
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 19 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.818241
This article is part of the Research Topic Genetic and Environmental Factors in the Occurrence of Neonatal Disorders - Volume I View all 10 articles
 Tong Yi1,2,3
Tong Yi1,2,3 Hairui Sun1,4Yuwei Fu5
Hairui Sun1,4Yuwei Fu5 Xiaoyan Hao1Lin Sun1Ye Zhang1Jiancheng Han1Xiaoyan Gu1
Xiaoyan Hao1Lin Sun1Ye Zhang1Jiancheng Han1Xiaoyan Gu1 Xiaowei Liu1Yong Guo1
Xiaowei Liu1Yong Guo1 Xin Wang1
Xin Wang1 Xiaoxue Zhou1
Xiaoxue Zhou1 Siyao Zhang1Qi Yang1
Siyao Zhang1Qi Yang1 Jiaqi Fan1
Jiaqi Fan1 Yihua He1*
Yihua He1*Objectives: Some genetic causes of heterotaxy have been identified in a small number of heterotaxy familial cases or animal models. However, knowledge on the genetic causes of heterotaxy in the fetal population remains scarce. Here, we aimed to investigate the clinical characteristics and genetic spectrum of a fetal cohort with heterotaxy.
Methods: We retrospectively investigated all fetuses with a prenatal diagnosis of heterotaxy at a single center between October 2015 and November 2020. These cases were studied using the genetic testing data acquired from a combination of copy number variation sequencing (CNV-seq) and whole-exome sequencing (WES), and their clinical phenotypes were also reviewed.
Result: A total of 72 fetuses diagnosed with heterotaxy and complete clinical and genetic results were enrolled in our research. Of the 72 fetuses, 18 (25%) and 54 (75%) had left and right isomerism, respectively. Consistent with the results of a previous study, intracardiac anomalies were more severe in patients with right atrial isomerism than in those with left atrial isomerism (LAI) and mainly manifested as atrial situs inversus, bilateral right atrial appendages, abnormal pulmonary venous connection, single ventricles or single atria, and pulmonary stenosis or atresia. In 18 fetuses diagnosed with LAI, the main intracardiac anomalies were bilateral left atrial appendages. Of the 72 fetuses that underwent CNV-seq and WES, 11 (15.3%) had positive genetic results, eight had definitive pathogenic variants, and three had likely pathogenic variants. The diagnostic genetic variant rate identified using WES was 11.1% (8/72), in which primary ciliary dyskinesia (PCD)-associated gene mutations (CCDC40, CCDC114, DNAH5, DNAH11, and ARMC4) accounted for the vast majority (n = 5). Other diagnostic genetic variants, such as KMT2D and FOXC1, have been rarely reported in heterotaxy cases, although they have been verified to play roles in congenital heart disease.
Conclusion: Thus, diagnostic genetic variants contributed to a substantial fraction in the etiology of fetal heterotaxy. PCD mutations accounted for approximately 6.9% of heterotaxy cases in our fetal cohort. WES was identified as an effective tool to detect genetic causes prenatally in heterotaxy patients.
Heterotaxy syndrome is a congenital disorder resulting from incorrect establishment of left–right (LR) patterning during embryogenesis. It is a relatively uncommon syndrome, with an incidence rate of approximately 1:7,000–1:5,000 (Lin et al., 2000; Reller et al., 2008). Classic heterotaxy is characterized by abnormally arranged thoracic and visceral organs and a relatively high mortality mainly due to cardiac defects. Owing to the complexity of this disease, there is no consensus regarding the precise nomenclature of heterotaxy and nosological relationships between clinical and phenotypic features of patients with heterotaxy; however, it is generally classified as left or right isomerism. Clinical outcomes are remarkably poorer in patients with right isomerism than in those with left isomerism (Baban et al., 2018). Moreover, despite surgical management, the occurrence of congenital heart defects associated with heterotaxy has a relatively poor survival associated compared to that of patients with similar congenital heart defects but without heterotaxy (Bartz et al., 2006; Kim et al., 2008). Hence, heterotaxy is not only a disease with significant phenotypic heterogeneity but also a disorder with a considerable medical and economic burden associated with it.
The molecular mechanisms of LR patterning have been extensively investigated in animal models, and numerous implicated genes have been identified. However, only a few of them are candidate genes for causing heterotaxy in humans (Ma et al., 2012; Bellchambers and Ware, 2018). In 1997, the zinc finger protein of the cerebellum 3 gene (ZIC3) was first identified as the cause of heterotaxy based on familial X-linked pedigrees. However, mutations in ZIC3 explain only 3–5% of sporadic heterotaxy cases (Bellchambers and Ware, 2018). Similarly, although the Nodal signaling pathway is a conserved and well-established pathway involved in embryonic LR development, point mutations in members of the Nodal pathway are found only in 5–10% of patients with heterotaxy (Dykes, 2014; Grimes and Burdine, 2017; Pelliccia et al., 2017). Currently, known genes account for only approximately 10–20% of sporadic cases (Sutherland and Ware, 2009; Fakhro et al., 2011). As its specific genetic etiology is currently identifiable in only a minority of patients, numerous novel genes and pathways implicated in the pathogeny of this disorder are expected to be discovered in the next years.
