 Yiming Qi
Yiming Qi Xueqi Ji1,3†
Xueqi Ji1,3† Yan Zhang
Yan Zhang Aihua Yin
Aihua Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 10 March 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.821587
This article is part of the Research TopicGenetic and Environmental Factors in the Occurrence of Neonatal Disorders - Volume IView all 10 articles
Recessive mutations in BRAT1 cause lethal neonatal rigidity and multifocal seizure syndrome (RMFSL), a phenotype characterized by neonatal microcephaly, hypertonia, and refractory epilepsy with premature death. Recently, attenuated disease variants have been described, suggesting that a wider clinical spectrum of BRAT1-associated neurodegeneration exists than was previously thought. Here, we reported a 10-year-old girl with severe intellectual disability, rigidity, ataxia or dyspraxia, and cerebellar atrophy on brain MRI; two BRAT1 variants in the trans configuration [c.1014A > C (p.Pro338 = ); c.706delC (p.Leu236Cysfs*5)] were detected using whole-exome sequencing. RNA-seq confirmed significantly decreased BRAT1 transcript levels in the presence of the variant; further, it revealed an intron retention between exon 7 and exon 8 caused by the synonymous base substitute. Subsequent prenatal diagnosis for these two variants guided the parents to reproduce. We expand the phenotypic spectrum of BRAT1-associated disorders by first reporting the pathogenic synonymous variant of the BRAT1 gene, resulting in clinical severity that is mild compared to the severe phenotype seen in RMFSL. Making an accurate diagnosis and prognostic evaluation of BRAT1-associated neurodegeneration is important for reproductive consultation and disease management.
Variations in BRAT1 (BRCA1-associated ataxia telangiectasia mutated activator 1) are initially recognized as the cause of lethal neonatal rigidity and multifocal seizure syndrome (RMFSL; OMIM#614498), which is characterized by neonatal microcephaly, intractable seizures, hypertonia, and early demise. Subsequently, neurodevelopmental disorder with cerebellar atrophy and with or without seizures (NEDCAS; OMIM#618056) caused by biallelic BRAT1 variants were reported and redefined the description of “lethality.” Recently, a milder clinical form that manifests as nonprogressive cerebellar ataxia (NPCA) was described in some childhood-onset patients (Mahjoub et al., 2019), suggesting that a wider phenotypic spectrum of BRAT1-associated neurodegeneration exists than was previously thought.
Physiological functions of the disease-causing gene BRAT1 are diverse (Fernandez-Jaen and Ouchi, 2016). It encodes a protein that interacts with the tumor suppressor gene BRCA1 at its C-terminus and binds to ATM1, considering a master controller of the cell cycle signaling pathways required for cellular responses to DNA damage (Ouchi and Ouchi, 2010). BRAT1 can form a complex with an ATPase domain-containing protein, BRP1 (BRAT1 Partner 1), and prevent transcriptional silencing at methylated genomic regions (Zhang et al., 2016). It may also be involved in cell growth and apoptosis (Straussberg et al., 2015). BRAT1 deficiency secondary to biallelic BRAT1 mutations may increase the glucose metabolism, reduce the mitochondrial reactive oxygen species (ROS) concentration, deteriorate cell growth and migration, and induce neuronal atrophy (Puffenberger et al., 2012; Saunders et al., 2012; So and Ouchi, 2014; Wolf et al., 2015). The complexity and extensiveness of the BRAT1 gene function are the biological basis for the huge phenotypic heterogeneity. However, the exact mechanism by which variations in BRAT1 trigger neurodegeneration and to what extent a defect in ATM function contributes to this disease are unknown.
In this study, we first reported the clinical course of a proband with NPCA caused by novel compound heterozygous BRAT1 variants, which include a negligible pathogenic synonymous variant. Functional studies confirmed the effect of them on transcription. The result provided a theoretical basis and guidance for this family in reproductive genetic counseling and prenatal diagnosis. Furthermore, we summarized all published cases with BRAT1 variation, which provide insight into the clinical–genetic correlation and the pathophysiology of the disease.
A 10-year-old girl who presented severe intellectual disability and poor motor ability was transferred to our genetics center for consultation due to the family’s reproductive plan.
The patient was the second child of nonconsanguineous parents of Chinese and had a healthy adolescent sister (pedigree in Figure 1A). There were unremarkable findings during her prenatal, perinatal, and neonatal courses. Physical examination at birth revealed a normal height, normal weight, and normal head 101 circumference (51 cm; 48th percentile).
