv between gene pairs located in synteny blocks were calculated using an in-house Perl script.
RNA-Seq Data Analysis
The raw reads of RNA-seq data were trimmed using Trimmomatic v0.32 (Bolger et al., 2014) to improve the quality. The adaptors and low-quality reads with <15 average quality per base and a 4-base wide sliding window were trimmed off. And then, the resulting clean data were aligned to the reference genome using the HISAT2 (Kim et al., 2015) software. The FPKM expression of target genes was calculated using the Stringtie (Pertea et al., 2016) transcript assembler.
Results
Genome Sequencing and Assembly
The M. tetraphylla was sequenced on the Oxford Nanopore Technologies (ONT) platform. A total of 68.17 Gb of data were generated with an average read length of 20.16 kb (Supplementary Table S2 and Supplementary Figure S1A) from the Nanopore platform, and 88.27 Gb of short reads from the HiSeq2500 platform (Illumina, CA, United States) with 500 bp insert size to genome survey and assembly polishing (Supplementary Table S3). After correcting, a total of 26.34 Gb clean data was obtained with an average length of 29.89 kb (Supplementary Figure S1B). The M. tetraphylla genome size was estimated by flow cytometry, and the deduced genome size was 740 Mb. We also evaluated the genome size using k-mer analysis. The cumulative k-mer count suggested a 758 Mb genome size (Supplementary Figure S2), similar to the flow cytometry results. A de novo assembly pipeline was used to alleviate the dual effects of the highly heterozygous genome and highly repetitive DNA sequences (Supplementary Figure S3). Finally, the M. tetraphylla genome was assembled into 4,335 contigs, with an N50 of 1,182,547 bp (Table 1). The assembly size (750.87 Mb) is consistent with the estimated genome size based on flow cytometry and k-mer analysis (740 and 758 Mb, respectively). The N50 contig showed a 335.8-fold increase compared to M. integrifolia (Figure 1A and Supplementary Table S4). Two Hi-C libraries were constructed from young leaves of M. tetraphylla. About 533 million 150-bp paired-end Illumina reads were produced and uniquely mapped onto the draft assembly contigs. The insertion size of Hi-C reads spanned predominantly from dozens to hundreds of kilobases, therefore providing efficient information for scaffolding (Figure 1B and Supplementary Table S5). Notably, 95.29% (715.17 Mb) of the genome anchored to 14 pseudo-chromosomes combined with the valid Hi-C data (Figure 1C, Supplementary Figure S4 and Supplementary Table S5). The chromosome lengths varied from ∼30.93 to ∼87.10 Mb with ∼51.11 Mb N50 size (Table 1, Supplementary Table S6).

TABLE 1. Summary of the M. tetraphylla genome assembly and annotation.
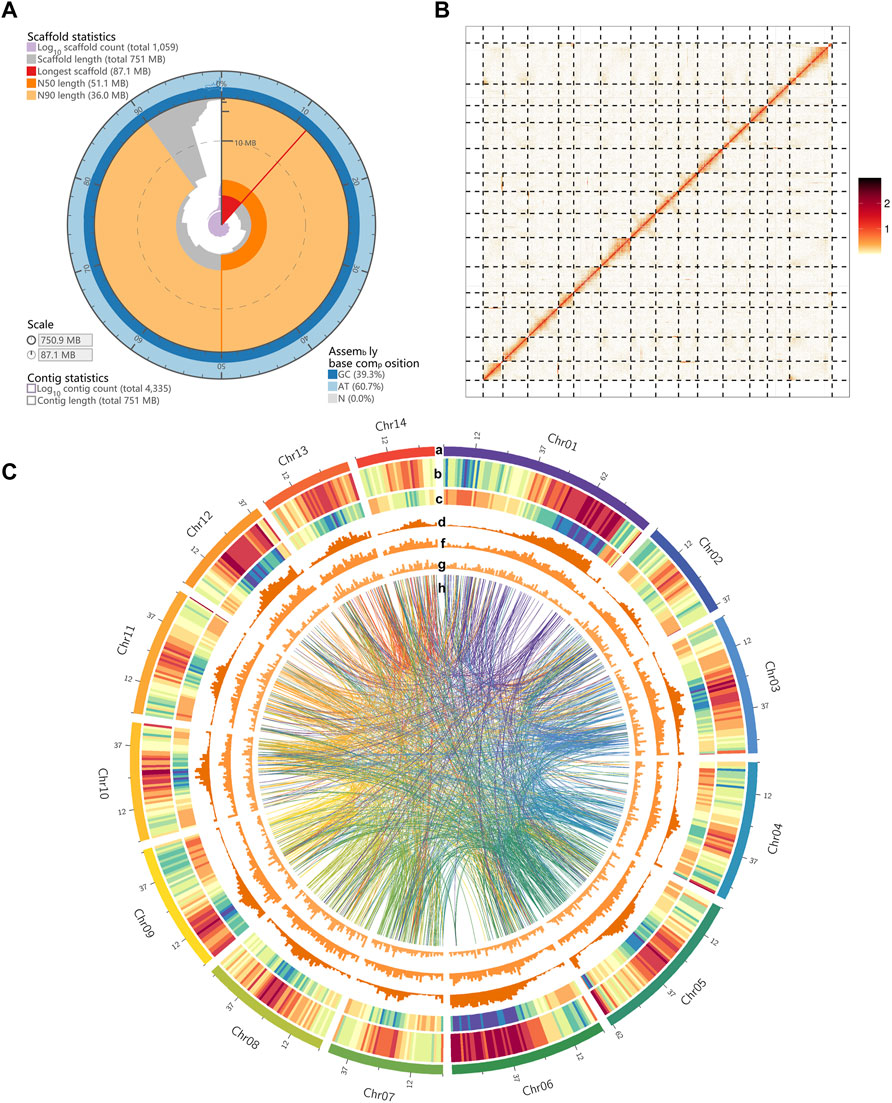
FIGURE 1. Landscape of macadimia genome. (A) Visualization of assembly stats (https://github.com/rjchallis/assembly-stats): the inner radius (highligthed in red color) represents the length of the longest scaffold, the radial axis originates at the circumference indicates the scaffold length, the N50 and N90 scaffold lengths are indicated respectively by dark and light orange arcs, respectively. The cumulative number of scaffolds within a given percentge of the genome is plotted in purple. The outermost circular layer shows the base composition at the given coverage of the genome. (B) Hic-contact map of macadimia genome. (C) Circos plot of macadimia genome. Tracks from outside to inside are the 14 chromosomes of M. tetraphylla, gene density (density measured in 1000-Kb sliding windows), transposable element (TE) density, Gypsy-type LTR retrotransposons density, Copia-type LTR retrotransposons density, DNA transposable element density. The syntenic blocks within chromosomes of macadimia genome are displayed with connecting lines in different colors.
