 Patrizia Mondello
Patrizia Mondello Stephen M. Ansell
Stephen M. Ansell Grzegorz S. Nowakowski
Grzegorz S. Nowakowski- Division of Hematology, Mayo Clinic, Rochester, MN, United States
Epigenetic reprogramming is a hallmark of lymphomagenesis, however its role in reshaping the tumor microenvironment is still not well understood. Here we review the most common chromatin modifier mutations in B cell lymphoma and their effect on B cells as well as on T cell landscape. We will also discuss precision therapy strategies to reverse their aberrant signaling by targeting mutated proteins or counterbalance epigenetic mechanisms.
Introduction
Among the highly recurrent mutations in diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL), almost 60% occur in transcription factors or epigenetic modifier proteins (Morin et al., 2011; Reddy et al., 2017; Chapuy et al., 2018; Schmitz et al., 2018). While mutations in chromatin modifier genes (CMGs) have been associated with lymphomagenesis, their role in reshaping the tumor microenvironment (TME) remains poorly characterized. Emerging evidence suggests that malignant B cells may influence the surrounding immune composition through a direct reprogramming of their immune receptors, which in turn disrupt the immune synapse between B and T cells, and indirectly by releasing of cytokines that impact immune response.
The notion of such an epigenetic circuit is plausible in the context of the germinal center (GC) B cells from which DLBCL and FL arise. Epigenetic mechanisms play a critical role in the entry and exit of B cells to the GC. Normally, antigen presentation and BCR signaling are attenuated in the GC dark zone (DZ) to enable B cells to freely proliferate and undergo somatic hypermutation. However, signals received by the B cells in the GC light zone (such as CD40L) induce expression of antigen presentation genes, so that high affinity B cells can be selected to exit the GC reaction and undergo terminal differentiation to plasma cells(Mesin et al., 2016). During selection, a fundamental role is played by T follicular helper (TFH) CD4+ cells - characterized by the expression of CXCR5+, PD1+ and BCL6+ - and follicular dendritic cells (FDCs), which engage GC B cells to promote clonal selection and affinity maturation(Crotty, 2014). In contrast, suppressive CD4+ FOXP3+ T regulatory (Treg) cells, called T follicular regulatory (TFR) cells, limit the function and output of GC reaction (Chung et al., 2011; Wollenberg et al., 2011). While TFR cells dominate the early-GC (Wing et al., 2017), a distinct Treg population prevails in the late-GC (Jacobsen et al., 2021). These recently described cells seem to arise from TFH cells, however it remains unclear what triggers their acquisition of regulatory properties (Jacobsen et al., 2021).
Somatic mutations of CMGs disrupt the fine-tuned mechanism governing GC (Figure 1), thus promoting lymphomagenesis and reprograming TME toward immune suppressive lymphoma niche (Ortega-Molina et al., 2015; Boice et al., 2016; Ennishi et al., 2019; Mondello et al., 2020; Krull et al., 2021; Amin and Braza, 2022). Despite retaining the characteristics of the primary site of development, the cellular composition and spatial arrangement of the TME mirror the genetic complexity and tumor type (Scott and Gascoyne, 2014). Accordingly, the TME has shown a close association with treatment failure and outcome (Dave et al., 2004; Kotlov et al., 2021; Mondello et al., 2021). Here we provide an overview of the most frequent epigenetic mechanisms associated with B cell lymphoma (Figure 1, 2 and Table 1) and discuss the therapeutic options to revert their oncogenic and immunosuppressive effect.
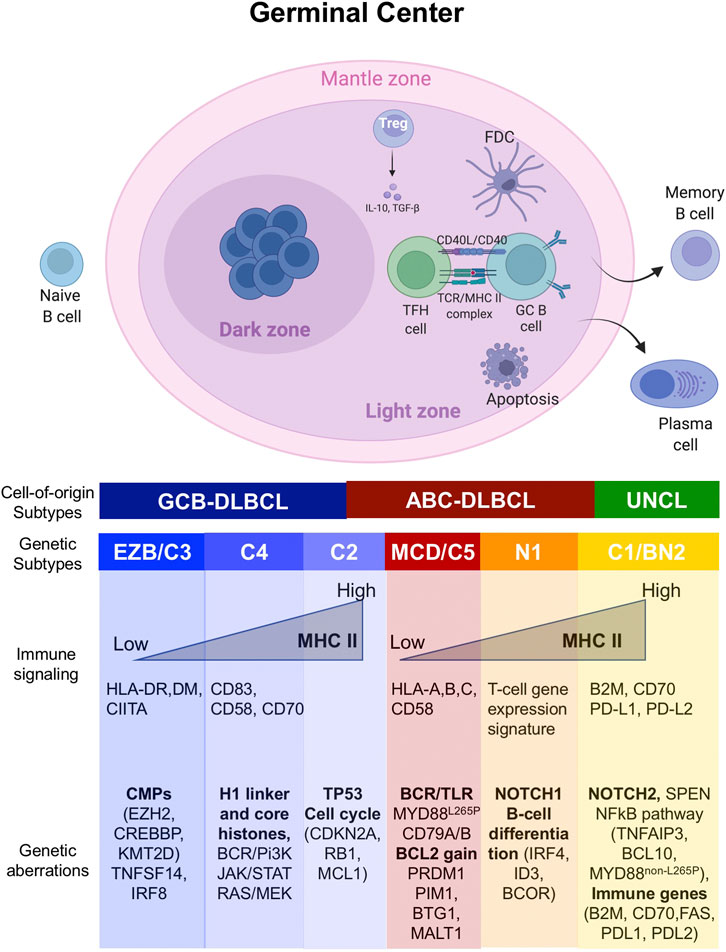
FIGURE 1. Genetic lesions associated with distinct transcription and molecular subtypes of DLBCL. Schematic representation of the germinal center (GC) reaction and its relationship with the major molecular and genetic subtypes of DLBCL. The GC forms upon encounter of a naïve B cell with an antigen, and it is divided in two distinct compartments: the dark zone (DZ) and the light zone (LZ). In the GC DZ, B cells freely proliferate and undergo somatic hypermutation. However, signals from the GC LZ induce expression of antigen presentation in B cells, so that high affinity B cells can be selected to exit the GC reaction and terminally differentiate into plasma cells or memory B cells. Cells that are not selected undergo apoptosis. As the B cells repeatedly cycle between DZ and LZ, they might acquire mutations in genes predominantly activated in the different GC phases, translating in a distinct genetic, transcriptional and phenotypic subtype which might shape the surrounding tumor microenvironment.