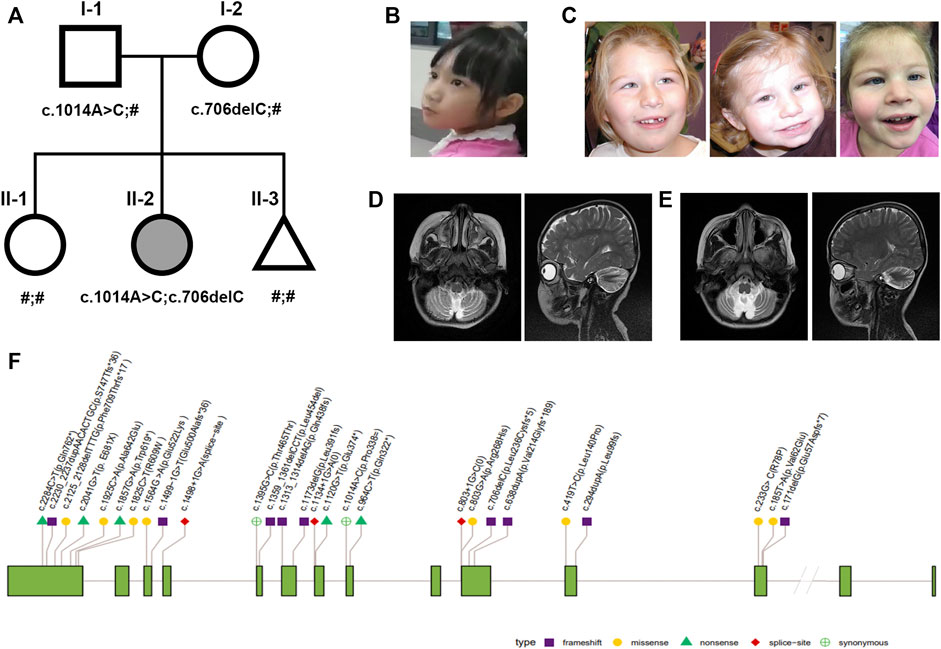
FIGURE 1. Nonprogressive cerebellar ataxia syndrome phenotype and pedigrees. (A) Pedigrees of the family with NPCA profiled in this study. The proband is marked in black. (B–C) Facial appearance of the proband is not specific (left); typical facial dysmorphisms in reported BRAT1-associated NPCA including epicanthal folds, a high arched palate, etc [right, Ref (Srivastava et al., 2016)]. (D–E) Brain MRI of the proband at the age of 4 showed typical cerebellar atrophy (left), which almost showed no progress at 9 years (right). (F) Distribution of reported pathogenic variants in BRAT1. Annotations include the following: exon (grass–green square); variation types: frameshift (purple square), missense (yellow circle), nonsense (green triangle), red diamond (splice site), and synonymous (green cross circle).
However, global developmental delay was presented initially a few months later. She developed head control at 6 months and could not sit until 15 months. At 2.5 years, she could only babble, make consonant sounds, and communicate her needs by crying and gazing. After an individualized neurorehabilitation therapy, she was able to sit briefly and pull to stand at 3 years. One year later, she could stand independently for a few minutes and walk with a walker. At 5 years, she could say 5–10 word phrases that were dysarthric, identify people who were in constant contact with her, and follow simple commands. Meanwhile, behavior with episodes of impulsivity and irritability began to appear since then. She attended special schooling for rehabilitation training but with poor performance until last re-evaluation and was considered to have severe intellectual disability with a mental age of less than 3 years.
Until now, she could stand alone and walk slowly with limited support in a rigid- and broad-based gait with flexed arm posture (Supplementary Videos S1,2). She also presented slight dysmetria, which interferes in fine motor skills during the performance of tasks (Supplementary Video S3). Although her facial expression shows relative paucity (Figure 1B), no evident BRAT1-assoicated dysmorphisms, such as epicanthal folds, high arched palate, fifth finger clinodactyly, or single palmar crease, were observed (Figure 1C). Her cranial nerves were intact except pendular nystagmus. Motor exam has shown mild hypertonia and resistance during extension, but tendon reflexes were normal.
Cranial magnetic resonance imaging (MRI) spectroscopy was first performed at the age of 4, which recorded cerebellar atrophy (Figure 1D). In the following years, she showed stable cerebellar atrophy on serial neuro-MRI (Figure 1E). Electroencephalography was uniformly negative.