Quality-filtered Illumina reads were mapped to the genome to validate the completeness and accuracy of the genome assembly (Supplementary Figure S5). Results showed that 94.25% of the short reads mapped to the genome, with an 87.84% properly-paired mapping rate (Supplementary Table S7). The accuracy and completeness of the assembly were also assessed by mapping the ONT long reads to the genome. Overall mapping rate of these long reads was 99.81% for our assembly (Supplementary Table S7). From the BUSCO software, ∼89.72% (1,292 out of 1,440) conserved genes in the embryophyta lineage were present in the assembly (Supplementary Table S7). Additionally, 92.00% of the RNA-Seq data independently aligned to the assembled genome. We have also calculated the GC content with a 2 kb non-overlapping sliding window established no obvious GC bias in the genome assembly (Supplementary Figure S5). Altogether, these results suggest a high-quality genome of M. tetraphylla.
Repeats and Gene Annotation
We identified 461 Mb of repetitive sequences, accounting for 61.42% of the genome. These repetitive sequences mainly comprised transposable elements, including RNA retrotransposons (Class I) and DNA transposons (Class II). Long terminal repeat (LTR) retrotransposons represent the most predominant class of transposable elements. The assembled M. tetraphylla contains 34.95% LTR retrotransposons, of which 22.00% are Gypsy-type elements, and 5.94% are Copia-type elements (Supplementary Table S8). A total of 510,893 SSRs were also identified in the M. tetraphylla genome (Supplementary Table S9). Among the repeat motifs, mono-nucleotide repeats were the most predominant, followed by di-, tri-, tetra-, penta-, and hexa-nucleotide (Supplementary Figure S6 and Supplementary Table S9). The identified SSR markers may serve as potential markers to M. tetraphylla breeding programs.
A total of 34.30 Gb RNA-seq data were obtained from five tissues representing major tissue types and developmental stages (Supplementary Table S3) to further aid in gene prediction. The 31,571 genes identified combine de novo, homology-based and EST-based evidence. The average length of the identified genes, exons, and introns were 6,055, 222, and 1,213 bp, respectively (Table 1 and Supplementary Table S10). Overall, 29,233 genes (92.59%) were functionally assigned to the public database, of which 22,869 (72.44%) genes had Swiss-Prot homologs, 8,303 (26.30%) had KEGG homologs, 29,052 (92.02%) had InterPro homologs, 17,864 (56.58%) had GO homologs and 21,925 (69.45%) had Pfam homologs (Supplementary Figure S7 and Supplementary Table S11). Up to 1,286 tRNAs, 542 rRNAs, 251 snRNAs, and 74 snoRNAs were identified in the M. tetraphylla genome (Supplementary Table S12).
Expanded Gene Families and Whole-Genome Duplication
The sequenced M. tetraphylla genome was further compared with seven other sequenced plant genomes, including A. chinensis, A. thaliana, C. canephora, N. nucifera, O. sativa, P. trichocarpa, and R. chinensis, to investigate the genetic basis underpinning the distinct traits of M. tetraphylla. A total of 24,346 orthologous gene families consisting of 204,948 genes were identified (Supplementary Figure S8 and Supplementary Table S13). The eight plant species shared a core set of 107,264 genes which belong to 6,823 gene families and represent ancestral gene families (Figure 2A and Supplementary Table S13). Besides, 965 gene families containing 4,337 genes were unique to M. tetraphylla (Figure 2A and Supplementary Table S13).