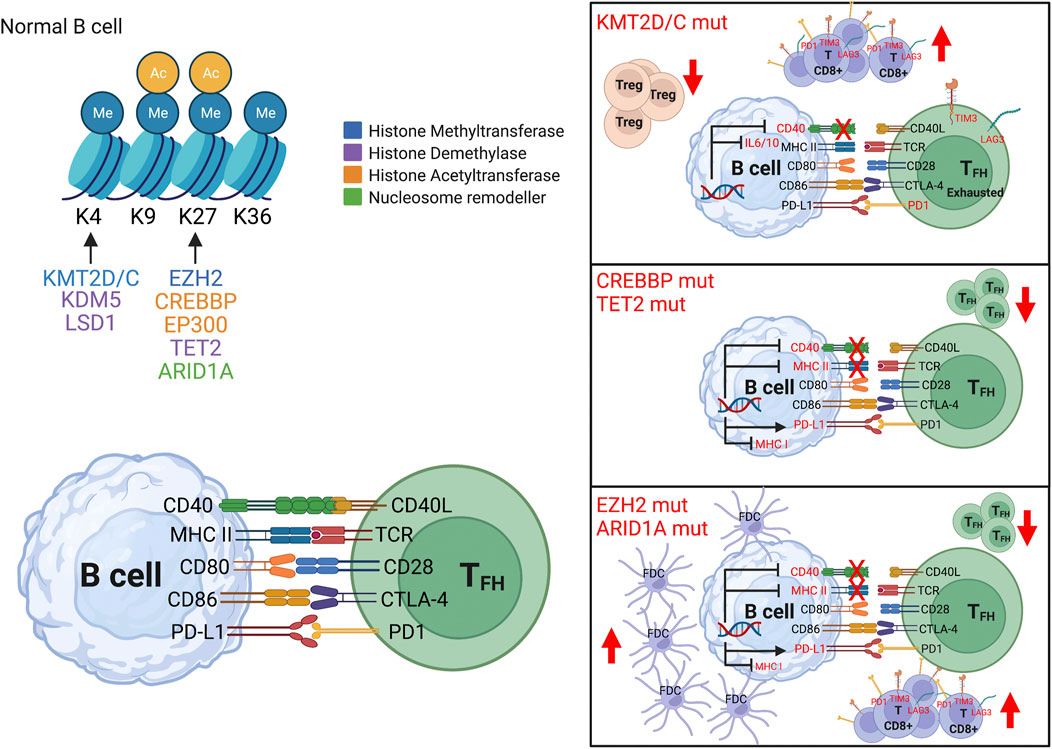
FIGURE 2. Epigenetic control on immune signaling. In normal B cells, epigenetic mechanisms (DNA methylation, histone modification and chromatin remodeling) control gene expression including immune signaling (left top panel), and in turn modulate immune receptors expression (left bottom panel). In B cell lymphoma, aberrant epigenetic programming can disrupt the immune synapse between B and T cells with concordant reshaping of the immune microenvironment. The most frequent epigenetic alterations are: 1) Mutations in KMT2D/C (right top panel) which suppress CD40, IL10-IL6, NF-kB signaling, with prevalence of exhausted CD8+ T cells and decrease of T regulatory (Treg) cells. 2) Mutations in CREBBP and TET2 (right middle panel) which repress antigen presentation genes (such as MHC class II, its transcription factor CIITA), increase inhibitory molecules (such as PDL1), with lower infiltration of CD4+ T cells. 3) Mutations in EZH2 and ARID1A (right lower panel) which repress CIITA (master regulator of MHC class II), NLRC5 (transactivator of MHC class I) and CD40, with decrease in T follicular helper (TFH) cells and increase in follicular dendritic cells (FDC).
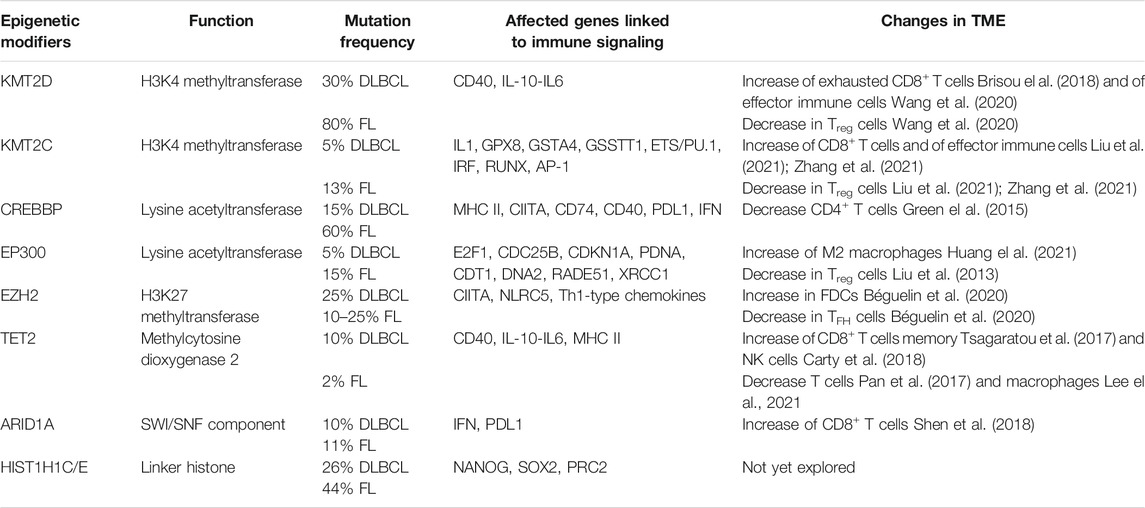
TABLE 1. The most common mutations of epigenetic factors and their effect on the tumor microenvironment.
KMT2D
The KMT2D gene (also known as MLL2 or MLL4) encodes a SET domain-containing lysine methyltransferase and is one of the most frequently mutated genes in B cell lymphomas, reported as being affected in up to 30% of DLBCL and 80% of FL (Morin et al., 2011; Pasqualucci et al., 2011; Ortega-Molina et al., 2015; Zhang et al., 2015; Chapuy et al., 2018; Schmitz et al., 2018). However, KMT2D mutations have not been associated with progression-free survival (PFS) or overall survival (OS) (Ortega-Molina et al., 2015). The majority of these mutations are nonsense or frameshift events that yield truncated proteins lacking the C-terminal SET domain, resulting in enzymatic loss of function (Morin et al., 2011; Pasqualucci et al., 2011; Ortega-Molina et al., 2015; Zhang et al., 2015). Loss of KMT2D in B cells in mice leads to the development of B-cell lymphomas, indicating that this gene is a bona fide tumor suppressor (Ortega-Molina et al., 2015; Zhang et al., 2015). The normal function of KMT2D is linked to its mediating H3K4 mono and demethylation (H4K3me1/2) primarily at gene enhancers (Hu et al., 2013; Lee et al., 2013; Wang et al., 2016). This histone mark is required for non-coding genomic elements to manifest functionality as enhancers (Kouzarides, 2007; Li et al., 2007). KMT2D loss of function was shown to result in focal loss of H3K4me1/2 primarily at gene enhancers, that results in aberrant repression of genes involved in immune signaling such as CD40, IL10-IL6, and NFkB signaling (Ortega-Molina et al., 2015; Zhang et al., 2015). These gene sets play a critical role also in the terminal differentiation of B cells towards the memory or plasma cell fate. Several tumor suppressor genes such as TNFAIP3 and SOCS3 also become silenced upon KMT2D loss of function (Ortega-Molina et al., 2015). Remarkably, KMT2D inactivation promotes remodeling of the TME with prevalence of exhausted CD8+ T cells strongly co-expressing inhibitory receptors (Lag3, Tim3, Pdcd1), suggesting that loss of KMT2D not only reprograms B cells, but also shapes a supportive lymphoma niche which contributes to immune escape (Brisou et al., 2018) Accordingly, a pooled mutagenic screening with CRISPR-mediated genetically engineered mouse models (CRISPR-GEMM) identified KMT2D as a major modulator of response to immune checkpoint blockade (ICB) with increase of effector immune cells (e.g., CD8+ T cells, NK cells and M1 macrophages) and decrease of Treg cells in the TME (Wang et al., 2020). Although KMT2D deficiency seems to sensitize tumors to ICB in solid cancers, this might not be the case for B cell lymphoma where concurrent mutations might alternatively lead to immune escape.
