 Yuan Fang1†
Yuan Fang1† Lian Chen
Lian Chen Jian-She Wang
Jian-She Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 24 March 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.833495
This article is part of the Research Topic Inherited Metabolic Diseases in Pediatrics: Clinical and Molecular Features View all 25 articles
Background: TMEM199-congenital disorder of glycosylation (TMEM199-CDG) is a rare autosomal recessive inherited disease characterized by chronically elevated serum transaminase, decreased serum ceruloplasmin, steatosis and/or fibrosis, TMEM199 mutation, reduced level of TMEM199 protein, and abnormal protein glycosylation.
Methods: The information of a Chinese patient with TMEM199-CDG in the Children’s Hospital of Fudan University was reviewed. The patient’s clinical, pathological, and molecular features were obtained by clinical data study, liver biopsy, immunohistochemistry, and molecular genetic analysis.
Results: A 4-year-old Chinese boy presented with hypertransaminasemia, hypercholesterolemia, elevated alkaline phosphatase, decreased serum ceruloplasmin and serum copper level, and coagulopathy since birth. To the best of our knowledge, novel findings included strabismus, cirrhosis by liver biopsy, reduced expression of TMEM199 by immunohistochemistry, and a frameshift variant of c.128delA/p.Lys43Argfs*25 in the TMEM199 gene.
Conclusion: This case added to the phenotypic and genotypic spectrum of TMEM199-CDG.
Congenital disorder of glycosylation (CDG) is an emerging group of metabolic diseases with autosomal recessive inheritance. One or more glycosylation enzyme defects lead to the abnormal glycosylation of protein or lipid in CDG, resulting in corresponding clinical symptoms (Yarema and Bertozzi, 2001; Jaeken, 2013). Common manifestations include dysmorphic features; diarrhea or malnutrition; abnormalities in the liver, coagulation, gland, and immune and nervous system; psychomotor disability; language retardation; ataxia; eye abnormalities; spinal or joint impairment; and cardiac disorders. Given the diversity of clinical performance, definitive diagnosis of CDG is difficult. With the development of whole-exome sequencing (WES), more than 140 CDG types have been identified. As a new subtype discovered in recent years, TMEM199-CDG (OMIM: 616829) is characterized by isolated or predominant liver involvement, which is different from most CDGs associated with multiple organ dysfunction (Marques-da-Silva et al., 2017). To date, only seven cases of TMEM199-CDG have been reported worldwide (Jansen et al., 2016; Vajro et al., 2018). The patients presented with cryptogenic elevated alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, creatine kinase, total cholesterol, low-density lipoprotein cholesterol, and decreased serum ceruloplasmin. Liver biopsies revealed steatosis and/or fibrosis, and an ultrastructure of dilation and vesiculation of the Golgi and/or endoplasmic reticulum (ER). Several mutation sites of TMEM199 were found by sequencing. Western blot confirmed reduced or undetectable levels of TMEM199 protein. Glycosylation analysis results were consistent with the type Ⅱ CDG pattern.
Herein, we reported a Chinese boy with TMEM199-CDG, presenting with abnormal liver function, strabismus, and mild psychomotor delay. The liver biopsy showed steatosis, severe fibrosis as cirrhosis at stage 4, and reduced expression of TMEM199 by immunohistochemistry. WES revealed two compound heterozygous variants, c.20C > A mutation and c.128delA deletion in TMEM199 exon 1, of which the latter had not been previously reported. Treatments with bicyclic alcohol, zinc sulfate, and penicillamine yielded no effect, while no significant progression in the course of the disease was observed after 1-year follow-up. To the best of our knowledge, he is the first Chinese patient described with TMEM199-CDG who carried a novel phenotype and genotype, which might expand the understanding of this rare disease.
The information of a Chinese pediatric patient with TMEM199-CDG in the Children’s Hospital of Fudan University was reviewed. Clinical, pathological, and molecular features of the patient were assessed by clinical manifestations, laboratory investigations, liver biopsy, immunohistochemistry, and molecular genetic analysis. This study conformed to the provisions of the institutional ethics committee and the Declaration of Helsinki (as revised in 2013). The patient’s parents shared all procedures including treatment and signed the written informed consent. The written informed consent was obtained for publication of any potentially identifiable images or data included in this article.
The specimen was obtained from core-needle biopsy, fixed in 10% buffered formalin, dehydrated in graded concentrations of ethyl alcohol, and embedded in paraffin. Then it underwent routine staining for hematoxylin and eosin (HE), and histochemical special staining for periodic acid–Schiff (PAS) with and without diastase, Masson, reticulin, copper, and iron.