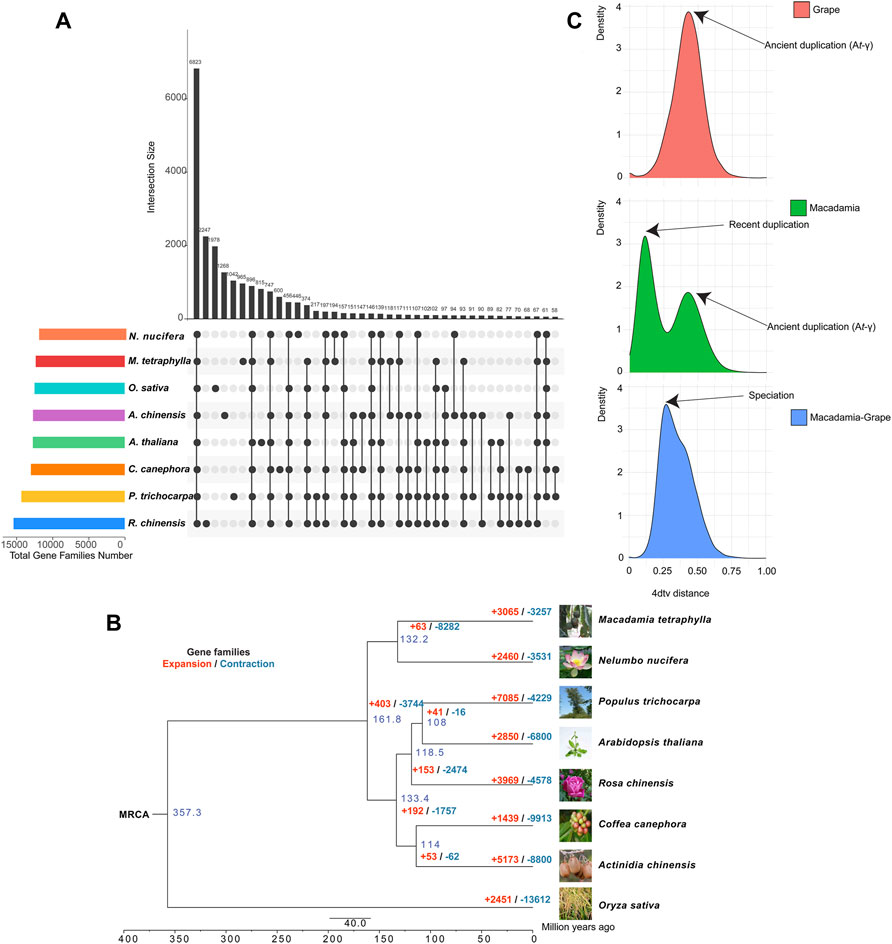
FIGURE 2. Evolution of macadimia genome. (A) Venn diagram showing shared and unique gene families among macadimia and other plant species. (B) Comparative genomic analysis of macadimia and other plant species. (C) Distribution of 4DTv for pairs of syntenic paralogs.
The GO annotation revealed that the M. tetraphylla specific gene families are highly enriched for the chitin catabolic process (GO:0006032, p < 1.23E-02), polysaccharide binding (GO:0030247, p < 3.99E-07), and ADP binding (GO:0043531, p < 5.94E-08). The M. tetraphylla GO enrichment is probably related to the thick shells of macadamia nuts (Supplementary Figure S9 and Supplementary Table S14). The M. tetraphylla specific gene families also mapped to the KEGG pathways of carbohydrate metabolism, metabolism of other amino acids, biosynthesis of other secondary metabolites, xenobiotics biodegradation, metabolism, and protein families for genetic information processing, signaling, and cellular processes (Supplementary Figure S10).
A phylogenomic tree constructed using 833 single-copy genes from the eight plant species showed that M. tetraphylla is closely related to N. nucifera but diverged ∼132.2 million years ago (MYA) (Supplementary Figure S11). The gene families with significant change are often associated with distinct traits (Zhang et al., 2017). A gene family evolutionary analysis of the eight plant species showed that 3,065 gene families underwent expansion, whereas 3,257 underwent contraction (Figure 2B, Supplementary Figures S12A,B).
The GO enrichment analysis revealed that the expansion of these families involved genes related to defense response (GO:0042742, GO:0006952), lipid transport (GO:0006869), steroid biosynthetic process (GO:0006694), triglyceride lipase activity (GO:0004806), and fatty acid metabolic process (GO:0006631) (Supplementary Table S15). On the KEGG pathway database, the expanded gene families were functionally associated with the oil biosynthesis pathway, including fatty acid biosynthesis (ko00061), fatty acid elongation (ko00062), fatty acid degradation (ko00071), and glycerolipid metabolism (ko00561) (Supplementary Table S16). These findings suggest that M. tetraphylla displayed an enhanced ability for oil biosynthesis, a critical trait for flavor and quality in macadamia.
WGD (whole-genome duplication) events are of great importance in generating species diversity during evolution (Fu et al., 2021). We used 4-fold synonymous third-codon transversion (4DTv) to detect the WGD events in the M. tetraphylla genome. In a self-alignment of M. tetraphylla, a total of 581 syntenic genomic blocks covering 8,953 genes were identified in the macadamia genome. The orthology within macadamia genome showed 4DTv distance peaks at ∼0.10 and ∼0.42, respectively (Figure 2C), suggesting that two rounds of WGD events occurred in the macadamia genome. We also compared the M. tetraphylla genome with grape genome sequences, and a peak (4DTv ∼0.14) was observed. These results indicated that M. tetraphylla has undergo a species-specific WGD event after the divergence between M. tetraphylla and grape (Figure 2C), and M. tetraphylla shared an ancient WGD event (At-γ) with grape (Figure 2C).
Analysis of Oil Metabolism Genes
The high fat content of macadamia kernels is the most prominent feature of this fruit tree, especially the high content of unsaturated fatty acids, which determines its benefits to human health. According to the determination of our research team, macadamia nuts contain up to 80% fatty acids consisting of 13 different types. The three most abundant fatty acids are oleic acid (57–66%), palmitoleic acid (10–18%), and palmitic acid (10–18%), and the other ten fatty acids constitute <4% of the total fatty acids content.