Laboratory examination included serum tests for alpha-fetoprotein (AFP), inborn errors of the metabolism, amino acids, very long-chain fatty acids, acylcarnitine and carnitine profiles, infectious work-up, TORCH (toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus) tests, urine organic acid analyses, and conventional indicators in cerebrospinal fluid (CSF) analysis, which were all within normal limits. We also ruled out the possibility of serine biosynthesis defects and nonketotic hyperglycinemia by normal serine and glycine. After precluding aneuploidy and copy number variations (CNVs), we conducted a Trio WES on the family.
Genomic DNA was extracted using a Qiagen DNA blood mini kit (Qiagen GmbH, Hilden, Germany). Library preparation and target enrichment were performed using a SureSelectXT Clinical Research Exome kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s specifications. Then, Trio WES was performed using 2 × 150 bp in the paired end mode of the NextSeq 500 platform (Illumina, San Diego, CA) to obtain an average coverage of above 110x, with 97.6% of target bases covered at least 10x. Sequence quality analysis and filtering of mapped target sequences were performed with the ‘varbank’ exome and genome analysis pipeline v.2.1 as described previously (Vetro et al., 2020). Analysis of genetic results was based on the genomic variation database (http://dgv.tcag.ca/dgv/app/home), DECIPHER database (https://decipher.sanger.ac.uk/), and OMIM database (http://www.ncbi.nlm.nih.gov/omim). The found variants were further verified by Sanger sequencing in fetuses and parents.
Total RNA was extracted from peripheral blood samples using Qiagen blood RNA extraction kit I (QIAGEN, United States); the procedures and standards were performed according to the manual. After complete property control of RNA and quality control of RNA concentration and purity are qualified, 1 μg of RNA is aspirated for mRNA library construction. The mRNA library was constructed using a TIANSeq Fast RNA Library Kit (Illumina, United States) according to the manufacturer’s instructions, where mRNA was purified and enriched from 1 μg of the total RNA samples and then fragmented about 250 bp, and the index adapter was added. Finally, using a high-fidelity enzyme amplifies the library. After quality control, the libraries were sequenced on an Illumina HiSeq 4,000 platform.
Fetal samples were collected by chorionic villus sampling at 13 weeks of gestation. The procedure was performed using the transabdominal approach. Under aseptic conditions, an 18 or 20 gauge spinal needle was inserted into the placenta under continuous ultrasound guidance. A 20 cc syringe containing the collection media is attached to the end of the needle once the stylet is removed. Negative pressure is created, and the needle is moved up and down through the placenta, collecting the tissue (Jones and Montero, 2021).
Whole-exome sequencing analysis identified compound heterozygous mutations in the BRAT1 gene, c.706delC (p.Leu236Cysfs*5), and a synonymous variant, c.1014A > C (p.Pro338 = ), which were confirmed by sanger sequencing and segregated with the disorder in her family (Figure 2A).
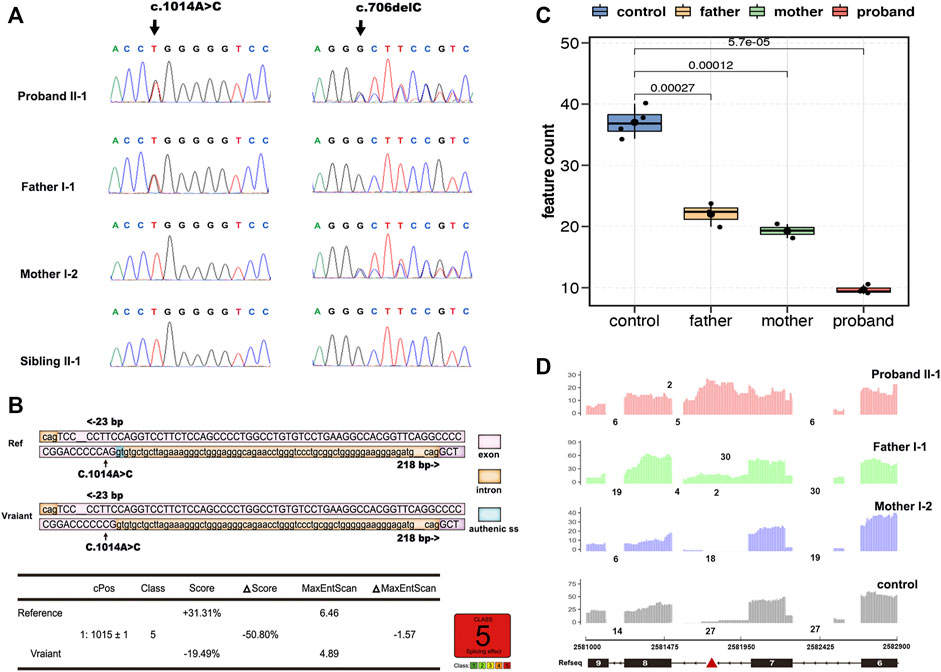
FIGURE 2. Overview of genetic testing results of BRAT1. Sanger sequencing of BRAT1 variants. (A) Chromatograms demonstrated the compound heterozygous status of c.1014A > C and c.706delC in the BRAT1 gene in the proband, the proband’s father, and the proband’s mother. The position of the variant or the corresponding wild-type nucleotide is labeled with black arrows. (B) Software predictions strongly suggest that the substitution of c.1014A > C might cause aberrant splicing. (C) Groups were significantly different from each other (controls vs. proband, p < 5.7*10−5; controls vs. father, p < 2.7*10−4; controls vs. mother, p < 1.2*10−4; and two-tailed unpaired t-test). (D) RNA-seq read coverage shows aberrant retention of intron 7 of BRAT1 in the proband and in the heterozygous (c.1014A > C) father relative to wild-type controls.