Additional 4-µm sections were deparaffinized, rehydrated, and pretreated with 3% H2O2 to eliminate endogenous peroxidase activity. Moreover, they were treated with EDTA (pH 9) or citrate buffer (pH 6) for heat-mediated antigen retrieval before commencing with the immunohistochemical staining protocol. The primary antibodies used included CK7 (clone MX053, ready-to-use solution), CK19 (clone MX054, ready-to-use solution), CD68 (clone MX075, ready-to-use solution), CD163 (clone MX081, ready-to-use solution), CD3 (clone MX036, ready-to-use solution), and CD8 (clone C8/144B, ready-to-use solution), which were purchased from http://www.maxim.com.cn (Fuzhou, China). In addition to the series of conventional antibodies, a rabbit polyclonal antibody against TMEM199 (purchased from http://www.abcam.cn, product code: ab121907) with a 1:50 dilution was utilized. The sections were incubated overnight at 4°C, followed by incubating with a general secondary antibody for 1 h at room temperature. Finally, the sections were developed with DAB and counterstained with hematoxylin. A normal liver sample (donated by a surgical patient) was prepared as the positive control, while omitting the first antibody as the negative control.
Genetic testing was performed by Running Gene Inc (http://www.mono-mz.com/auth, Beijing, China). EDTA-anticoagulated whole blood specimens were collected from the patient and his parents. DNA was isolated from peripheral blood using the DNA Isolation Kit (Blood DNA Kit V2, CW2553). Concentrations were determined on a Qubit fluorometer (Invitrogen, Q33216) using the Qubit dsDNA HS Assay Kit (Invitrogen, Q32851). Agarose gel (1%) electrophoresis was performed for the quality control. A total of 1 μg isolated DNA was sheared using the following parameters: duty cycle 10%, intensity 5, cycles per burst 200, and time six cycles per 60 s. This step was performed using a Bioruptor UCD-200 (Diagenode). All samples were sheared very consistently, and the size distribution peak was around 200 bp. In total, 3 μl of the sheared DNA was electrophoresed in a 2% agarose gel to confirm the presence of fragments of the desired size range. The multiplex ligation-dependent probe amplification assay (MLPA) was performed using the ATP7B MLPA kit (SALSA MLPA Probemix P098-D1, MRC-Holland, the Netherlands) (Chen et al., 2019). However, MLPA analysis excluded copy number variations in ATP7B. Then IDT-WES was performed to dig for potential genetic abnormalities. DNA libraries were prepared using the KAPA Library Preparation Kit (Kapa Biosystems, KR0453) following the manufacturer’s instructions. The procedure comprised three standard steps: end-repair of fragmented DNA, A-tailing, adapter ligation, and amplification. Purifications between steps were carried out using Agencourt AMPure XP beads. The libraries were estimated using the Qubit dsDNA HS Assay kit (Invitrogen, Q32851). Hybridization of pooled libraries to the capture probes and removal of non-hybridized library molecules were carried out according to the SeqCap hybrid Mix system. DNA libraries were sequenced using the Illumina novaseq platform (Illumina, SanDiego, United States) as paired-end 200-bp reads. For data analysis, adapters and low-quality sequences were removed to obtain clean and basic data. Genome Analysis Toolkit (GATK) (https://gatk.broadinstitute.org) was used for variation calling to summarize single-nucleotide variants (SNVs) and indels. ANNOVAR software and Enliven® Variants Annotation Interpretation System were employed for annotation and interpretation. Data were aligned against the Human Gene Mutation Database (HGMD) Professional (http://www.hgmd.cf.ac.uk), 1,000 Genome Database (www.1000 genomes.org), Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org), dbSNP152 (https://www.ncbi.nlm.nih.gov/snp), and Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org). The variants were filtered out only with a population frequency of <5% in 1,000 genomes and <2% in gnomAD. Damage prediction of the genetic variants was conducted by Combined Annotation-Dependent Depletion (CADD) (https://cadd.gs.washington.edu) for scoring and mutation significance cutoff (MSC) (https://lab.rockefeller.edu/casanova/MSC) for further comparison. The MSC server was applied to CADD, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (https://sift.bii.a-star.edu.sg/www/SIFT_indels2.html), with a confidence interval of 99% and database source of HGMD and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). Genomics England PanelApp (https://panelapp.genomicsengland.co.uk), a crowdsourcing tool, was utilized for analysis based on variant–disease and gene–disease associations. Human Phenotype Ontology (HPO) (https://hpo.jax.org), Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org), and HGMD database were used to match the phenotype descriptions with variant and gene prioritization results. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, genetic variants were classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, and benign. Thereafter, two pathogenic variants of TMEM199 were identified with the transcript of NM_152464.1, which was validated by using Sanger sequencing. The pathogenicity of amino acid changes caused by gene mutations was predicted using MutationTaster (http://www.mutationtaster.org). We conducted protein modeling using the SWISS-model (https://www.swissmodel.expasy.org) with a UniProtKB code Q8N511, and the mutated structures were analyzed and visualized using PyMol (http://www.pymol.org).