Since mutations in KMT2D cause loss of function and are replicated by genetic deletion of this gene, there is no immediately obvious opportunity for development of precision targeted therapies to reverse their effect. Lymphomas with KMT2D somatic mutations manifest reduction (but not complete loss) of enhancer H3K4 methylation (Ortega-Molina et al., 2015). The reasons for this could be simply due to loss of histone methyltransferase activity, or possibly due to the actions of putative histone demethylases that might normally counteract the actions of KMT2D. This latter scenario is intriguing since it would imply that KMT2D forms part of a reversible epigenetic circuit with histone methyltransferases and histone demethylases competing to set enhancers into a more active or repressed configuration. Therefore, it is possible that in the presence of KMT2D mutation a histone demethylase could act relatively unopposed to maintain silencing of the GC differentiation program. If this was the case it would follow that KMT2D mutant DLBCL cells would become biologically dependent on such a demethylase, which would thus represent a potential precision epigenetic therapy target for these patients. There are two families of enzymes that specifically demethylate H3K4me1/2 (Højfeldt et al., 2013). The first to be discovered were the FAD dependent amino-oxidases KDM1A (LSD1). LSD1 is highly expressed in GC B-cells and DLBCLs. However, previous work ruled out LSD1 as the KMT2D antagonist in DLBCL and FL cells, since specific inhibitors against this protein do not revert silencing of KMT2D target genes nor cause any kind of proliferation arrest or differentiation in DLBCL cells (Hatzi et al., 2019). The second family of H3K4 demethylases are the KDM5 family of jumonji proteins consisting of KDM5A (JARID1A/RBP2), KDM5B (JARID1B/PLU1), KDM5C (JARID1C/SMCX) and KDM5D (JARID1D/SMCY). Recently, inhibition of the demethylases KDM5 (KDM5i) has showed to increase H3K4me3, restore the gene expression repressed on loss of KMT2D and cause antiproliferative effect in vitro and in vivo. (Targeting KDM5 demethylases counteracts KMT2D loss of function in diffuse large B-cell lymphoma; Heward et al., 2021) Additionally, reactivation of silenced CD40 pathway genes by KDM5i yields a powerful synthetic lethal effect in combination with CD40 agonist antibodies (Targeting KDM5 demethylases counteracts KMT2D loss of function in diffuse large B-cell lymphoma), which can open the opportunity for novel combination targeted therapies.
KMT2C
The KMT2C gene (also known as MLL3) is another of the four members in the mixed lineage leukemia (MLL) family. It also encodes a histone methyltransferase that mediates mono- and tri-methylation of histone H3 at lysine K4 (H3K4me1 and H3K4me3), suggesting a partial redundancy with KMT2D in lymphomagenesis (Lee et al., 2013). However, mutations in KMT2C are found at a much lower frequency with 5–13% incidence in DLBCL and FL, respectively (Reddy et al., 2017; Chapuy et al., 2018; Schmitz et al., 2018). The KMT2C mutations are not mutually exclusive with those involving KMT2D, suggesting that their function may not completely overlap. Nevertheless, their redundancy remains a matter of debate due to the contrasting results on the effect of KMT2D on genome-wide distribution of H3K4 methylation (Ortega-Molina et al., 2015; Zhang et al., 2015). Therefore, future studies are required to clarify the relation between these two enzymes in GC B cells and in lymphomagenesis. Interestingly, KMT2C mutations showed a positive association with tumor infiltration of CD8+ T cells, M1 macrophages, neutrophils and NK cells, while they negatively correlated with Treg cells, and in turn predicted response ICB and favorable outcome (Liu et al., 2021; Zhang et al., 2021). However, the putative immune signaling responsible for the remodeling of the TME remains elusive.
CREBBP
Approximately 30–40% of DLBCL and FL manifest somatic mutations of the CREBBP histone acetyltransferase (Morin et al., 2011; Pasqualucci et al., 2011). These occur early during pathogenesis and are more frequent in GCB DLBCL (Green et al., 2015; Jiang et al., 2017). Inactivating missense mutation of the histone acetyltransferase (HAT) domain account for 50–70% of cases, whereas most remaining alleles cause truncation or loss of expression (Morin et al., 2011; Pasqualucci et al., 2011). CREBBP mutations have been associated with adverse outcome in FL (Pastore et al., 2015; Mondello et al., 2020) and DLBCL (Huang et al., 2021), however when discriminating point mutations from nonsense/frameshift mutations only failure-free survival remained significant, but not OS (Mondello et al., 2020). The normal function of CREBBP is to mediate H3K27 acetylation at specific gene enhancers required for terminal differentiation and immune signaling. In the GC dark zone these enhancers are temporarily repressed by the BCL6/SMRT/HDAC3 complexes through H3K27 deacetylation (Hatzi et al., 2013). Then, signals for the GC light zone dissociates HDAC3 from BCL6, and drives CREBBP to “toggle” these enhancers back to the active state by restoring H3K27 acetylation (Hatzi et al., 2013; Jiang et al., 2017), which induces expression of antigen presentation genes (such as MHC class II, its transcription factor CIITA, and its co-receptor CD74), so that high affinity B-cells can be selected to exit the GC reaction.
CREBBP mutation induces focal loss of enhancer H3K27 acetylation and concordant repression of antigen presentation, CD40 and BCR signaling genes. As a consequence, CREBBP mutant FLs featured lower infiltration of CD4+ T cells and were impaired in activating autologous T cells as compared to CREBBP wild type (WT) lymphoma ex vivo (Green et al., 2015; Jiang et al., 2017). This is the result of impaired acetyltransferase activity and unopposed deacetylation by BCL6/SMRT/HDAC3 complexes at enhancer of B cell signaling and immune response genes (Jiang et al., 2017). Hence, lymphomas with CREBBP mutation become extremely dependent on HDAC3. Similarly, CREBBP knockout downregulated MHC class II (García-Ramírez et al., 2017; Hashwah et al., 2017) while MHC class II deletion phenocopies the effect of CREBBP knockout and CD4+ depletion on lymphomagenesis (Hashwah et al., 2017). These data support the notion that immune evasion is the key effect of CREBBP mutation. Recently, HDAC3 inhibition showed to counteract aberrant CREBBP by reverting the unopposed deacetylation of BCL6/SMRT/HDAC3 complexes on enhancers genes involved in antigen presentation and terminal differentiation. Targeting HDAC3 caused more profound growth suppression and cell death in CREBBP mutant than WT DLBCL cell lines (Mondello et al., 2020). Reciprocally, conditional deletion of HDAC3 in GC B cells impaired GC formation and induced upregulation of genes repressed in CREBBP mutant patients (Jiang et al., 2017). Furthermore, selective HDAC3 inhibitors restored immune surveillance by reactivating BCL6-repressed INF pathway and antigen presentation genes in lymphoma cells, enabling T-cells to recognize and kill them, especially in the presence of CREBBP mutation (Mondello et al., 2020). As HDCA3 is a crucial repressor of PDL1 transcription (Deng et al., 2019), HDAC3 inhibition increases PDL1 expression on B cells (Deng et al., 2019; Mondello et al., 2020). Thus, combining HDAC3 inhibitors with PDL1 checkpoint blockade enhances tumor regression through direct cell autonomous activity and enhanced antitumor immune response(Mondello et al., 2020).
EP300
EP300 is a paralog of CREBBP gene and similarly encodes for histone acetyltransferase. However, it is found mutated only in 5–15% of DLBCL and FL and largely in mutually exclusivity with CREBBP (Morin et al., 2011; Pasqualucci et al., 2011; Chapuy et al., 2018; Schmitz et al., 2018). Interestingly, EP300 and CREBBP display structural and functional similarity with 60% shared amino acid identity (Chan and La Thangue, 2001), suggesting their potential compensatory function. However, despite having common transcriptional signatures, they also present distinct transcriptional targets in GC B cells (Meyer et al., 2019). Specifically, mice with conditional deletion of EP300 showed down-regulation for genes involved in cell cycle (e.g. E2F1, CDC25B, CDKN1A), DNA replication (PCNA, CDT1, DNA2) and DNA repair (RAD51 and XRCC1) while those deficient for CREBBP were reduced for genes involved in antigen presentation (e.g., CIITA, H2-DM) and terminal differentiation (e.g., IRF4, SPIB, NFKB2 and CD40). The difference in dysregulated transcriptional programs is reflection of the two distinct GC compartments largely involved - the dark zone and the light zone—whose enrichment was confirmed by gene set enrichment analysis (Meyer et al., 2019). Remarkably, combining genetic deletion of both genes prevented GC formation in vivo and impaired DLBCL proliferation in vitro, suggesting that DLBCLs with CREBBP mutations become biologically dependent on EP300 (Meyer et al., 2019). Additionally, EP300 mutation seems to increase infiltration of M2 macrophages via FCXW7-NOTCH-CCL2/CSF1 axis and associate with high level of the immunosuppressive cytokine IL-10 (Huang et al., 2021), while its inhibition impairs Treg cells, therefore promoting anti-lymphoma immune response (Liu et al., 2013).