Neither both the variants were reported in public databases nor their functional impact was examined. The deletion variant c.706delC (p.Leu236Cysfs*5) in exon 5 was predicted to lead to a frameshift and to cause loss of the full-length protein (821 amino acids) due to truncation after the first 236 residues. It is predicted to be pathogenic (VarSome, https://varsome.com/ and ClinVar https://www.ncbi.nlm.nih.gov/clinvar/).
The other variant, c.1014A > C (p.Pro338 = ) in exon 7/8, is not conserved among species, and the amino acid pro at the position 338 of BRAT1 protein is not changed as well; however, varSEAK analysis predicts that the mutation may cause the classic splicing site c.1015 + 1 to be skipped (Figure 2B). Thus, RNA-seq was exerted to identify potential splicing defects associated with the variants in the NPCA case.
BRAT1 expression levels in RNA-seq showed a trend toward lower expression in heterozygous parents and the compound heterozygous proband and confirmed significantly decreased BRAT1 transcript levels in the presence of the variant (Figure 2C), consistent with NMD of the mutant transcript. Meanwhile, BRAT1 expression was higher in the synonymous variants’ father than in the frameshift variants’ mother, although the difference was not statistically significant (Figure 2C). Furthermore, RNA-seq analyses identified significantly increased retention of intron 7 of BRAT1 in the proband and the heterozygous father relative to the heterozygous mother and wild-type control samples (Figure 2D). It was presumed to be the effect of NM_152743.3 c.1014A > C (p.Pro338 = ) mutation. Altogether, these results suggest that both the variants participate in the pathogenesis of NPCA.
The mother of the proband underwent prenatal diagnosis at 12 weeks of gestation during her third pregnancy using chorionic villus sampling. Fortunately, Sanger sequencing showed that the fetus inherited neither of the above sites (Figure 1A). Prenatal and neonatal courses of the fetus were uncomplicated. A male neonate 2,560 g in weight and 51 cm high was delivered at the term, without microcephaly (HC 348 mm). In the following years, his head circumference has grown consistently within the 95th centile. Other growth parameters, strength, reflexes, and sensation were normal until the last visit at 18 months.
A total of 30 patients with clinical manifestations ranging from RMFSL to NPCA had been identified as homozygous or compound heterozygous variants in BRAT1 (Table 1, Figure 1F). In BRAT1-related disorders, cerebellar hypoplasia seemed not congenital (Wolf et al., 2015; Srivastava et al., 2016; Celik et al., 2017). Clinical classifications depend on the rate of progression of atrophy after birth determined. Nearly 75% of them present with severe RMFSL, with almost all accompanied by multifocal or refractory epilepsy or even intrauterine jerks (Celik et al., 2017). Epilepsy occurs in 41.6% of NEDCAS/NPCA patients, mostly before 3 years of age. It had been reported that a few NPCA cases, which mapped to the SCA15 locus on chromosome 3pter, might develop spasticity or focal dystonia with increasing age (Dudding et al., 2004). However, in NPCA caused by BRAT1, including the case here, later-onset spasticity is extremely rare on long-term follow-up.