The 187 genes encoding 33 crucial oil biosynthesis enzymes, including those involved in de novo fatty acid synthesis, elongation, and TAG assembly, were manually annotated to expound on the evolution of oil metabolism genes of M. tetraphylla (Figure 3). The RNA-seq data from leaves, young flowers, mature flowers, roots, and barks identified member genes from key enzyme gene families (Supplementary Table S17). The acetyl-CoA carboxylase (ACCase, EC: 6.4.1.2) is the key enzyme determining the metabolic pathways that lead to oil or protein biosynthesis in the seed (Chen et al., 1999). Ten ACCase genes were identified in the M. tetraphylla genome but exhibited diverse expression patterns in different tissues (Figure 3 and Supplementary Table S17). Gene MTE004907 of the ACCase gene family was highly expressed in all tissues, but the expression levels were substantially higher in leaves and flowers than roots and barks.
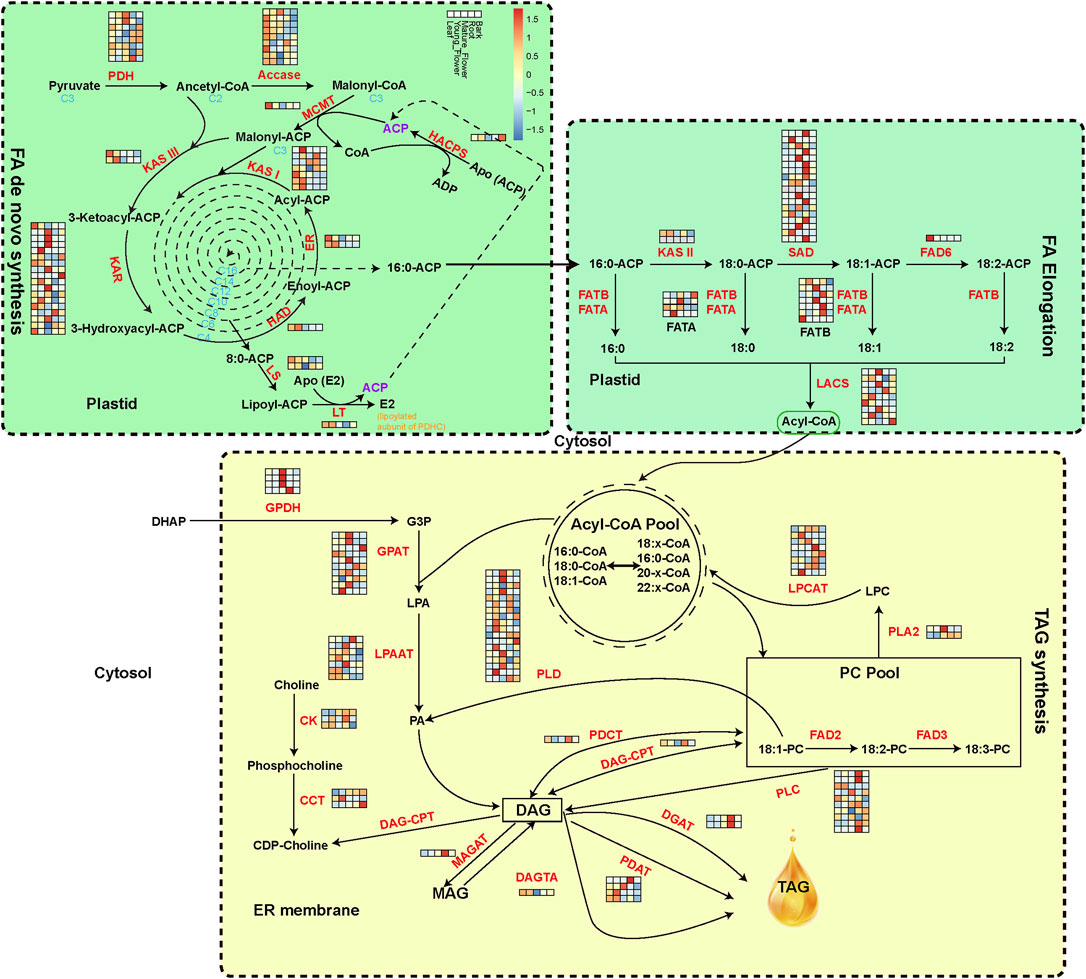
FIGURE 3. Expression level of oil biosynthesis-related genes. Acetyl-CoA is converted into C16 and C18 fatty acids in the plastid. TAG is synthesized in the endoplasmic reticulum and packed in the oil bodies. The isozymes and metabolites involved in oil biosynthesis were colored in red and black, respectively. The expression levels of oil-biosynthesis genes from leaf, young flower, mature flower, root and bark, are presented with the heat map.
The FAD protein family catalyzes the desaturation of fatty acids (Park et al., 2008). FAD2 and FAD3 are the main enzymes responsible for linoleic acid desaturation. FAD2 is reported to be accountable for polyunsaturated lipid synthesis in the developing seed of oil crops. Two FAD2 genes were identified in the M. tetraphylla genome, but none was expressed in the five sequenced tissues. Other gene families, including KAR, SAD, and PLD, contained the maximum number of gene copies, implying their central role in the oil formation pathway.