EZH2
EZH2 is a histone methyltransferase important for bivalency at key promoters that regulate the GC B cell phenotype (Velichutina et al., 2010; Béguelin et al., 2013; Caganova et al., 2013). EZH2 mutations are present in about 10–25% of FL, 25% of GCB DLBCLs while virtually absent in the ABC subtype (Morin et al., 2011; Béguelin et al., 2013). Most frequently the mutations are gain-of-function events that occur in the protein’s SET domain, resulting in increased trimethylation at H3K27 compared with wild-type. EZH2 mediates its effect by suppressing genes involved in proliferation check point, GC exit and terminal differentiation (Béguelin et al., 2013). This translates into persistent centroblast proliferation, which in presence of additional oncogenic cooperator events enable transformation(Sneeringer et al., 2010; Yap et al., 2011; McCabe et al., 2012a; Béguelin et al., 2013). Furthermore, EZH2 mutation represses CIITA, which is master regulator of MHC class II, NLRC5, which is a transactivator of MHC class I, and Th1-type chemokines (Peng et al., 2015). The epigenetic reprogramming of GC B cells modifies the surrounding microenvironment toward immune suppression which further boosts lymphomagenesis (Beguelin et al., 2020). In Ezh2-Y641F conditional knock-in mice there was silencing of genes (CD69, ICOSL, ICAM1, ICAM2, SLAM, LY108 and BASP1) involved in the immune synapse between GC B cells and TFH cells with concordant disruption of CD40/CD40L signaling and of B-T cells immune module (Liu et al., 2015; Zaretsky et al., 2017; Ise et al., 2018; Beguelin et al., 2020). In the absence of TFH interaction, GC persisted only in EZH2 mutant, but not EZH2 WT GC cells due to the dense network with FDCs which seems to play a supportive role. This network is sustained in part by the aberrant expression of genes involved in the homeostasis of FCDs, such as TNFRSF13C and LTB (Beguelin et al., 2020).
Clinically, EZH2 mutation has been associated with favorable outcome in FL patients who received R-CHOP or chemo-free regimens (Pastore et al., 2015; Huet et al., 2017; Lockmer et al., 2020), while no prognostic association was observed with those who received rituximab plus bendamustine (Jurinovic et al., 2019), suggesting a potential role for therapy decision-making. On the contrary, aberrant EZH2 seemed associated with an inferior prognosis in the novel molecular subtypes C3 (p = 0.05) (Chapuy et al., 2018) and EZB (p = 0.06) (Schmitz et al., 2018) compared to other GCB DLBCL. Nevertheless, it should be noted that the genetic clusters include other mutations which may impact on the survival. Several EZH2 inhibitors (EZH2i) arrest proliferation and induce apoptosis of EZH2-mutant B cells by blocking H3K27me3, resulting in the reactivation of silenced gene targets (McCabe et al., 2012b; Knutson et al., 2012; Béguelin et al., 2013). In line with these data, the clinical development of EZH2i has demonstrated a superior efficacy in FL patients with EZH2 mutations compared to those with wild-type EZH2, however a modest activity was observed also in the latter likely due to other concordant mutations that indirectly activate EZH2 and/or conserved baseline EZH2 activity (Morschhauser et al., 2020; Mondello and Ansell, 2022). Additionally, treatment with EZH2i restored antigen presentation including MHC class I and II and increased infiltration of T cells in the TME as well as the expression of immune checkpoint markers. This negative feedback opens the possibility for combination therapy with ICB (Ennishi et al., 2019).
TET2
TET2 encodes a dioxygenase that converts 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (Ito et al., 2011). 5mC is required for DNA demethylation in tandem with the base excision repair machinery, however it also activates transcription independently (Cimmino et al., 2011; Wu et al., 2011; Rampal et al., 2014). Somatic mutations of TET2 occur in about 10% of DLBCL (Asmar et al., 2013; Reddy et al., 2017) and display a tumor suppressor role (Dominguez et al., 2018). TET2 loss leads to focal loss of enhancer hydroxymethylation and repression of genes involved in GC exit. Accordingly, a transgenic mouse model with deletion of TET2 showed GC hyperplasia, block of plasma cell differentiation and preneoplastic phenotype (Dominguez et al., 2018). Notably, the enhancers and genes dysregulated in tumors with aberrant TET2 are the same as those repressed in CREBBP-mutant lymphoma. These include CD40, cytokines (IL6 and IL10) and antigen presentation gene sets (Dominguez et al., 2018). TET2 and CREBBP mutations are mutually exclusive. TET2-dependent loss in hydromethylation impairs H3K27 acetylation, suggesting that CREBBP requires TET2 and the 5hmC to mediate H3K27 acetylation. In support of this, a superior sensitivity to treatment with HDAC3 inhibitor was observed in cells with TET2 loss (Dominguez et al., 2018). Furthermore, mouse studies have shown that TET2 reshapes chromatin accessibility at genomic binding regions of key transcription factors involved in immune signaling (e.g., BATF and ETS1) favoring disfunction of tumor infiltrating lymphocytes (TIL) (Lee et al., 2021). Conversely, TET2 deletion augments CD8+ T cell memory (Carty et al., 2018), induces cell proliferation of natural killer T cells (Tsagaratou et al., 2017) and significantly enhanced anti-tumor activity of the TIL in a similar manner as that observed with anti-PDL1 blockade, however no synergistic effect was observed combining TET2 depletion and ICB (Lee et al., 2021). Additionally, loss of TET2 significantly enhanced immune response to CAR T cell therapy and suppressed tumor growth (Pan et al., 2017; Fraietta et al., 2018).
ARID1A
ARID1A (also known as BAF250a) is a critical component of the multi-subunit remodeling complexes SWI/SNF (SWItch/Sucrose Non-Fermentable), which control chromatin condensation and accessibility. Mutations in ARID1A are more commonly frameshift mutations and present in mutual exclusivity with its paralogous subcomponent SMARCA4. Normally, this complex modulates gene expression involving cell proliferation and differentiation (Mittal and Roberts, 2020). ARID1A can also bind directly to p53, promoting its activation as well as the expression of the cell cycle inhibitor CDKN1A (Guan et al., 2012). Additionally, ARID1A seems to be involved in FAS-mediated apoptosis, which is a critical mechanism for clonal deletion of GC B cells (Luo et al., 2008). However, the role of ARID1A in lymphomagenesis remains unclear. Mutations in ARID1A compromises mismatch repair resulting in increased tumor mutational load and genetic instability (Mittal and Roberts, 2020). In line with these findings, a remarkable efficacy was observed using inhibitors of the DNA damage checkpoint kinase ATR as well as PARP inhibitors in ARID1A mutant tumors (Shen et al., 2015). Additionally, aberrant ARID1A expression impairs IFN signaling, leading to immunosuppressive TME (Li et al., 2020) which is characterized by increased PDL1 expression and cytotoxic CD8+ T cell infiltration (Shen et al., 2018). Mechanistically, ARID1A displays an antagonistic effect with EZH2 and functionally competes for IFN signaling, which may account for immune response (Li et al., 2020). This novel mechanism ushers the opportunity for therapeutic vulnerabilities. For example, mutations of SWI/SNF genes have been associated with increased sensitivity to ICB (Shen et al., 2018).