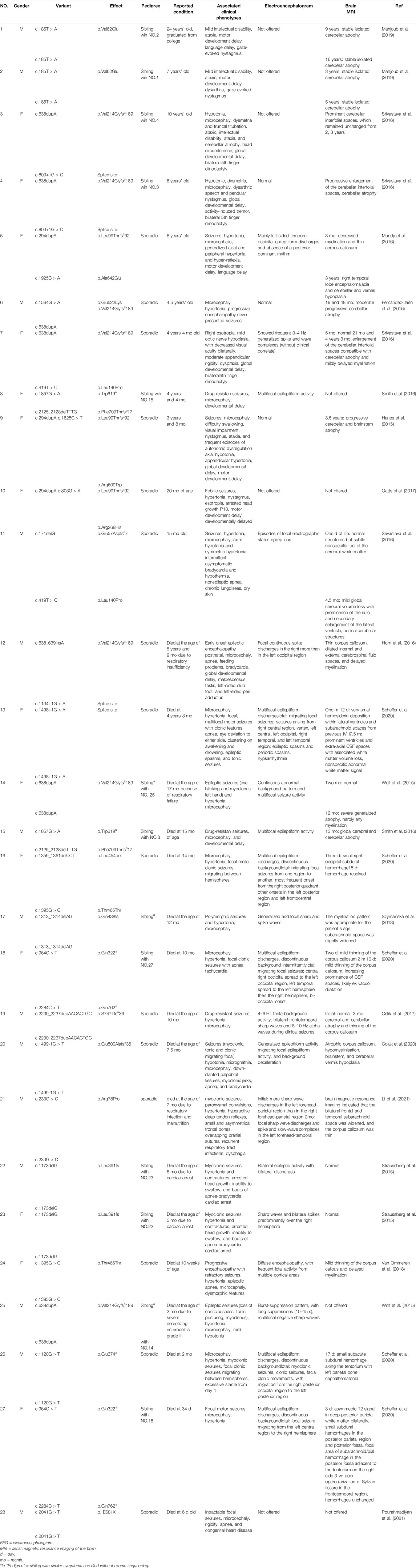
TABLE 1. Clinical features of individuals with previously described BRAT1 variants besides the present study.
Divergence is much huger for the clinical–genetic correlation. It has been mentioned that the phenotypic spectrum of BRAT1-associated disorders is associated with the domain, localization, type, and zygosity of the identified variant (13) (Dudding et al., 2004). According to Table 1, the variant type rather than the variant domain is closely related to the phenotype severity. For example, homozygous variants R78P and p. V62E, both located on the apoptosis-related N-terminal CIDE (cell death-inducing DFF-45-like effector) (Lugovskoy et al., 1999; Choi et al., 2017), caused typical RMFSL (Li et al., 2021) and mild NPCA (Lugovskoy et al., 1999), respectively. Complex mechanisms such as the mutation affecting one or more as unidentified activities of this protein (Kurosaki and Maquat, 2016) or associated with BRAT1-related pathways might be involved. It may modulate the severity rather than simply disrupt the mitochondrial function and the ATM kinase activity. As to the variant type, individuals with BRAT1 biallelic null variants usually lead to severe symptoms and are mostly fatal at the early stage. However, in biallelic BRAT1 missense variants, phenotypic variability is huge, ranging from RMFSL to NPCA. Interestingly, we noticed that in individuals with biallelic BRAT1 gene null variants, females always exhibited a milder phenotype than males (Mundy et al., 2016; Srivastava et al., 2016), even for siblings who carried the same variants. The special phenotypic divergence cannot be explained simply by variable penetrance or genetic backdrop heterogeneity. It is hypothesized that other sub-equivalent genes located on the X chromosome have some effects of the “female protective model” in neurodevelopmental disorders (Jacquemont et al., 2014). The mechanism is yet to be further confirmed.
The variant p. Leu236Cysfs*5 resulted truncation after the first 236 residues, and half of the protein was most likely to lose the function. The trans variant c.1014A > C translates to p. Pro338Pro, which has not been functionally silent. It generated an aberrant transcript with intron 7 retention by affecting the splicing accuracy. Intron retaining can reduce gene expression at the post-transcriptional level and thereby impose an additional level of gene regulation, such as the degradation of mRNA transcripts via nonsense-mediated decay (NMD) and the regulation of nuclear mRNA export (Low et al., 2015; Schmitz et al., 2017). Wild-type BRAT1 was diffusely expressed in the cytoplasm and the nucleus (Li et al., 2021). As-retained intron transcripts accumulate in the nucleus (Boutz et al., 2015), which might also reduce the amount of cytoplasmic BRAT1 available for the downstream events.