Genome-Wide Investigation of WRKY Gene Family
The WRKY transcription factors (TFs) are among the most widespread gene families in higher plants (ülker and Somssich, 2004). The WRKY proteins play a crucial role in plant defense against biotic stress (Levée et al., 2009; Pandey and Somssich, 2009; Kloth et al., 2016). Fifty-five WRKY proteins were identified based on a WRKY domain and BLAST searches (Supplementary Table S18). The identified WRKY proteins were 134 aa (MTE002361) to 1,050 aa (MTE011780) long (Supplementary Table S18). Multiple sequence alignment was performed to check the phylogenetic relationship of the WRKY proteins. The WRKY domains covered three groups corresponding to the groups I, II, and III. Group II was dominant which contained 30 members (Figure 4A and Supplementary Table S18). As previously reported, the WRKY domains from the N-termini and C-termini were grouped into different clades, indicating that the two domains underwent parallel evolution (Tao et al., 2018). The WRKY genes possessed at least two exons, including the optimal number of exons to a maximum of twenty (Figure 4B). To further confirm whether different tissues influenced the expression level of WRKY genes, we calculated the FPKM (Fragments Per Kilobase per Million) value of each gene. Forty-one WRKY genes were expressed in all five sampled tissues, with 20 genes showing constitutive expression (FPKM >1 in all samples) (Supplementary Table S19). However, gene MTE017431 was not expressed in any of the five sampled tissues, suggesting a pseudogene (Figure 4C). Interestingly, the WRKY genes showed higher transcript abundance in roots than other tissues (Figure 4C), suggesting that defense in macadamia primarily occurs in this organ, probably against soil microorganisms and pathogens.
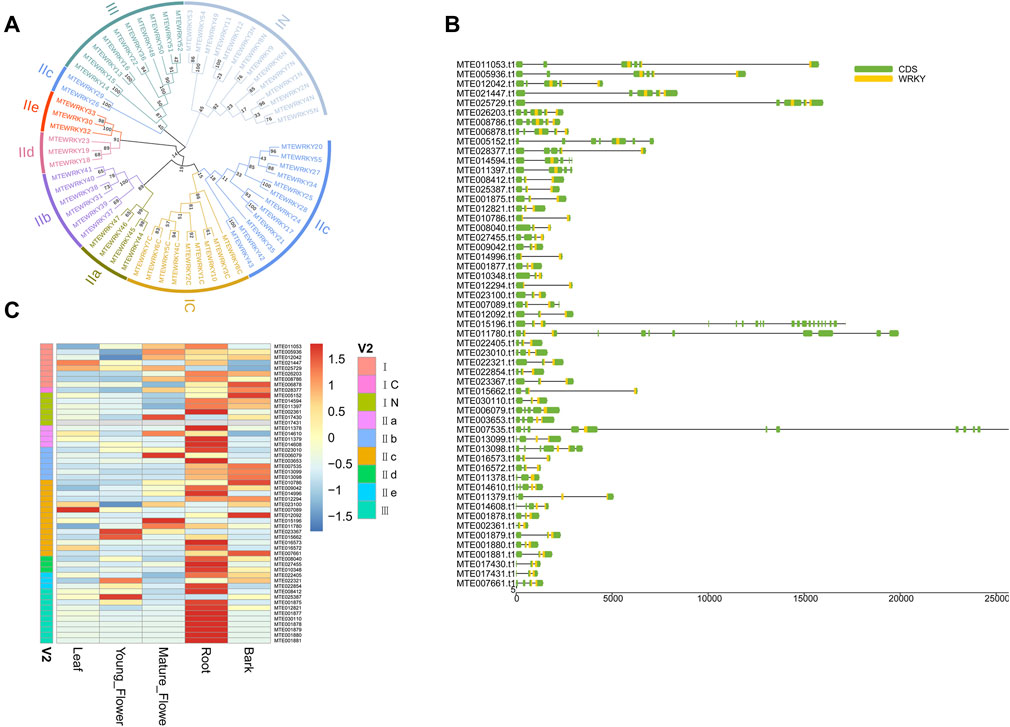
FIGURE 4. Genome-wide investigation of WRKY gene family. (A) Unrooted phylogenetic tree among WRKY domains from macadimia genome. (B) Exon-intron structure of WRKY genes. (C) Expression profiles of the WRKY genes.
Discussion
M. tetraphylla L. Johnson, is a tropical to subtropical tree that has its origins in southeastern Queensland and northeastern New South Wales in Australia (Mulwa and Bhalla, 2000). It is highly valued for its versatile nut. However, production of macadamia is hampered by low yield. Here, we generated a chromosome-scale assembly of M. tetraphylla genome combing Oxford Nanopore Technologies and Hi-C technology. The N50 contig showed a striking 335.8-fold increase compared to M. integrifolia. Repeated sequence insertion has been proved to be a main force for the expansion of plant genome as observed in tea tree and rubber tree (Liu et al., 2020; Zhang et al., 2020). Similar to these species, Gypsy-type elements contributed the most to the macadamia tree genome.
We have identified lineage-specific genes that likely control the thick shells of macadamia, in particular genes encoding enzymes involved in the chitin catabolic process. Our comparative analyses indicate that M. tetraphylla is closely related to N. nucifera and diverged ∼132.2 million year ago. A high proportion of gene families related to fatty acid metabolism were expanded in the macadamia tree genome, indicating M. tetraphylla displayed an enhanced ability for oil biosynthesis. A similar result was observed in the tung tree, which is an economically important woody oil plant that produces tung oil rich in eleostearic acid (Zhang et al., 2019). A recent WGD event was also detected in the M. tetraphylla genome.