Histone 1C/E
Linker histone H1 proteins are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes (Fyodorov et al., 2018). They repress transcription by limiting chromatin accessibility (Fan et al., 2005) either directly through condensation of chromatin fiber, or indirectly via recruitment of transcriptional repressors or impairment of transcriptional activators (Fyodorov et al., 2018). Their function is poorly understood, however H1c/d/e knockout mice exhibited impaired embryonic stem cell differentiation (Zhang et al., 2012), suggesting a critical role in cellular phenotyping. Histone H1 mutations are highly recurrent in B cell lymphoma, with H1C and H1E mutations being the most frequent and occurring in ∼30% of DLBCL and FL (Li et al., 2014; Okosun et al., 2014). Recently, Yusufova et al. showed how disruption of H1 proteins results in architectural remodeling with focal opening of chromatin throughout the all genome. This decompaction was associated with gain of H3K36me2 and/or loss of repressive H3K27me3, and concordant re-expression of the normally silent developmental genes. Loss of H1c and H1e in mice also associated with increased proliferation of the GC B cells, ultimately leading to an aggressive lymphoma with T cell infiltrates as reminiscence of ABC subtype of DLBCL (Yusufova et al., 2021). These data establish H1 as a bona fide tumor suppressor since its aberrant expression drives lymphomagenesis through architectural reorganization of genomic compartments, which in turn leads to epigenetic reprogramming and subsequent reactivation of “stemness” genes. Future studies will be needed to explore the impact of GC B cells differently expressing H1 linkers on the immune components of the microenvironment.
Conclusion and Perspective
Over the last 2 decades we have witnessed the breakthrough of immunotherapy. However, this therapeutic success has been damped in non-Hodgkin B cell lymphoma. Part of the reason is the presence of intrinsic and/or extrinsic immune escape strategies that lymphoma cells develop to survive. The most frequent mechanisms are 1) “hide” from the immune system through genetic or epigenetic alterations that mask molecules involved in antigen presentation or immune receptors, or 2) “defend” from immune eradication through activation of anti-apoptotic mechanisms (e.g., perforin/granzyme pathway, extrinsic pathway including FAS and TRAIL death receptors, and intrinsic pathway including BL2 family proteins) and/or expression of inhibitory molecules (PDL1/L2, CD47, FASL) and/or release of immunosuppressive cytokines (IL10, IL4, TGF-β) (de Charette and Houot, 2018). Despite the remarkable scientific improvements, we are still far from a deep understanding of the cancer immune surveillance. Particularly, the driving mechanisms that are responsible for the distinct tumor microenvironment remain unknown. Several studies have pointed towards aberrant epigenetic programming as a critical player in shaping the lymphoma immune niche. Interestingly, many of the above-mentioned CMGs control—with some degree of overlap - the same GC B cell transcriptional programs by targeting either the enhancers (CREBBP/EP300, KMT2D/KMT2C, TET2) or the promoters (EZH2, ARID1A). Similarly, these genes control a “GC B cell exit” immune module involving multiple immune recognition molecules, including MHC class II, CD40 and PDL1, as well as induce cytokines release which further influence the composition of the TME. It is possible that mutations of CMGs could promote lymphomagenesis while causing aberrant silencing of these immune signaling, which leads, for example, to disruption of the immune synapse between B and T cells, ultimately impairing the immune surveillance.
To decipher the crosstalk between B and T cells, future work must further elucidate the epigenetic circuits regulating immune signaling, and the different ways of modulating the acquisition or removal of histone markers. Additional knowledge on the role of CMGs in normal and malignant B cell and immune cells is mandatory and must be supplemented by functional evidence of a possible inter-influence. For instance, structural rearrangement of immune receptor including downregulation of stimulatory immune molecules and upregulation of inhibitory molecules in B cells could lead to decrease and/or exhaustion of TFH cells. Identification of these mechanisms might be quite impactful as it would open opportunity for precision therapy with a dual mechanism of action by targeting synthetic vulnerabilities while restoring immune response. This novel therapeutic approach might also be exploited to overcome the existing limitation with cellular therapy, with the potential to yield maximal anti-tumor immunity.
Author Contributions
PM contributed to the manuscript design, reviewed the literature and wrote the manuscript. SMA and GSN contributed to the manuscript design and manuscript writing. All the authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amin, R., and Braza, M. S. (2022). The Follicular Lymphoma Epigenome Regulates its Microenvironment. J. Exp. Clin. Cancer Res. 41, 21. doi:10.1186/S13046-021-02234-9
Asmar, F., Punj, V., Christensen, J., Pedersen, M. T., Pedersen, A., Nielsen, A. B., et al. (2013). Genome-wide Profiling Identifies a DNA Methylation Signature that Associates with TET2 Mutations in Diffuse Large B-Cell Lymphoma. Haematologica 98, 1912–1920. doi:10.3324/HAEMATOL.2013.088740
Béguelin, W., Popovic, R., Teater, M., Jiang, Y., Bunting, K. L., Rosen, M., et al. (2013). EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 23, 677–692. doi:10.1016/j.ccr.2013.04.011
Béguelin, W., Teater, M., Meydan, C., Hoehn, K. B., Phillip, J. M., Soshnev, A. A., et al. (2020). Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer cell 37, 655–673. e11. doi:10.1016/J.CCELL.2020.04.004
Boice, M., Salloum, D., Mourcin, F., Sanghvi, V., Amin, R., Oricchio, E., et al. (2016). Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 167, 405–418. e13. doi:10.1016/J.CELL.2016.08.032