The poor efficiency of mRNA transcription, rather than completely abolished BRAT1 protein in a truncating genetic backdrop, made the BRAT1 protein synthesis in the early life insufficient to maintain the good development of the cerebellum, which may underlie the etiology of the mild, nonprogress phenotypic form of BRAT1-related neurodevelopmental disorders. c.1014A > C is the first pathogenic synonymous variant identified in the BRAT1 gene, which is associated with autosomal recessive NPCA. Before this, there was a synonymous substitution at c.1395G > C (p.Thr465 = ), which had been reported in the severe phenotypic form of RMFSL (24). Although the mechanism has not been clarified, it conjectured that there exist multiple exon skipping, mRNA degradation, and complete deletion of BRAT1 protein. Novel synonymous variants in BRAT1 should never be ignored as silent sound in diagnosis, which occasionally shed light on the underlying pathogenesis of the disease.
Our results not only broaden the mutation/phenotype spectrum of BRAT1 but also contribute to comprehend possible pathogenic mechanisms of BRAT1. It is beneficial to specific genetic counseling and timely perinatal management.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Written informed consent was obtained from the individual(s)' and minor(s)' legal guardian/next to kin for the publication of any potentially identifiable images or data included in this article.
All authors have materially participated in the study and manuscript preparation. YQ carried out all the data analyses, participated in the design of the work, and wrote the draft; XJ, HD, and YZ collected all clinical data; LL participated in molecular genetic test work; AY designed the work and revised the manuscript. All authors critically reviewed the manuscript and provided final approval for submission. All authors agree to be accountable for all aspects of the work, ensuring the accuracy and integrity of the publication.
The study was financially supported by the Guangdong Medical Research Foundation (A2020257) and the Science and Technology Planning Project of Guangdong Province (2021A1414020008). This funding supported us in completing the experiments, sequencing, data analysis, and publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
We would like to thank all the members in the family of the patient for their interest and cooperation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.821587/full#supplementary-material
Boutz, P. L., Bhutkar, A., and Sharp, P. A. (2015). Detained Introns Are a Novel, Widespread Class of post-transcriptionally Spliced Introns. Genes Dev. 29 (1), 63–80. doi:10.1101/gad.247361.114
Celik, Y., Okuyaz, C., Arslankoylu, A. E., and Ceylaner, S. (2017). Lethal Neonatal Rigidity and Multifocal Seizure Syndrome with a New Mutation in BRAT1. Epilepsy Behav. Case Rep. 8, 31–32. doi:10.1016/j.ebcr.2017.05.003
Choi, J. Y., Qiao, Q., Hong, S.-H., Kim, C. M., Jeong, J.-H., Kim, Y.-G., et al. (2017). CIDE Domains Form Functionally Important Higher-Order Assemblies for DNA Fragmentation. Proc. Natl. Acad. Sci. USA 114 (28), 7361–7366. doi:10.1073/pnas.1705949114
Colak, F. K., Guleray, N., Azapagasi, E., Yazıcı, M. U., Aksoy, E., and Ceylan, N. (2020). An Intronic Variant in BRAT1 Creates a Cryptic Splice Site, Causing Epileptic Encephalopathy without Prominent Rigidity. Acta Neurol. Belg. 120 (6), 1425–1432. doi:10.1007/s13760-020-01513-0
Dudding, T. E., Friend, K., Schofield, P. W., Lee, S., Wilkinson, I. A., and Richards, R. I. (2004). Autosomal Dominant Congenital Non-progressive Ataxia Overlaps with the SCA15 Locus. Neurology 63 (12), 2288–2292. doi:10.1212/01.wnl.0000147299.80872.d1
Fernández-Jaén, A., Álvarez, S., Young So, E., Ouchi, T., Jiménez de la Peña, M., Duat, A., et al. (2016). Mutations in BRAT1 Cause Autosomal Recessive Progressive Encephalopathy: Report of a Spanish Patient. Eur. J. Paediatric Neurol. 20 (3), 421–425. doi:10.1016/j.ejpn.2016.02.009
Hanes, I., Kozenko, M., and Callen, D. J. A. (2015). Lethal Neonatal Rigidity and Multifocal Seizure Syndrome-A Misnamed Disorder? Pediatr. Neurol. 53 (6), 535–540. doi:10.1016/j.pediatrneurol.2015.09.002
Horn, D., Weschke, B., Knierim, E., Fischer-Zirnsak, B., Stenzel, W., Schuelke, M., et al. (2016). BRAT1mutations Are Associated with Infantile Epileptic Encephalopathy, Mitochondrial Dysfunction, and Survival into Childhood. Am. J. Med. Genet. 170 (9), 2274–2281. doi:10.1002/ajmg.a.37798
Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., et al. (2014). A Higher Mutational burden in Females Supports a "female Protective Model" in Neurodevelopmental Disorders. Am. J. Hum. Genet. 94 (3), 415–425. doi:10.1016/j.ajhg.2014.02.001
Jones, T. M., and Montero, F. J. (2021). “Chorionic Villus Sampling,” in StatPearls [Internet] (Treasure Island (FL): StatPearls Publishing).