The patient was born to healthy non-consanguineous parents as the second child of the family, whose older sister was healthy. At the age of 1 month, the patient was hospitalized for jaundice. During a routine blood test, remarkably elevated aspartate aminotransferase (253.0 IU/L, reference range 0–40 IU/L), γ-glutamyl transferase (540.0 IU/L, reference range 7–50 IU/L) and total bilirubin (303.8 μmol/L, reference range 5.1–17.1 μmol/L), and decreased globulin (9.6 g/L, reference range 20–30 g/L) and ceruloplasmin (0.04 g/L, reference range 0.22–0.58 g/L) were noticed. The patient was discharged after jaundice was treated. However, the hypertransaminasemia, hypercholesterolemia, elevated alkaline phosphatase, decreased serum ceruloplasmin and serum copper level, and coagulopathy persisted during the approximate 4-year follow-up.
Then the 4-year-old boy with abnormal liver function since birth was admitted to our in-patient department. Physical examination showed that his height was 108 cm (87th percentile by the WHO standard) and weight was 19 kg (88th percentile). Ocular strabismus was present, while the Kayser–Fleischer ring was absent. The liver was located 3 cm below the right costal and 4 cm below the xiphoid process, with a soft texture. The examination of the skin, abdomen, and nervous system showed no abnormalities. Abdominal ultrasonography indicated that the echogenicity of the liver was inhomogeneous, showing mild fatty liver manifestation. Cranial magnetic resonance imaging revealed that the anterior part of the right ventricle was slightly plentiful, and part of the sinus mucosa was thickened. The patient was assessed as having a mild intellectual disability by the development screening test (DST). The measured intellectual age was below his actual 51 months, showing 36 months in exercise, 27 months in social adaptation, and 36 months in intelligence. The majority of liver enzymes were elevated, including alanine aminotransferase (58.80 IU/L, reference range 0–40 IU/L), aspartate aminotransferase (123.20 IU/L, reference range 0–40 IU/L), alkaline phosphatase (1296.00 IU/L, reference range 42–383 IU/L), and creatine kinase isoenzyme (30.8 IU/L, reference range 0–30 IU/L). The lipid profile revealed increased total cholesterol (7.92 mmol/L, reference range 3.1–5.2 mmol/L), high-density lipoprotein cholesterol (1.61 mmol/L, reference range 0.91–2.05 mmol/L), and low-density lipoprotein cholesterol (5.72 mmol/L, reference range 1.30–3.90 mmol/L). Serum ceruloplasmin (0.04 g/L, reference range 0.22–0.58 g/L) and serum copper (2.80umol/L, reference range 10.99–21.98 μmol/L) levels were decreased. Transferrin (2.7 g/L, reference range 2.5–4.3 g/L) and ferritin (48.02 ng/ml, reference range 30–400 ng/ml) levels were normal. With the exception of prolonged activated partial thromboplastin time (46.6 s, reference range 28.0–44.5 s), increased D-dimer (0.57 mg/L, reference range 0–0.3 mg/L) and decreased fibrinogen (1.43 g/L, reference range 2–4 g/L), other coagulation indicators were normal. Table 1 summarizes the predominant laboratory investigations of the patient in different phases. Subsequently, the patient was treated with bicyclic alcohol, zinc sulfate, and penicillamine for 1 year. To date, there has been no significant improvement or disease progression during the surveillance period. The follow-up showed elevated serum transaminases, alkaline phosphatase, and lipid profiles, and persistent low ceruloplasmin.

TABLE 1. Predominant changes in laboratory investigations of the Chinese patient.
Liver biopsy showed that the architecture of liver lobules was disordered, and pseudo-lobule was formed (Figure 1A). High power exhibited ground-glass-like hepatic cells, with a lightly stained cytoplasm and clear cell membrane, and vesicular steatosis, which were more evident in the portal area, rather than the regions around the central vein (Figure 1B). Infiltration of few lymphocytes in the portal area was also observed (Figure 1C). PAS staining identified small vacuoles in partial hepatocytes (Figure 1D), while PAS with diastase, copper, and iron staining were negative. The hyperplastic fibrous tissue in the portal area separated liver lobules, formatting bridging-like fibrosis and pseudo-lobules, which was highlighted by Masson staining (Figure 1E). The preserved reticular scaffold structure was displayed by reticulin staining. It presented cirrhosis with stage 4 assessment according to the Scheurer histopathologic scoring system (Scheuer, 1991).