Brisou, G., Zala, M., Gil, L., Pagano, G., Bru, A., Cervera-Marzal, I., et al. (2018). Desynchronization of the Germinal Center Dynamics and Remodeling of the Tumor Microenvironment Characterize KMT2D-Driven Lymphomagenesis. Blood 132, 670. doi:10.1182/BLOOD-2018-99-116617
Caganova, M., Carrisi, C., Varano, G., Mainoldi, F., Zanardi, F., Germain, P.-L., et al. (2013). Germinal center Dysregulation by Histone Methyltransferase EZH2 Promotes Lymphomagenesis. J. Clin. Invest. 123, 5009–5022. doi:10.1172/JCI70626
Carty, S. A., Gohil, M., Banks, L. B., Cotton, R. M., Johnson, M. E., Stelekati, E., et al. (2018). The Loss of TET2 Promotes CD8+ T Cell Memory Differentiation. J. Immunol. 200, 82–91. doi:10.4049/JIMMUNOL.1700559
Chan, H. M., and La Thangue, N. B. (2001). p300/CBP Proteins: HATs for Transcriptional Bridges and Scaffolds. J. Cel Sci. 114, 2363–2373. doi:10.1242/JCS.114.13.2363
Chapuy, B., Stewart, C., Dunford, A. J., Kim, J., Kamburov, A., Redd, R. A., et al. (2018). Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 24, 679–690. doi:10.1038/s41591-018-0016-8
Chung, Y., Tanaka, S., Chu, F., Nurieva, R. I., Martinez, G. J., Rawal, S., et al. (2011). Follicular Regulatory T Cells Expressing Foxp3 and Bcl-6 Suppress Germinal center Reactions. Nat. Med. 17, 983–988. doi:10.1038/nm.2426
Cimmino, L., Abdel-Wahab, O., Levine, R. L., and Aifantis, I. (2011). TET Family Proteins and Their Role in Stem Cell Differentiation and Transformation. Cell stem cell 9, 193–204. doi:10.1016/J.STEM.2011.08.007
Crotty, S. (2014). T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity 41, 529–542. doi:10.1016/j.immuni.2014.10.004
Dave, S. S., Wright, G., Tan, B., Rosenwald, A., Gascoyne, R. D., Chan, W. C., et al. (2004). Prediction of Survival in Follicular Lymphoma Based on Molecular Features of Tumor-Infiltrating Immune Cells. N. Engl. J. Med. 351, 2159–2169. doi:10.1056/NEJMoa041869
de Charette, M., and Houot, R. (2018). Hide or Defend, the Two Strategies of Lymphoma Immune Evasion: Potential Implications for Immunotherapy. Haematologica 103, 1256–1268. doi:10.3324/HAEMATOL.2017.184192
Deng, S., Hu, Q., Zhang, H., Yang, F., Peng, C., and Huang, C. (2019). HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti-PD-L1 Therapy. Mol. Cancer Ther. 18, 900–908. doi:10.1158/1535-7163.MCT-18-1068
Dominguez, P. M., Ghamlouch, H., Rosikiewicz, W., Kumar, P., Béguelin, W., Fontan, L., et al. (2018). TET2 Deficiency Causes Germinal center Hyperplasia, Impairs Plasma Cell Differentiation and Promotes B-Cell Lymphomagenesis. Cancer Discov. 8, CD–18. doi:10.1158/2159-8290.CD-18-0657
Ennishi, D., Takata, K., Béguelin, W., Duns, G., Mottok, A., Farinha, P., et al. (2019). Molecular and Genetic Characterization of MHC Deficiency Identifies Ezh2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 9, 546–563. doi:10.1158/2159-8290.CD-18-1090
Fan, Y., Nikitina, T., Zhao, J., Fleury, T. J., Bhattacharyya, R., Bouhassira, E. E., et al. (2005). Histone H1 Depletion in Mammals Alters Global Chromatin Structure but Causes Specific Changes in Gene Regulation. Cell 123, 1199–1212. doi:10.1016/J.CELL.2005.10.028
Fraietta, J. A., Nobles, C. L., Sammons, M. A., Lundh, S., Carty, S. A., Reich, T. J., et al. (2018). Disruption of TET2 Promotes the Therapeutic Efficacy of CD19-Targeted T Cells. Nature 558, 307–312. doi:10.1038/S41586-018-0178-Z
Fyodorov, D. V., Zhou, B.-R., Skoultchi, A. I., and Bai, Y. (2018). Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat. Rev. Mol. Cel Biol 19, 192–206. doi:10.1038/NRM.2017.94
García-Ramírez, I., Tadros, S., González-Herrero, I., Martín-Lorenzo, A., Rodríguez-Hernández, G., Moore, D., et al. (2017). Crebbp Loss Cooperates with Bcl2 Overexpression to Promote Lymphoma in Mice. Blood 129, 2645–2656. doi:10.1182/blood-2016-08-733469
Green, M. R., Kihira, S., Liu, C. L., Nair, R. V., Salari, R., Gentles, A. J., et al. (2015). Mutations in Early Follicular Lymphoma Progenitors Are Associated with Suppressed Antigen Presentation. Proc. Natl. Acad. Sci. USA 112, E1116–E1125. doi:10.1073/pnas.1501199112
Guan, B., Gao, M., Wu, C.-H., Wang, T.-L., and Shih, I.-M. (2012). Functional Analysis of In-Frame Indel ARID1A Mutations Reveals New Regulatory Mechanisms of its Tumor Suppressor Functions. Neoplasia 14, 986–993. doi:10.1593/NEO.121218
Hashwah, H., Schmid, C. A., Kasser, S., Bertram, K., Stelling, A., Manz, M. G., et al. (2017). Inactivation of CREBBP Expands the Germinal center B Cell Compartment, Down-Regulates MHCII Expression and Promotes DLBCL Growth. Proc. Natl. Acad. Sci. USA 114, 9701–9706. doi:10.1073/pnas.1619555114
Hatzi, K., Geng, H., Doane, A. S., Meydan, C., LaRiviere, R., Cardenas, M., et al. (2019). Histone Demethylase LSD1 Is Required for Germinal center Formation and BCL6-Driven Lymphomagenesis. Nat. Immunol. 20, 86–96. doi:10.1038/s41590-018-0273-1
Hatzi, K., Jiang, Y., Huang, C., Garrett-Bakelman, F., Gearhart, M. D., Giannopoulou, E. G., et al. (2013). A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cel Rep. 4, 578–588. doi:10.1016/j.celrep.2013.06.016
Heward, J., Konali, L., D’Avola, A., Close, K., Yeomans, A., Philpott, M., et al. (2021). KDM5 Inhibition Offers a Novel Therapeutic Strategy for the Treatment of KMT2D Mutant Lymphomas. Blood 138, 370–381. doi:10.1182/BLOOD.2020008743
Højfeldt, J. W., Agger, K., and Helin, K. (2013). Histone Lysine Demethylases as Targets for Anticancer Therapy. Nat. Rev. Drug Discov. 12, 917–930. doi:10.1038/nrd4154
Hu, D., Gao, X., Morgan, M. A., Herz, H.-M., Smith, E. R., and Shilatifard, A. (2013). The MLL3/MLL4 Branches of the COMPASS Family Function as Major Histone H3K4 Monomethylases at Enhancers. Mol. Cel Biol 33, 4745–4754. doi:10.1128/MCB.01181-13
Huang, Y.-H., Cai, K., Xu, P.-P., Wang, L., Huang, C.-X., Fang, Y., et al. (2021). CREBBP/EP300 Mutations Promoted Tumor Progression in Diffuse Large B-Cell Lymphoma through Altering Tumor-Associated Macrophage Polarization via FBXW7-NOTCH-Ccl2/csf1 axis. Sig Transduct Target. Ther. 6, 10. doi:10.1038/S41392-020-00437-8