Kurosaki, T., and Maquat, L. E. (2016). Nonsense-mediated mRNA Decay in Humans at a Glance. J. Cell Sci 129 (3), 461–467. doi:10.1242/jcs.181008
Li, W., Wu, S., Xu, H., Zhao, X., Pan, Y., Huang, H., et al. (2021). Novel Variant in BRAT1 with the Lethal Neonatal Rigidity and Multifocal Seizure Syndrome. Pediatr. Res. 2021, 1. doi:10.1038/s41390-021-01468-9
Low, L.-H., Chow, Y.-L., Li, Y., Goh, C.-P., Putz, U., Silke, J., et al. (2015). Nedd4 Family Interacting Protein 1 (Ndfip1) Is Required for Ubiquitination and Nuclear Trafficking of BRCA1-Associated ATM Activator 1 (BRAT1) during the DNA Damage Response. J. Biol. Chem. 290 (11), 7141–7150. doi:10.1074/jbc.M114.613687
Lugovskoy, A. A., Zhou, P., Chou, J. J., McCarty, J. S., Li, P., and Wagner, G. (1999). Solution Structure of the CIDE-N Domain of CIDE-B and a Model for CIDE-N/CIDE-N Interactions in the DNA Fragmentation Pathway of Apoptosis. Cell 99 (7), 747–755. doi:10.1016/s0092-8674(00)81672-4
Mahjoub, A., Cihlarova, Z., Tétreault, M., MacNeil, L., Sondheimer, N., Caldecott, K. W., et al. (2019). Homozygous Pathogenic Variant in BRAT1 Associated with Nonprogressive Cerebellar Ataxia. Neurol. Genet. 5 (5), e359. doi:10.1212/NXG.0000000000000359
Mundy, S. A., Krock, B. L., Mao, R., and Shen, J. J. (2016). BRAT1-related Disease-Identification of a Patient without Early Lethality. Am. J. Med. Genet. 170 (3), 699–702. doi:10.1002/ajmg.a.37434
Oatts, J. T., Duncan, J. L., Hoyt, C. S., Slavotinek, A. M., and Moore, A. T. (2017). Inner Retinal Dystrophy in a Patient with Biallelic Sequence Variants in BRAT1. Ophthalmic Genet. 38 (6), 559–561. doi:10.1080/13816810.2017.1290118
Ouchi, M., and Ouchi, T. (2010). Regulation of ATM/DNA-PKcs Phosphorylation by BRCA1-Associated BAAT1. Genes & Cancer 1 (12), 1211–1214. doi:10.1177/1947601911404222
Pourahmadiyan, A., Heidari, M., Shojaaldini Ardakani, H., Noorian, S., and Savad, S. (2021). A Novel Pathogenic Variant of BRAT1 Gene Causes Rigidity and Multifocal Seizure Syndrome, Lethal Neonatal. Int. J. Neurosci. 131 (9), 875–878. doi:10.1080/00207454.2020.1759589
Puffenberger, E. G., Jinks, R. N., Sougnez, C., Cibulskis, K., Willert, R. A., Achilly, N. P., et al. (2012). Genetic Mapping and Exome Sequencing Identify Variants Associated with Five Novel Diseases. PLoS One 7 (1), e28936. doi:10.1371/journal.pone.0028936
Saunders, C. J., Miller, N. A., Soden, S. E., Dinwiddie, D. L., Noll, A., Alnadi, N. A., et al. (2012). Rapid Whole-Genome Sequencing for Genetic Disease Diagnosis in Neonatal Intensive Care Units. Sci. Transl. Med. 4 (154), 154ra135. doi:10.1126/scitranslmed.3004041
Scheffer, I. E., Boysen, K. E., Schneider, A. L., Myers, C. T., Mehaffey, M. G., Rochtus, A. M., et al. (2020). BRAT 1 Encephalopathy: a Recessive Cause of Epilepsy of Infancy with Migrating Focal Seizures. Dev. Med. Child. Neurol. 62 (9), 1096–1099. doi:10.1111/dmcn.14428