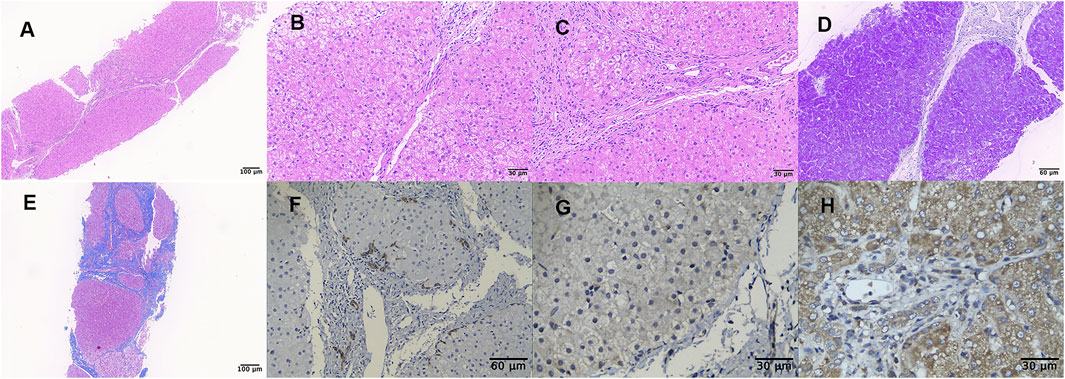
FIGURE 1. Liver biopsy (all originally magnified principal images). Hematoxylin and eosin staining showed the discorded architecture of liver lobules and the formation of pseudo-lobules (A×100), ground-glass-like hepatic cells with lightly stained cytoplasm and obvious vesicular steatosis in portal area (B×400), and lymphocytic infiltration in the portal area (C×400). PAS staining identified small vacuoles in partial hepatocytes (D×200). Formation of bridging-like fibrosis and pseudo-lobule was highlighted by Masson staining (E×100). Mild hyperplasia of bile ducts along the margins of pseudo-lobule was showed by immunohistochemical staining of CK7 (F×200), while weaker staining in the index patient (G×400) than in normal control (H×400) was observed by TMEM199 immunohistochemical detection.
Immunohistochemical staining of CK7 and CK19 showed mild hyperplasia of bile ducts along the margins of pseudo-lobules (Figure 1F). Slightly proliferated Kupffer cells in hepatic sinusoids were indicated by CD68 and CD163 staining, and lymphocytes infiltrating in the portal area were detected by CD3 and CD8 staining. It was worth noting that the expression of TMEM199 was reduced in this index patient sample (Figure 1G), compared with the normal liver sample (donated by a surgical patient), which displayed a cytoplasmic positive pattern in hepatic and bile duct cells (Figure 1H).
With a suspected diagnosis of Wilson disease according to the clinical manifestation, a multi-gene panel was screened. However, the absence of ATP7B mutation and normal DNA copy number variant (CNV) ruled out Wilson disease. Thereafter, two compound heterozygous variations, c.20C > A and c.128delA, were found in TMEM199 gene exon 1 (Figure 2A) by WES. The c.20C > A was inherited from the healthy father, leading to a change of alanine to glutamate at amino acid position 7 (p.Ala7Glu), which was reported in the HGMD (Jansen et al., 2016). The c.128delA as a deletion mutation was inherited from the healthy mother, which caused a frameshift with an early termination (p.Lys43Argfs*25), which was not reported in the HGMD. The amino acid residue of the splice-site mutation (p.Ala7Glu) was conserved across various species, while the frameshift mutation (p.Lys43Argfs*25) was not conserved (Figure 2B). The CADD score of c.20C > A and c.128delA was 14.560 and 11.410 respectively, both predicted as a high impact by MSC. Both mutations of c.20C > A (functional test PS3 + absent from controls PM2 + homozygous variants and distribution in trans PM3-Strong + phenotype match PP4) and c.128delA (frameshift variant with LOF PVS1 + absent from controls PM2 + phenotype match PP4) were evaluated to be pathogenic according to the guideline of the ACMG. The nomenclature of variants was based on the recommendations of the Human Genome Variation Society (HGVS, http://www.hgvs.org/varnomen). Wild-type and mutated TMEM199 were modeled by PyMol (http://www.pymol.org) which showed no effect of Ala7Glu residue change on the polar contact with them around amino acid Arg53 (Figure 2C). Spatial conformation of the frameshift mutation (p.Lys43Argfs*25) predicted by protein modeling showed that the amino acid at position 43 moved backward 25 positions and then terminated, resulting in protein truncation, which contained a loss of the Vam12 domain (Figure 2D). Figure 2E illustrates the TMEM199 protein domains, location of amino acid changes of the reported variants so far.