Huet, S., Xerri, L., Tesson, B., Mareschal, S., Taix, S., Mescam-Mancini, L., et al. (2017). EZH2 Alterations in Follicular Lymphoma: Biological and Clinical Correlations. Blood Cancer J. 7, e555. doi:10.1038/BCJ.2017.32
Ise, W., Fujii, K., Shiroguchi, K., Ito, A., Kometani, K., Takeda, K., et al. (2018). T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 48, 702–715. e4. doi:10.1016/J.IMMUNI.2018.03.027
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet Proteins Can Convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi:10.1126/SCIENCE.1210597
Jacobsen, J. T., Hu, W., R. Castro, T. B., Solem, S., Galante, A., Lin, Z., et al. (2021). Expression of Foxp3 by T Follicular Helper Cells in End-Stage Germinal Centers. Science 373, eabe5146. doi:10.1126/SCIENCE.ABE5146
Jiang, Y., Ortega-Molina, A., Geng, H., Ying, H.-Y., Hatzi, K., Parsa, S., et al. (2017). CREBBP Inactivation Promotes the Development of HDAC3-dependent Lymphomas. Cancer Discov. 7, 38–53. doi:10.1158/2159-8290.CD-16-0975
Jurinovic, V., Passerini, V., Oestergaard, M. Z., Knapp, A., Mundt, K., Araf, S., et al. (2019). Evaluation of the M7-FLIPI in Patients with Follicular Lymphoma Treated within the Gallium Trial: EZH2 Mutation Status May Be a Predictive Marker for Differential Efficacy of Chemotherapy. Blood 134, 122. doi:10.1182/BLOOD-2019-130208
Knutson, S. K., Wigle, T. J., Warholic, N. M., Sneeringer, C. J., Allain, C. J., Klaus, C. R., et al. (2012). A Selective Inhibitor of EZH2 Blocks H3K27 Methylation and Kills Mutant Lymphoma Cells. Nat. Chem. Biol. 8, 890–896. doi:10.1038/nchembio.1084
Kotlov, N., Bagaev, A., Revuelta, M. V., Phillip, J. M., Cacciapuoti, M. T., Antysheva, Z., et al. (2021). Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 11, 1468–1489. doi:10.1158/2159-8290.CD-20-0839
Kouzarides, T. (2007). Chromatin Modifications and Their Function. Cell 128, 693–705. doi:10.1016/j.cell.2007.02.005
Krull, J., Wenzl, K., Manske, M. K., Hopper, M. A., Larson, M. C., Sarangi, V., et al. (2021). Somatic Alterations in Follicular Lymphoma Associate with Unique Tumor‐cell Trancriptional States and Tumor‐immune Microenvironments. Hematological Oncol. 39. doi:10.1002/HON.41_2879
Lee, J.-E., Wang, C., Xu, S., Cho, Y.-W., Wang, L., Feng, X., et al. (2013). H3K4 Mono- and Di-methyltransferase MLL4 Is Required for Enhancer Activation during Cell Differentiation. eLife 2, e01503. doi:10.7554/eLife.01503
Lee, M., Li, J., Li, J., Fang, S., Zhang, J., Vo, A. T. T., et al. (2021). Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 81, 1965–1976. doi:10.1158/0008-5472.CAN-20-3213
Li, B., Carey, M., and Workman, J. L. (2007). The Role of Chromatin during Transcription. Cell 128, 707–719. doi:10.1016/j.cell.2007.01.015
Li, H., Kaminski, M. S., Li, Y., Yildiz, M., Ouillette, P., Jones, S., et al. (2014). Mutations in Linker Histone Genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A Underlying the Pathogenesis of Follicular Lymphoma. Blood 123, 1487–1498. doi:10.1182/BLOOD-2013-05-500264
Li, J., Wang, W., Zhang, Y., Cieślik, M., Guo, J., Tan, M., et al. (2020). Epigenetic Driver Mutations in ARID1A Shape Cancer Immune Phenotype and Immunotherapy. J. Clin. Invest. 130, 2712–2726. doi:10.1172/JCI134402
Liu, D., Xu, H., Shih, C., Wan, Z., Ma, X., Ma, W., et al. (2015). T-B-cell Entanglement and ICOSL-Driven Feed-Forward Regulation of Germinal centre Reaction. Nature 517, 214–218. doi:10.1038/NATURE13803
Liu, R., Niu, Y., Zhang, X., and Ma, T. (2021). Association of KMT2C/D Loss-Of-Function Mutations with Tumor Infiltrating Lymphocytes and Response to Immune Checkpoint Inhibitors in Solid Tumors. J. Clin. Oncol. 39, 2587. doi:10.1200/JCO.2021.39.15_SUPPL.2587
Liu, Y., Wang, L., Predina, J., Han, R., Beier, U. H., Wang, L.-C. S., et al. (2013). Inhibition of P300 Impairs Foxp3+ T Regulatory Cell Function and Promotes Antitumor Immunity. Nat. Med. 19, 1173–1177. doi:10.1038/NM.3286
Lockmer, S., Ren, W., Brodtkorb, M., Østenstad, B., Wahlin, B. E., Pan‐Hammarström, Q., et al. (2020). M7‐FLIPI Is Not Prognostic in Follicular Lymphoma Patients with First‐line Rituximab Chemo‐free Therapy. Br. J. Haematol. 188, 259–267. doi:10.1111/BJH.16159
Luo, B., Cheung, H. W., Subramanian, A., Sharifnia, T., Okamoto, M., Yang, X., et al. (2008). Highly Parallel Identification of Essential Genes in Cancer Cells. Proc. Natl. Acad. Sci. 105, 20380–20385. doi:10.1073/PNAS.0810485105
McCabe, M. T., Graves, A. P., Ganji, G., Diaz, E., Halsey, W. S., Jiang, Y., et al. (2012a). Mutation of A677 in Histone Methyltransferase EZH2 in Human B-Cell Lymphoma Promotes Hypertrimethylation of Histone H3 on Lysine 27 (H3K27). Proc. Natl. Acad. Sci. 109, 2989–2994. doi:10.1073/pnas.1116418109
McCabe, M. T., Ott, H. M., Ganji, G., Korenchuk, S., Thompson, C., Van Aller, G. S., et al. (2012b). EZH2 Inhibition as a Therapeutic Strategy for Lymphoma with EZH2-Activating Mutations. Nature 492, 108–112. doi:10.1038/nature11606
Mesin, L., Ersching, J., and Victora, G. D. (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. doi:10.1016/j.immuni.2016.09.001
Meyer, S. N., Scuoppo, C., Vlasevska, S., Bal, E., Holmes, A. B., Holloman, M., et al. (2019). Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 51, 535–547. e9. doi:10.1016/J.IMMUNI.2019.08.006
Mittal, P., and Roberts, C. W. M. (2020). The SWI/SNF Complex in Cancer - Biology, Biomarkers and Therapy. Nat. Rev. Clin. Oncolclinical Oncology 17, 435–448. doi:10.1038/S41571-020-0357-3
Mondello, P., and Ansell, S. M. (2022). Tazemetostat: a Treatment Option for Relapsed/Refractory Follicular Lymphoma. Expert Opin. Pharmacother. 23 (3), 295–301. doi:10.1080/14656566.2021.2014815
Mondello, Patrizia., Toska, Eneda., Teater, Matt., Fontan, Lorena., Durant, Matthew., Casalena, Gabriella., et al. (2018). TargetingTargeting KDM5 Demethylases Counteracts KMT2D Loss of Function in Diffuse Large B-Cell Lymphoma. Available at: http://www.abstractsonline.com/pp8/#!/5739/presentation/434 (Accessed December 17, 2018).