During the past decade, copy number variants (CNVs) and single-nucleotide polymorphisms (SNPs) that are associated with heterotaxy have been identified using G-banding and chromosome microarray analysis (Cowan et al., 2016). Moreover, whole-exome sequencing (WES) has been slowly implemented in the clinical setting in recent years as it can detect intragenic variants and lead to a more comprehensive genetic diagnosis (Li et al., 2019). WES enables the rapid analysis of a large number of gene sequences and has permitted the discovery of genes involved in numerous genetic diseases. To date, only a few studies have used WES to uncover the genetic etiology of congenital abnormalities, and most of them have focused on the study of postnatal cases (Lord et al., 2019; Diderich et al., 2021). To the best of our knowledge, no studies have used WES for the prenatal genetic diagnosis of heterotaxy.
In this study, the combination of CNV sequencing (CNV-seq) and WES was used to detect causative genes and mutations associated with heterotaxy in a prenatal cohort. We aimed to determine the characteristics of the genetic variants identified using WES and to evaluate the diagnostic value of WES in patients with heterotaxy. To this end, fetus, mother, and father trios were sequenced simultaneously. The detected variants were then interpreted following the guidelines recommended by the American College of Medical Genetics (ACMG).
We retrospectively investigated the previously diagnosed cases of fetuses with heterotaxy at the Beijing Anzhen Hospital, Capital Medical University, from October 2015 to November 2020. Clinical data, including gestational age at diagnosis, family history, indications for fetal echocardiography, and pregnancy outcomes, were collected. The available postnatal echocardiograms and autopsy studies were reviewed to confirm the diagnosis of heterotaxy. This study was approved by the research ethics board of Anzhen Hospital, and informed consent was obtained from the parents.
Fetal echocardiography was performed using a segmental approach with standardized anatomical planes and pulse-wave and color Doppler imaging. All cases were diagnosed by two independent investigators with extensive experience in the field of fetal cardiology (Carvalho et al., 2013). Heterotaxy was diagnosed when fetuses exhibited visceral situs ambiguity and complex congenital heart disease (CHD) (Primo et al., 2020).
Right atrial isomerism (RAI) was diagnosed in the presence of characteristic cardiac anomalies, including atrioventricular septal defect, conotruncal lesions, anomalous pulmonary venous return, and atrial appendages on both sides of the body that have the appearance of a morphologically right atrial appendage, and with at least one of the following findings: 1) juxtaposition of the abdominal aorta and inferior vena cava (IVC) or 2) visceral heterotaxy, namely, discordant laterality of the stomach, portal sinus, or gallbladder. Left atrial isomerism (LAI) was suspected in the presence of azygos continuation of the interrupted IVC and at least one of the common features of LAI: 1) cardiac defects, 2) heart block, and 3) visceral heterotaxy. The spleen was also assessed to classify the type of atrial isomerism, namely, asplenia in RAI and polysplenia in LAI.
Fetal samples, including dermal biopsies, umbilical cord blood, and parent blood, were collected. All fetus–mother–father trios underwent CNV-seq and WES. Both CNV-seq and WES were performed using a research-based protocol. CNV-seq was performed as described previously (Kearney et al., 2011; Liu et al., 2015). Briefly, RNA-free high-molecular-weight genomic DNA (gDNA) was isolated from the umbilical cord using a QIAGEN DNA Blood Midi/Mini Kit (QIAGEN GmbH, Hilden, Germany) following the manufacturer’s protocol. The quality and concentration of gDNA from the samples were assessed using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States). Approximately 5 million sequencing reads from each sample were mapped to the NCBI human reference genome (hg19/GRCh37) using the Burrows–Wheeler Aligner and allocated to 20 kb sequencing bins with a 5 kb sliding window to achieve a high resolution in identifying CNVs. The expected resolution was 100 kb. The CNV profiles of each chromosome were represented as log2 of the mean sequencing reads of each sequencing bin along the chromosome.
Detected CNVs were evaluated based on a scientific literature review and the following public databases: DECIPHER (https://decipher.sanger.ac.uk/), DGV (http://dgv.tcag.ca/), 1000 Genomes Project (http://www.internationalgenome.org/), OMIM (http://omim.org/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), ClinGen (https://www.clinicalgenome.org/), and ISCA CNV (https://www.iscaconsortium.org). Following the ACMG and Genomics standards and guidelines for the interpretation of CNVs, CNVs were classified into three categories: benign, uncertain clinical significance, and pathogenic (Riggs et al., 2021). In this study, we only report pathogenic CNVs.
gDNA was extracted from the umbilical cord and parental blood using a QIAGEN DNA Blood Midi/Mini Kit. DNA libraries were prepared using an Agilent liquid capture system (Agilent SureSelect Human All Exon V6) according to the manufacturer’s protocol. The size distribution and concentration of the libraries were determined using an Agilent 2100 Bioanalyzer and quantified using real-time polymerase chain reaction. The DNA library was sequenced on an Illumina Hiseq 4000 or Illumina Novaseq for paired-end 150 bp-long reads according to the manufacturer’s protocol. The mean sequencing coverage on target regions of whole-exome sequencing was 103-fold. Raw image files were processed using bcl2fastq (Illumina) for base calling and generating raw data. Low-quality sequencing reads were filtered using a quality score of 20 (Q20). The average read depths were 103X for each case. The reads were aligned with the NCBI human reference genome (hg19/GRCh37) using the Burrows–Wheeler Aligner. The BAM files were subjected to SNP analysis, duplication marking, indel realignment, and recalibration using GATK and Picard.