Like many other nuts, macadamia nuts are high in monounsaturated fatty acids. Monounsaturated fatty acids have been linked to reduced cholesterol in the blood (https://www.medicinalfoodnews.com/). A total of 187 genes encoding 33 crucial oil biosynthesis enzymes were identified. ACCase is a key enzyme determining the metabolic pathways toward oil or protein biosynthesis (Chen et al., 1999). Ten ACCase genes were identified but the expression levels varied, which may be a cause of different fatty acids content in different tissues. We have also observed the KAR, SAD, and PLD gene families contained the maximum number of gene copies, implying their central role in the oil formation pathway. A total of 55 WRKY genes were identified in the M. tetraphylla genome. We have found that most of the WRKY members showed higher expression level in roots, indicating that this tissue may play a crucial role in plant defense against soil microorganisms and pathogens. We believe that the genome sequencing efforts summarized in this study would facilitate the breeding of this elite nut tree.
Conclusion
This paper presents the sequencing, assembly, and annotation of the M. tetraphylla genome. The extensive datasets and analyses presented will provide novel insights into the genome evolution of this species and facilitate the breeding strategies for genetic improvement. As a woody plant, breeding for any new variety often takes decades, and molecular marker-assisted screening offers a practical approach to shorten the breeding cycle. The genomic data obtained in this study will also provide the primary data for mining genes and developing molecular markers hence the foundation for molecular breeding of macadamia.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
YN and JL conceived the study and managed the project. GL, SN, and XH designed the scientific objectives. CZ, ZL, LG, and GK collected the samples and extracted the genomic DNA. JL and WL estimated the genome size, assembled the genome and carried out the gene annotation. YN wrote the manuscript, and all authors contributed to writing and editing the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31760215), the Technology Innovation Talents Project of Yunnan Province (2018HB086), and Sci-Tech Innovation System Construction for Tropical Crops Grant of Yunnan Province (RF 2022).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.835363/full#supplementary-material
References
Ahmad Termizi, A., Hardner, C., Batley, J., Nock, C. J., Hayashi, S., Montenegro, J., et al. (2014). SNP Analysis of Macadamia Integrifolia Chloroplast Genomes to Determine the Genetic Structure of Wild Populations. XXIX Int. Hortic. Congress Hortic. Sustaining Lives 1109, 175–180. doi:10.17660/actahortic.2016.1109.29
Akinsanmi, O. A., Neal, J., Drenth, A., and Topp, B. (2017). Characterization of Accessions and Species ofMacadamiato Stem Infection byPhytophthora Cinnamomi. Plant Pathol. 66, 186–193. doi:10.1111/ppa.12566
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT Protein Sequence Database and its Supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi:10.1093/nar/28.1.45
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mobile Dna 6, 11. doi:10.1186/s13100-015-0041-9
Benson, G. (1999). Tandem Repeats Finder: a Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14, 988–995. doi:10.1101/gr.1865504
Chagné, D. (2015). “Whole Genome Sequencing of Fruit Tree Species,” in Advances in Botanical Research. Editors C. Plomion, and A. F. Adam-Blondon (Academic Press), 1–37. doi:10.1016/bs.abr.2015.04.004
Chen, J. Q., Lang, C. X., Hu, Z. H., Liu, Z. H., and Huang, R. Z. (1999). Antisense PEP Gene Regulates to Ratio of Protein and Lipid Content in Brassica Napus Seeds. J. Agric. Biotechnol. 7, 316–320. doi:10.3969/j.issn.1674-7968.1999.04.003
Daehwan, K., Ben, L., and Salzberg, S. L. (2015). HISAT: a Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12, 357–360. doi:10.1038/nmeth.3317
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a Computational Tool for the Study of Gene Family Evolution. Bioinformatics 22, 1269–1271. doi:10.1093/bioinformatics/btl097
Doležel, J., Greilhuber, J., and Suda, J. (2007). Estimation of Nuclear DNA Content in Plants Using Flow Cytometry. Nat. Protoc. 2, 2233–2244. doi:10.1038/nprot.2007.310
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De Novo assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 356, 92–95. doi:10.1126/science.aal3327
Edgar, R. C. (2004). MUSCLE: a Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinformatics 5, 113. doi:10.1186/1471-2105-5-113
Edgar, R. C., and Myers, E. W. (2005). PILER: Identification and Classification of Genomic Repeats. Bioinformatics 21, i152–i158. doi:10.1093/bioinformatics/bti1003
Fu, A., Wang, Q., Mu, J., Ma, L., Wen, C., Zhao, X., et al. (2021). Combined Genomic, Transcriptomic, and Metabolomic Analyses Provide Insights into Chayote (Sechium Edule) Evolution and Fruit Development. Hortic. Res. 8, 35. doi:10.1038/s41438-021-00487-1