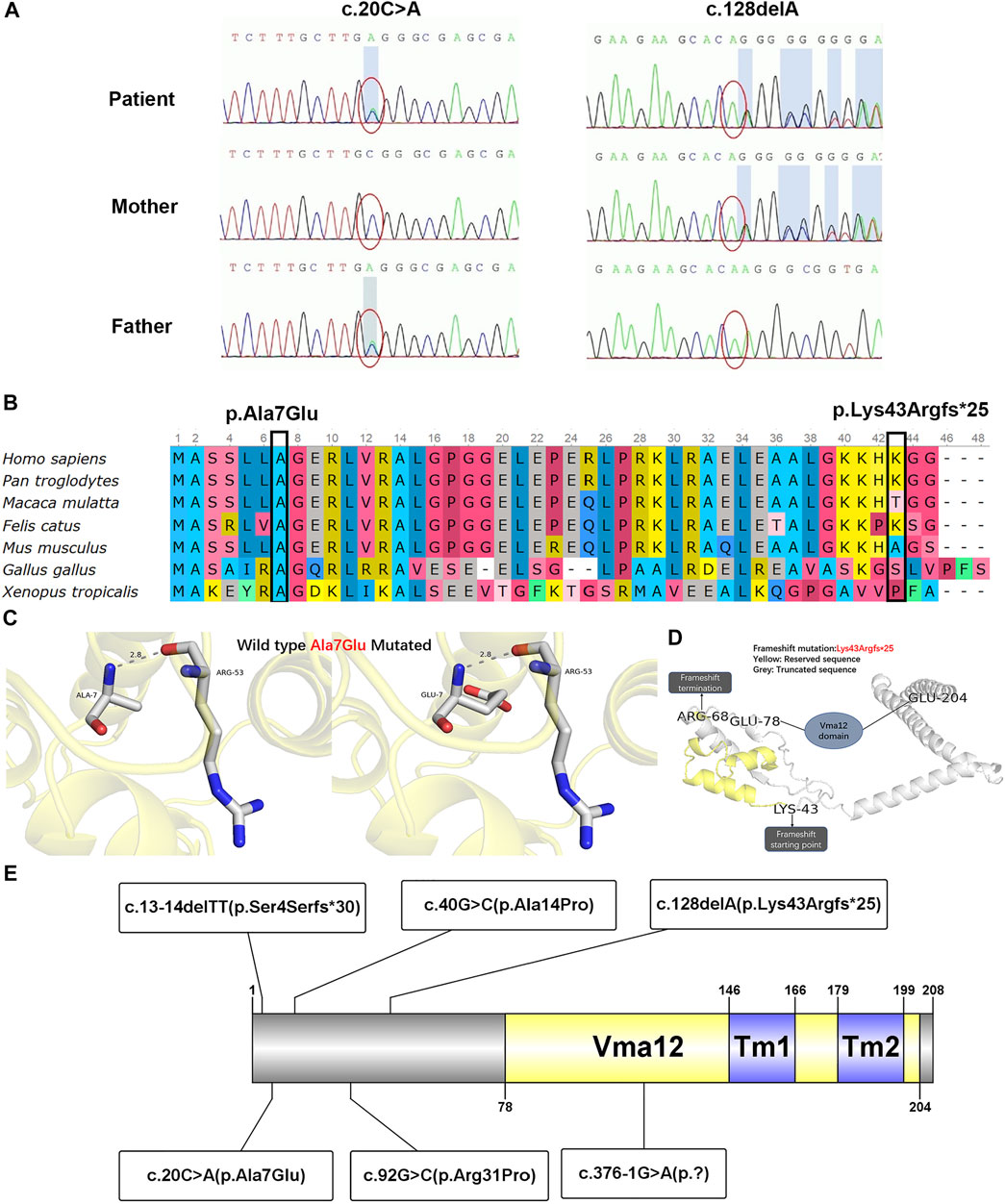
FIGURE 2. Genetic testing results, conservation of amino acid residues, protein modulation, and distribution of reported variants with the TMEM199 protein. (A) Sanger sequencing confirmation of c.20C > A and c.128delA mutation of TMEM199 in the index case and his parents. (B) Conservation status of amino acid residues of the two variants across various species. (C) Wild and mutated types of the p.Ala7Glu variant compared by PyMol. (D) Spatial conformation of the frameshift mutation (p.Lys43Argfs*25) showed by protein modeling. (E) Illustration of TMEM199 protein domains, location of amino acid changes of the reported variants so far.