Schmitz, U., Pinello, N., Jia, F., Alasmari, S., Ritchie, W., Keightley, M.-C., et al. (2017). Intron Retention Enhances Gene Regulatory Complexity in Vertebrates. Genome Biol. 18 (1), 216. doi:10.1186/s13059-017-1339-3
Smith, N. J., Lipsett, J., Dibbens, L. M., and Heron, S. E. (2016). BRAT1-associated Neurodegeneration: Intra-familial Phenotypic Differences in Siblings. Am. J. Med. Genet. 170 (11), 3033–3038. doi:10.1002/ajmg.a.37853
So, E. Y., and Ouchi, T. (2014). BRAT1 Deficiency Causes Increased Glucose Metabolism and Mitochondrial Malfunction. BMC Cancer 14, 548. doi:10.1186/1471-2407-14-548
Srivastava, S., Olson, H. E., Cohen, J. S., Gubbels, C. S., Lincoln, S., Davis, B. T., et al. (2016). BRAT1mutations Present with a Spectrum of Clinical Severity. Am. J. Med. Genet. 170 (9), 2265–2273. doi:10.1002/ajmg.a.37783
Straussberg, R., Ganelin-Cohen, E., Goldberg-Stern, H., Tzur, S., Behar, D. M., Smirin-Yosef, P., et al. (2015). Lethal Neonatal Rigidity and Multifocal Seizure Syndrome - Report of Another Family with a BRAT1 Mutation. Eur. J. Paediatric Neurol. 19 (2), 240–242. doi:10.1016/j.ejpn.2014.11.004
Szymańska, K., Laure-Kamionowska, M., Szczałuba, K., Koppolu, A., Furmanek, M., Kuśmierska, K., et al. (2018). Clinico-pathological Correlation in Case of BRAT1 Mutation. fn 56 (4), 362–371. doi:10.5114/fn.2018.80870
Van Ommeren, R. H., Gao, A. F., Blaser, S. I., Chitayat, D. A., and Hazrati, L.-N. (2018). BRAT1Mutation: The First Reported Case of Chinese Origin and Review of the Literature. J. Neuropathol. Exp. Neurol. 77 (12), 1071–1078. doi:10.1093/jnen/nly093
Vetro, A., Pisano, T., Chiaro, S., Procopio, E., Guerra, A., Parrini, E., et al. (2020). Early Infantile Epileptic-Dyskinetic Encephalopathy Due to Biallelic PIGP Mutations. Neurol. Genet. 6 (1), e387. doi:10.1212/NXG.0000000000000387
Wolf, N., van Weissenbruch, M., Stam, C., Weiss, J., Waisfisz, Q., Kevelam, S., et al. (2015). Early-Onset Severe Encephalopathy with Epilepsy: The BRAT1 Gene Should Be Added to the List of Causes. Neuropediatrics 46 (6), 392–400. doi:10.1055/s-0035-1564791
Keywords: BRAT1, nonprogressive cerebellar ataxia syndrome, synonymous variant, intron retention, prenatal diagnosis
Citation: Qi Y, Ji X, Ding H, Liu L, Zhang Y and Yin A (2022) Novel Biallelic Variant in the BRAT1 Gene Caused Nonprogressive Cerebellar Ataxia Syndrome. Front. Genet. 13:821587. doi: 10.3389/fgene.2022.821587
Received: 24 November 2021; Accepted: 07 January 2022;
Published: 10 March 2022.
Edited by:
Maria Elisabetta Baldassarre, University of Bari Aldo Moro, ItalyReviewed by:
Saud Alsahli, King Faisal Specialist Hospital and Research Centre, Saudi ArabiaCopyright © 2022 Qi, Ji, Ding, Liu, Zhang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aihua Yin, eWluYWl3YUB2aXAuMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.