Garg, M. L., Blake, R. J., Wills, R. B. H., and Clayton, E. H. (2007). Macadamia Nut Consumption Modulates Favourably Risk Factors for Coronary Artery Disease in Hypercholesterolemic Subjects. Lipids 42, 583–587. doi:10.1007/s11745-007-3042-8
Garg, M. L., Blake, R. J., and Wills, R. B. H. (2003). Macadamia Nut Consumption Lowers Plasma Total and LDL Cholesterol Levels in Hypercholesterolemic Men. J. Nutr. 133, 1060–1063. doi:10.1093/jn/133.4.1060
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: Annotating Non-coding RNAs in Complete Genomes. Nucleic Acids Res. 33, D121–D124. doi:10.1093/nar/gki081
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucleic Acids Res. 31, 5654–5666. doi:10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi:10.1186/gb-2008-9-1-r7
Hardner, C. (2016). Macadamia Domestication in Hawai'i. Genet. Resour. Crop Evol. 63, 1411–1430. doi:10.1007/s10722-015-0328-1
Hibrand Saint-Oyant, L., Ruttink, T., Hamama, L., Kirov, I., Lakhwani, D., Zhou, N. N., et al. (2018). A High-Quality Genome Sequence of Rosa Chinensis to Elucidate Ornamental Traits. Nat. Plants 4, 473–484. doi:10.1038/s41477-018-0166-1
Initiative, A. G. (2000). Analysis of the Genome Sequence of the Flowering Plant Arabidopsis thaliana. Nature 408, 796–815. doi:10.1038/35048692
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 30, 1236–1240. doi:10.1093/bioinformatics/btu031
Kaijser, A., Dutta, P., and Savage, G. (2000). Oxidative Stability and Lipid Composition of Macadamia Nuts Grown in New Zealand. Food Chem. 71, 67–70. doi:10.1016/s0308-8146(00)00132-1
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12, 357–360. doi:10.1038/nmeth.3317
Kloth, K. J., Wiegers, G. L., Busscher-Lange, J., Van Haarst, J. C., Kruijer, W., Bouwmeester, H. J., et al. (2016). AtWRKY22 Promotes Susceptibility to Aphids and Modulates Salicylic Acid and Jasmonic Acid Signalling. Exbotj 67, 3383–3396. doi:10.1093/jxb/erw159
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi:10.1101/gr.215087.116
Korf, I. (2004). Gene Finding in Novel Genomes. BMC Bioinformatics 5, 59. doi:10.1186/1471-2105-5-59
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 35, 3100–3108. doi:10.1093/nar/gkm160
Langmead, B., and Salzberg, S. L. (2012). Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923
Levée, V., Major, I., Levasseur, C., Tremblay, L., Mackay, J., and Séguin, A. (2009). Expression Profiling and Functional Analysis of Populus WRKY23 Reveals a Regulatory Role in Defense. New Phytol. 184, 48–70. doi:10.1111/j.1469-8137.2009.02955.x
Li, H. (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189. doi:10.1101/gr.1224503
Liu, J., Niu, Y.-F., Ni, S.-B., He, X.-Y., and Shi, C. (2017). Complete Chloroplast Genome of a Subtropical Fruit Tree Macadamia Ternifolia (Proteaceae). Mitochondrial DNA B 2, 738–739. doi:10.1080/23802359.2017.1390401
Liu, J., Niu, Y.-F., Ni, S.-B., He, X.-Y., Zheng, C., Liu, Z.-Y., et al. (2018). The Whole Chloroplast Genome Sequence of Macadamia Tetraphylla (Proteaceae). Mitochondrial DNA Part B 3, 1276–1277. doi:10.1080/23802359.2018.1532836
Liu, J., Shi, C., Shi, C.-C., Li, W., Zhang, Q.-J., Zhang, Y., et al. (2020). The Chromosome-Based Rubber Tree Genome Provides New Insights into Spurge Genome Evolution and Rubber Biosynthesis. Mol. Plant 13, 336–350. doi:10.1016/j.molp.2019.10.017
Lowe, T. M., and Eddy, S. R. (1999). A Computational Screen for Methylation Guide snoRNAs in Yeast. Science 283, 1168–1171. doi:10.1126/science.283.5405.1168
Mario, S., Rasmus, S., Stephan, W., and Burkhard, M. (2004). AUGUSTUS: a Web Server for Gene Finding in Eukaryotes. Nucleic Acids Res. 32, 309–312. doi:10.1093/nar/gkh379
Marçais, G., and Kingsford, C. (2011). A Fast, Lock-free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27, 764–770. doi:10.1093/bioinformatics/btr011
Ming, R., Vanburen, R., Liu, Y., Yang, M., Han, Y., Li, L. T., et al. (2013). Genome of the Long-Living Sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 14, R41. doi:10.1186/gb-2013-14-5-r41
Mulwa, R., and Bhalla, P. (2000). In Vitro shoot Multiplication ofMacadamia tetraphyllaL. Johnson. J. Hortic. Sci. Biotechnol. 75, 1–5. doi:10.1080/14620316.2000.11511192
Navarro, S. L. B., and Rodrigues, C. E. C. (2018). Macadamia Oil Extraction with Alcoholic Solvents: Yield and Composition of Macadamia Oil and Production of Protein Concentrates from Defatted Meal. Eur. J. Lipid Sci. Technol. 120, 1800092. doi:10.1002/ejlt.201800092
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: Inference of RNA Alignments. Bioinformatics 25, 1335–1337. doi:10.1093/bioinformatics/btp157
Neal, J. M., Hardner, C. M., and Gross, C. L. (2010). Population Demography and Fecundity Do Not Decline with Habitat Fragmentation in the Rainforest Tree Macadamia Integrifolia (Proteaceae). Biol. Conservation 143, 2591–2600. doi:10.1016/j.biocon.2010.06.029
Nock, C. J., Baten, A., Barkla, B. J., Furtado, A., Henry, R. J., and King, G. J. (2016). Genome and Transcriptome Sequencing Characterises the Gene Space of Macadamia Integrifolia (Proteaceae). BMC Genomics 17, 937. doi:10.1186/s12864-016-3272-3