CDGs are clinically and genetically heterogenous disorders sharing a primary defect of glycosylation. Mandato et al. (2006) detected aberrant glycosylation of transferrin (Tf) by electrospray ionization mass spectrometry (ESI-MS) in four children with cryptogenic chronic hypertransaminasemia and/or liver steatosis and fibrosis, leading to the discovery of a new type of CDG-X with unknown disease-causing genetic alterations. With the development of WES, researchers found two cases of CDG-X and one unrelated case with TMEM199 heterozygous mutations (Vajro et al., 2018). In addition, Jansen et al. (2016) confirmed that the abnormal glycosylation of serum protein was caused by Golgi homeostasis defects, and mutations at different sites in the TMEM199 gene were found in four patients. Two heterozygous mutations in TMEM199 were detected in our patient with the abnormal liver function since birth. Based on the mutations of the TMEM199 gene, all the eight patients were diagnosed with TMEM199-CDG. The main features of the eight patients are listed in Table 2.
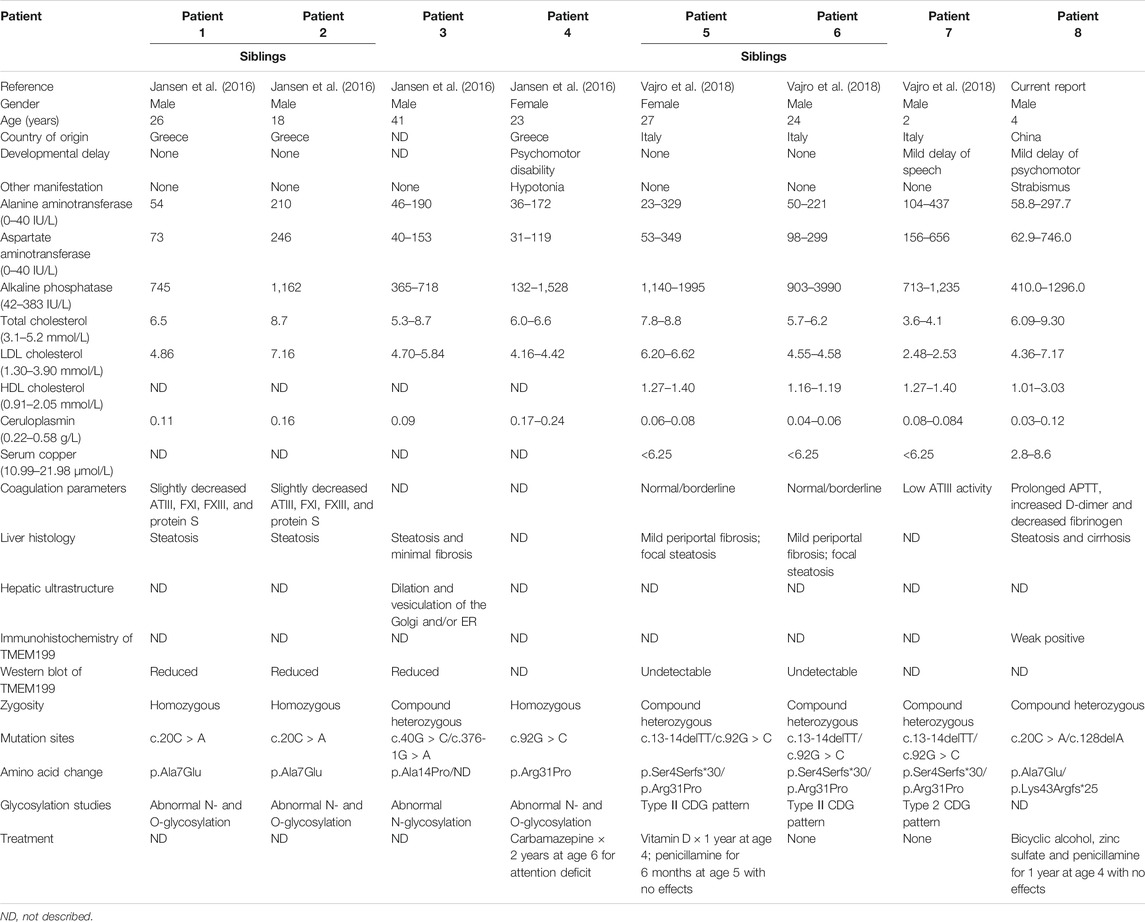
TABLE 2. Main findings of the eight patients with TMEM199-CDG.