Mondello, P., Fama, A., Larson, M. C., Feldman, A. L., Villasboas, J. C., Yang, Z.-Z., et al. (2021). Lack of Intrafollicular Memory CD4 + T Cells Is Predictive of Early Clinical Failure in Newly Diagnosed Follicular Lymphoma. Blood Cancer J. 11, 130. doi:10.1038/S41408-021-00521-4
Mondello, P., Tadros, S., Teater, M., Fontan, L., Chang, A. Y., Jain, N., et al. (2020). Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 10, 440–459. doi:10.1158/2159-8290.CD-19-0116
Morin, R. D., Mendez-Lago, M., Mungall, A. J., Goya, R., Mungall, K. L., Corbett, R. D., et al. (2011). Frequent Mutation of Histone-Modifying Genes in Non-hodgkin Lymphoma. Nature 476, 298–303. doi:10.1038/nature10351
Morschhauser, F., Tilly, H., Chaidos, A., McKay, P., Phillips, T., Assouline, S., et al. (2020). Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: an Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 21, 1433–1442. doi:10.1016/S1470-2045(20)30441-1
Okosun, J., Bödör, C., Wang, J., Araf, S., Yang, C.-Y., Pan, C., et al. (2014). Integrated Genomic Analysis Identifies Recurrent Mutations and Evolution Patterns Driving the Initiation and Progression of Follicular Lymphoma. Nat. Genet. 46, 176–181. doi:10.1038/NG.2856
Ortega-Molina, A., Boss, I. W., Canela, A., Pan, H., Jiang, Y., Zhao, C., et al. (2015). The Histone Lysine Methyltransferase KMT2D Sustains a Gene Expression Program that Represses B Cell Lymphoma Development. Nat. Med. 21, 1199–1208. doi:10.1038/nm.3943
Pan, W., Zhu, S., Qu, K., Meeth, K., Cheng, J., He, K., et al. (2017). The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 47, 284–297. doi:10.1016/J.IMMUNI.2017.07.020
Pasqualucci, L., Dominguez-Sola, D., Chiarenza, A., Fabbri, G., Grunn, A., Trifonov, V., et al. (2011). Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 471, 189–195. doi:10.1038/nature09730
Pastore, A., Jurinovic, V., Kridel, R., Hoster, E., Staiger, A. M., Szczepanowski, M., et al. (2015). Integration of Gene Mutations in Risk Prognostication for Patients Receiving First-Line Immunochemotherapy for Follicular Lymphoma: a Retrospective Analysis of a Prospective Clinical Trial and Validation in a Population-Based Registry. Lancet Oncol. 16, 1111–1122. doi:10.1016/S1470-2045(15)00169-2
Peng, D., Kryczek, I., Nagarsheth, N., Zhao, L., Wei, S., Wang, W., et al. (2015). Epigenetic Silencing of TH1-type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 527, 249–253. doi:10.1038/NATURE15520
Rampal, R., Alkalin, A., Madzo, J., Vasanthakumar, A., Pronier, E., Patel, J., et al. (2014). DNA Hydroxymethylation Profiling Reveals that WT1 Mutations Result in Loss of TET2 Function in Acute Myeloid Leukemia. Cel Rep. 9, 1841–1855. doi:10.1016/J.CELREP.2014.11.004
Reddy, A., Zhang, J., Davis, N. S., Moffitt, A. B., Love, C. L., Waldrop, A., et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494. e15. doi:10.1016/j.cell.2017.09.027
Schmitz, R., Wright, G. W., Huang, D. W., Johnson, C. A., Phelan, J. D., Wang, J. Q., et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 378, 1396–1407. doi:10.1056/NEJMoa1801445
Scott, D. W., and Gascoyne, R. D. (2014). The Tumour Microenvironment in B Cell Lymphomas. Nat. Rev. Cancer 14, 517–534. doi:10.1038/nrc3774
Shen, J., Ju, Z., Zhao, W., Wang, L., Peng, Y., Ge, Z., et al. (2018). ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat. Med. 24, 556–562. doi:10.1038/S41591-018-0012-Z
Shen, J., Peng, Y., Wei, L., Zhang, W., Yang, L., Lan, L., et al. (2015). ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 5, 752–767. doi:10.1158/2159-8290.CD-14-0849
Sneeringer, C. J., Scott, M. P., Kuntz, K. W., Knutson, S. K., Pollock, R. M., Richon, V. M., et al. (2010). Coordinated Activities of Wild-type Plus Mutant EZH2 Drive Tumor-Associated Hypertrimethylation of Lysine 27 on Histone H3 (H3K27) in Human B-Cell Lymphomas. Proc. Natl. Acad. Sci. 107, 20980–20985. doi:10.1073/pnas.1012525107
Tsagaratou, A., González-Avalos, E., Rautio, S., Scott-Browne, J. P., Togher, S., Pastor, W. A., et al. (2017). TET Proteins Regulate the Lineage Specification and TCR-Mediated Expansion of iNKT Cells. Nat. Immunol. 18, 45–53. doi:10.1038/NI.3630
Velichutina, I., Shaknovich, R., Geng, H., Johnson, N. A., Gascoyne, R. D., Melnick, A. M., et al. (2010). EZH2-mediated Epigenetic Silencing in Germinal center B Cells Contributes to Proliferation and Lymphomagenesis. Blood 116, 5247–5255. doi:10.1182/blood-2010-04-280149
Wang, C., Lee, J.-E., Lai, B., Macfarlan, T. S., Xu, S., Zhuang, L., et al. (2016). Enhancer Priming by H3K4 Methyltransferase MLL4 Controls Cell Fate Transition. Proc. Natl. Acad. Sci. USA 113, 11871–11876. doi:10.1073/pnas.1606857113
Wang, G., Chow, R. D., Zhu, L., Bai, Z., Ye, L., Zhang, F., et al. (2020). CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 10, 1912–1933. doi:10.1158/2159-8290.CD-19-1448
Wing, J. B., Kitagawa, Y., Locci, M., Hume, H., Tay, C., Morita, T., et al. (2017). A Distinct Subpopulation of CD25− T-Follicular Regulatory Cells Localizes in the Germinal Centers. Proc. Natl. Acad. Sci. USA 114, E6400–E6409. doi:10.1073/PNAS.1705551114
Wollenberg, I., Agua-Doce, A., Hernández, A., Almeida, C., Oliveira, V. G., Faro, J., et al. (2011). Regulation of the Germinal Center Reaction by Foxp3+ Follicular Regulatory T Cells. J. Immunol. 187, 4553–4560. doi:10.4049/jimmunol.1101328
Wu, H., D'Alessio, A. C., Ito, S., Wang, Z., Cui, K., Zhao, K., et al. (2011). Genome-wide Analysis of 5-hydroxymethylcytosine Distribution Reveals its Dual Function in Transcriptional Regulation in Mouse Embryonic Stem Cells. Genes Dev. 25, 679–684. doi:10.1101/GAD.2036011
Yap, D. B., Chu, J., Berg, T., Schapira, M., Cheng, S.-W. G., Moradian, A., et al. (2011). Somatic Mutations at EZH2 Y641 Act Dominantly through a Mechanism of Selectively Altered PRC2 Catalytic Activity, to Increase H3K27 Trimethylation. Blood 117, 2451–2459. doi:10.1182/blood-2010-11-321208
Yusufova, N., Kloetgen, A., Teater, M., Osunsade, A., Camarillo, J. M., Chin, C. R., et al. (2021). Histone H1 Loss Drives Lymphoma by Disrupting 3D Chromatin Architecture. Nature 589, 299–305. doi:10.1038/S41586-020-3017-Y
Zaretsky, I., Atrakchi, O., Mazor, R. D., Stoler-Barak, L., Biram, A., Feigelson, S. W., et al. (2017). ICAMs Support B Cell Interactions with T Follicular Helper Cells and Promote Clonal Selection. J. Exp. Med. 214, 3435–3448. doi:10.1084/JEM.20171129
Zhang, J., Dominguez-Sola, D., Hussein, S., Lee, J.-E., Holmes, A. B., Bansal, M., et al. (2015). Disruption of KMT2D Perturbs Germinal center B Cell Development and Promotes Lymphomagenesis. Nat. Med. 21, 1190–1198. doi:10.1038/nm.3940
Zhang, L., Song, J., Wang, Y., and Chen, Y. (2021). KMT2C as a Positive Predictor for Treatment of Immune Checkpoint Inhibitor and Correlation with Immune Infiltrates in Colorectal Cancer (CRC). J. Clin. Oncol. 39, 3538. doi:10.1200/JCO.2021.39.15_SUPPL.3538
Keywords: epigenetics, tumor microenvironment, immune crosstalk, B cell lymphoma, T cells, diffuse large B cell lymphoma, follicular lymphoma
Citation: Mondello P, Ansell SM and Nowakowski GS (2022) Immune Epigenetic Crosstalk Between Malignant B Cells and the Tumor Microenvironment in B Cell Lymphoma. Front. Genet. 13:826594. doi: 10.3389/fgene.2022.826594
Received: 01 December 2021; Accepted: 24 January 2022;
Published: 14 February 2022.
Edited by:
Mounia S. Braza, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Margarita Sánchez-Beato, Hospital Universitario Puerta de Hierro Majadahonda, SpainMakoto Yamagishi, University of Tokyo, Japan
Copyright © 2022 Mondello, Ansell and Nowakowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrizia Mondello, bW9uZGVsbG8ucGF0cml6aWFAbWF5by5lZHU=, cGF0cml6aWFtb25kZWxsb0Bob3RtYWlsLml0; Grzegorz S. Nowakowski, bm93YWtvd3NraS5HcnplZ29yekBtYXlvLmVkdQ==