Nock, C. J., Baten, A., and King, G. J. (2014). Complete Chloroplast Genome of Macadamia Integrifoliaconfirms the Position of the Gondwanan Early-Diverging Eudicot Family Proteaceae. BMC Genomics 15, S13. doi:10.1186/1471-2164-15-s9-s13
Pandey, S. P., and Somssich, I. E. (2009). The Role of WRKY Transcription Factors in Plant Immunity. Plant Physiol. 150, 1648–1655. doi:10.1104/pp.109.138990
Park, J.-Y., Kim, D.-K., Wang, Z.-M., Lu, P., Park, S.-C., and Lee, J.-S. (2008). Production and Characterization of Biodiesel from Tung Oil. Appl. Biochem. Biotechnol. 148, 109–117. doi:10.1007/s12010-007-8082-2
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi:10.1038/nprot.2016.095
Pisanu, P. C., Gross, C. L., and Flood, L. (2009). Reproduction in Wild Populations of the Threatened TreeMacadamia Tetraphylla: Interpopulation Pollen Enriches Fecundity in a Declining Species. Biotropica 41, 391–398. doi:10.1111/j.1744-7429.2008.00484.x
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De Novo identification of Repeat Families in Large Genomes. Bioinformatics 21, i351–i358. doi:10.1093/bioinformatics/bti1018
Ruan, J., and Li, H. (2020). Fast and Accurate Long-Read Assembly with Wtdbg2. Nat. Methods. 17 (2), 155–158. doi:10.1038/s41592-019-0669-3
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, Snoscan and snoGPS Web Servers for the Detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi:10.1093/nar/gki366
Shapcott, A., and Powell, M. (2011). Demographic Structure, Genetic Diversity and Habitat Distribution of the Endangered, Australian Rainforest Tree Macadamia Jansenii Help Facilitate an Introduction Program. Aust. J. Bot. 59, 215–225. doi:10.1071/bt10132
She, R., Chu, J. S.-C., Wang, K., Pei, J., and Chen, N. (2009). GenBlastA: Enabling BLAST to Identify Homologous Gene Sequences. Genome Res. 19, 143–149. doi:10.1101/gr.082081.108
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 31, 3210–3212. doi:10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML Version 8: a Tool for Phylogenetic Analysis and post-analysis of Large Phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Tao, X., Chengjie, C., Chuhao, L., Jiarou, L., Chaoyang, L., and Yehua, H. (2018). Genome-wide Investigation of WRKY Gene Family in Pineapple: Evolution and Expression Profiles during Development and Stress. BMC Genomics 19, 490. doi:10.1186/s12864-018-4880-x
Tarailo‐Graovac, M., and Chen, N. (2009). Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics 25, 4. doi:10.1002/0471250953.bi0410s25
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum Vulgare L.). Theor. Appl. Genet. 106, 411–422. doi:10.1007/s00122-002-1031-0
Toft, B. D., Alam, M., and Topp, B. (2018). Estimating Genetic Parameters of Architectural and Reproductive Traits in Young Macadamia Cultivars. Tree Genet. Genomes 14, 50. doi:10.1007/s11295-018-1265-x
Trueman, S., and Turnbull, C. (1994). Effects of Cross-Pollination and Flower Removal on Fruit Set in Macadamia. Ann. Bot 73, 23–32. doi:10.1006/anbo.1994.1003
Ülker, B., and Somssich, I. E. (2004). WRKY Transcription Factors: from DNA Binding towards Biological Function. Curr. Opin. Plant Biol. 7, 491–498. doi:10.1016/j.pbi.2004.07.012
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The Genome of the Domesticated Apple (Malus × Domestica Borkh.). Nat. Genet. 42, 833–839. doi:10.1038/ng.654
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: Fast Reference-free Genome Profiling from Short Reads. Bioinformatics 33, 2202–2204. doi:10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9, e112963. doi:10.1371/journal.pone.0112963
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Yuki, M., Masumi, I., Shujiro, O., Yoshizawa, A. C., and Minoru, K. (2007). KAAS: an Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 35, 182–185. doi:10.1093/nar/gkm321
Zhang, L., Li, X., Ma, B., Gao, Q., Du, H., Han, Y., et al. (2017). The Tartary Buckwheat Genome Provides Insights into Rutin Biosynthesis and Abiotic Stress Tolerance. Mol. Plant 10, 1224–1237. doi:10.1016/j.molp.2017.08.013
Zhang, L., Liu, M., Long, H., Dong, W., Pasha, A., Esteban, E., et al. (2019). Tung Tree (Vernicia Fordii) Genome Provides a Resource for Understanding Genome Evolution and Improved Oil Production. Genomics, Proteomics & Bioinformatics 17, 558–575. doi:10.1016/j.gpb.2019.03.006
Keywords: Macadamia tetraphylla, nanopore sequencing, Hi-C, whole genome duplication, fatty acid biosynthesis
Citation: Niu Y, Li G, Ni S, He X, Zheng C, Liu Z, Gong L, Kong G, Li W and Liu J (2022) The Chromosome-Scale Reference Genome of Macadamia tetraphylla Provides Insights Into Fatty Acid Biosynthesis. Front. Genet. 13:835363. doi: 10.3389/fgene.2022.835363
Received: 14 December 2021; Accepted: 08 February 2022;
Published: 23 February 2022.
Edited by:
Valentino Ruggieri, Sequentia Biotech, SpainReviewed by:
Xiyin Wang, Agricultural University of Hebei, ChinaHamed Bostan, National Institute of Environmental Health Sciences (NIH), United States
Copyright © 2022 Niu, Li, Ni, He, Zheng, Liu, Gong, Kong, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Li, bGl3ZWlzZGF1QDEyNi5jb20=; Jin Liu, bGl1amluMjQxNkAxNjMuY29t
†These authors have contributed equally to this work