The eight patients with TMEM199-CDG included seven Europeans (originating from Greece and Italy) and one Chinese. There were six male and two female patients, with an age range of 2–41 years, four of whom were two pairs of siblings. Seven patients had the disease onset since early childhood. Intellectual disability or mild language retardation was found in three patients, among whom one also presented with hypotonia, while one Chinese patient also presented with strabismus. All patients reported to date presented high fluctuating levels of alanine aminotransferase (23–437 IU/L, reference range 0–40 IU/L), aspartate aminotransferase (31–746 IU/L, reference range 0–40 IU/L), and alkaline phosphatase (132–3990 IU/L, reference range 42–383IU/L). Total cholesterol (5.3–9.3 mmol/L, reference range 3.1–5.2 mmol/L) and low-density lipoprotein cholesterol (1.16–7.17 mmol/L, reference range 1.30–3.90 mmol/L) were elevated in seven patients, while high-density lipoprotein cholesterol (1.01–3.03 mmol/L, reference range 0.91–2.05 mmol/L) was normal or slightly increased in four patients. Low levels of serum ceruloplasmin (0.03–0.24 g/L, reference range 0.22–0.58 g/L) and serum copper (2.80–8.60 umol/L, reference range 10.99–21.98 μmol/L) were found. The coagulation function was normal, close to borderline or slightly weakened in six patients. These findings indicate that TMEM199-CDG mainly affects the liver, and nervous and muscular systems may be involved as well.
Liver biopsies were performed in six patients, four of whom showed various degrees of steatosis and periportal fibrosis, and two only showed steatosis. Among them, one patient underwent the assessment of the ultrastructural characteristics of hepatocytes using a transmission electron microscope. Diffuse and severe vacuolization were observed to most likely derive from the rough ER, smooth ER, and/or Golgi apparatus (Jansen et al., 2016). Unlike the mild or minimal fibrosis manifested in European patients, the liver biopsy of the Chinese patient presented cirrhosis with bridging-like fibrosis and pseudo-lobule formation, which was assessed as stage 4 according to the Scheurer histopathologic scoring system (Scheuer, 1991). The presentation of cirrhosis in early childhood indicates that the severity of liver impairment caused by TMEM199-CDG may require more attention and early intervention.
WES demonstrated heterozygous TMEM199 mutations in five patients and homozygous TMEM199 mutations in three patients, including c.13-14delTT (p.Ser4Serfs*30), c.20C > A (p.Ala7Glu), c.40G > C (p.Ala14Pro), c.92G > C (p.Arg31Pro), c.128delA (p.Lys43Argfs*25), and c.376-1G > A (p.?) (Figure 2E). TMEM199 (NM_152464.1, ENST00000292114) is located on chromosome 17 (chr17:26,684,604–26,690,705), containing six exons encoding a protein of 208 amino acids. The encoded TMEM199 (previously known as C17orf32), a transmembrane protein, is mainly located in the endoplasmic reticulum–Golgi apparatus intermediate compartment (ERGIC) and is involved in Golgi homeostasis. Moreover, as a human homolog of yeast Vma12p, TMEM199 participates in the formation of V-ATPase, which is the proton pump in the process of vesicle acidification via the secretory pathway (Miles et al., 2017). The homeostasis of Golgi apparatus cannot be maintained in TMEM199 deficiency due to the pH imbalance, leading to the abnormal glycosylation of serum proteins, which is closely related to the clinical manifestations of liver dysfunction in patients with TMEM199-CDG. In previously reported patients, glycosylation and functional studies showed aberrant N- and O-glycosylation as a type Ⅱ CDG pattern, and deficient incorporation of galactose and sialic acid as seen in other Golgi homeostasis defects, which was confirmed by metabolic labeling and restored by lentiviral transduction with wild-type TMEM199 (Jansen et al., 2016; Vajro et al., 2018). Hence, the hepatic phenotype with abnormal glycosylation is considered to be resulted from TMEM199 deficiency. Glycosylation defects and the abnormal protein function of the Chinese patient need to be determined in future studies. Additionally, the expression level of TMEM199 in the Chinese patient’s liver specimen detected by immunohistochemical staining was lower than normal control, which was consistent with the reduced or unmeasurable level of TMEM199 protein in skin fibroblasts detected using the Western blot in the European patients (Jansen et al., 2016; Vajro et al., 2018). The TMEM199 antibody (ab121907) is designed with a recombinant fragment corresponding to human TMEM199 amino acids at positions 7–143. Therefore, the unique c.128delA (p.Lys43Argfs*25) in TMEM199 exon 1 in our patient caused a frameshift with an early termination, which was mainly responsible for the weak immunohistochemical signal.
Treatment and prognosis were reported in five patients, two of whom did not receive any medication. Despite the 2 years of carbamazepine usage for attention deficit starting at age 6, no other drugs were given to a Greek patient (Jansen et al., 2016). Vitamin D usage for 1 year starting at age 4 and penicillamine usage for 6 months starting at age 5 were reported in an Italian patient (Vajro et al., 2018), while the Chinese patient in the present study was given bicyclic alcohol, zinc sulfate, and penicillamine for 1 year initiating at age 4. However, it seemed that medical treatments had no definite effect on improving the liver function of these patients. Nevertheless, the previous patients were reported to have no deterioration over several decades, and the Chinese patient presented no significant disease progression during 1-year follow-up. Therefore, there was a view that TMEM199-CDG had a more favorable course to avoid worries of the patients and their parents and unnecessary repeated blood sampling (Vajro et al., 2018). As far as our case is concerned, the early emergence of cirrhosis makes the outcome of the disease not too optimistic; more cohort observations are needed to verify our findings.
This report expands the phenotypic and genotypic spectra of TMEM199-CDG by presenting the first Chinese patient carrying strabismus, severe fibrosis, reduced expression of TMEM199, and a novel truncating mutation. Like the previously reported patients, this Chinese boy was monitored, without significant disease progression. But the appearance of cirrhosis in early childhood needs to be paid attention to, and more cases will help better understand the unclear course of this rare condition.
The data presented in this study is included in the article/Supplementary Material. The DNA datasets are not readily available due to privacy restrictions, further inquiries should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Children’s Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YF and KA drafted and revised the work, Y-ZW and S-ML analyzed and interpreted the data for the work, and LC and J-SW designed the work and approved the final version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Running Gene Inc., for performing next-generation sequencing and Sanger sequencing and providing details of the genetic testing. We thank the patient and his parents for their participation in this study.
Chen, Y.-C., Yu, H., Wang, R.-M., Xie, J.-J., Ni, W., and Zhang, Y. (2019). Contribution of Intragenic Deletions to Mutation Spectrum in Chinese Patients with Wilson's Disease and Possible Mechanism Underlying ATP7B Gross Deletions. Parkinsonism Relat. Disord. 62, 128–133. doi:10.1016/j.parkreldis.2019.01.001
Jaeken, J. (2013). Congenital Disorders of Glycosylation. Handb Clin. Neurol. 113, 1737–1743. doi:10.1016/B978-0-444-59565-2.00044-7
Jansen, J. C., Timal, S., van Scherpenzeel, M., Michelakakis, H., Vicogne, D., Ashikov, A., et al. (2016). TMEM199 Deficiency Is a Disorder of Golgi Homeostasis Characterized by Elevated Aminotransferases, Alkaline Phosphatase, and Cholesterol and Abnormal Glycosylation. Am. J. Hum. Genet. 98 (2), 322–330. doi:10.1016/j.ajhg.2015.12.011
Mandato, C., Brive, L., Miura, Y., Davis, J. A., Di Cosmo, N., Lucariello, S., et al. (2006). Cryptogenic Liver Disease in Four Children: a Novel Congenital Disorder of Glycosylation. Pediatr. Res. 59 (2), 293–298. doi:10.1203/01.pdr.0000196378.30165.26
Marques-da-Silva, D., Dos Reis Ferreira, V., Monticelli, M., Janeiro, P., Videira, P. A., Witters, P., et al. (2017). Liver Involvement in Congenital Disorders of Glycosylation (CDG). A Systematic Review of the Literature. J. Inherit. Metab. Dis. 40 (2), 195–207. doi:10.1007/s10545-016-0012-4
Miles, A. L., Burr, S. P., Grice, G. L., and Nathan, J. A. (2017). The Vacuolar-ATPase Complex and Assembly Factors, TMEM199 and CCDC115, Control HIF1alpha Prolyl Hydroxylation by Regulating Cellular Iron Levels. Elife 6. doi:10.7554/eLife.22693
Scheuer, P. J. (1991). Classification of Chronic Viral Hepatitis: a Need for Reassessment. J. Hepatol. 13 (3), 372–374. doi:10.1016/0168-8278(91)90084-o
Vajro, P., Zielinska, K., Ng, B. G., Maccarana, M., Bengtson, P., Poeta, M., et al. (2018). Three Unreported Cases of TMEM199-CDG, a Rare Genetic Liver Disease with Abnormal Glycosylation. Orphanet J. Rare Dis. 13 (1), 4. doi:10.1186/s13023-017-0757-3
Keywords: congenital disorders of glycosylation, TMEM199-CDG, inherited metabolic disease, liver disease, mutation
Citation: Fang Y, Abuduxikuer K, Wang Y-Z, Li S-M, Chen L and Wang J-S (2022) TMEM199-Congenital Disorder of Glycosylation With Novel Phenotype and Genotype in a Chinese Boy. Front. Genet. 13:833495. doi: 10.3389/fgene.2022.833495
Received: 11 December 2021; Accepted: 14 February 2022;
Published: 24 March 2022.
Edited by:
Hui Xiong, Peking University First Hospital, ChinaReviewed by:
Dario Ronchi, University of Milan, ItalyCopyright © 2022 Fang, Abuduxikuer, Wang, Li, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lian Chen, ZG9jdGNoZW5saWFuQDE2My5jb20=; Jian-She Wang, anNod2FuZ0BzaG11LmVkdS5jbg==
†The authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.