After variant detection, wANNOVAR (http://wannovar.wglab.org/) was used for annotation. Variant frequencies were determined using dbSNP150 (https://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project, Exome Variant Server (http://evs.gs.washington.edu/EVS), ExAC (http://exac.broadinstitute.org/), and in-house databases. Common SNPs (minor allele frequency > 0.1%) were removed. Nonsynonymous, spliced, frameshift, nonframeshift variants and variants located in splice sites within 10 bp of an exon were prioritized for the study. SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster (http://www.mutationtaster.org), and CADD (http://cadd.gs.washington.edu) were used to predict the pathogenicity of missense variants, whereas Human Splicing Finder (http://www.umd.be/HSF) and MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) were used to evaluate the splicing effects. Moreover, databases such as OMIM, ClinVar, and Human Gene Mutation Database (http://www.hgmd.org) were used to determine variant harmfulness and pathogenicity when appropriate. ACMG variant classification recommendations were utilized for all reported variants (Li et al., 2017). Variants were classified into one of the following five categories: pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign.
Sanger sequencing was performed to confirm all potentially diagnostic genetic variants identified.
Categorical variables are presented as frequencies (percentages) and were compared using the Pearson χ2 test or Fisher’s exact test. Statistical analyses were performed using SPSS version 23 (SPSS Inc., Chicago, IL, United States). Statistical significance was set at p < 0.05.
From October 2015 to November 2021, 135 pregnant women found to have fetuses with heterotaxy were screened for inclusion in our study. Among them, eight chose to continue pregnancy without genetic testing and 127 chose to terminate their pregnancy (of which 49 women chose not to undergo genetic testing or to do it at other medical centers or companies). Hence, 78 parents chose to terminate the pregnancy and underwent CNV-seq and WES sequentially at our center. Six cases were excluded because of incomplete imaging or the lack of evidence of heterotaxy. Thus, 72 pregnancies were eligible for the analysis in our study (Figure 1). Autopsy was refused by the parents for 41 fetuses.
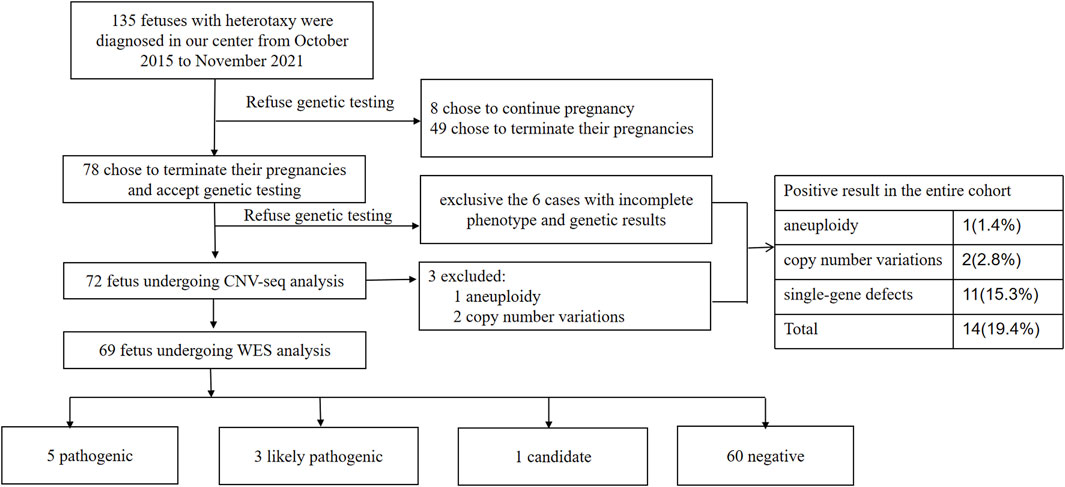
FIGURE 1. Flowchart summarizing detection of genetic variants by CNV-Seq and WES performed in 72 prenatal samples with heterotaxy. CNV, copy number variation; WES, whole-exome sequencing.
All parents were healthy according to their physical examination reports and were nonconsanguineous. The mean maternal age at diagnosis was 28.2 ± 4.11 years, and the median gestational age was 23.74 ± 2.84 weeks. Left isomerism was present in 18 (25%) fetuses, and right isomerism was present in 54 (75%) among all fetuses analyzed (72). The spectrum of cardiovascular abnormalities is shown in Table 1. The main intracardiac anomalies detected among the 54 fetuses with prenatally diagnosed RAI were atrial situs inversus, bilateral right atrial appendages, abnormal pulmonary venous connection, single ventricles or atria, and pulmonary stenosis or atresia. Among the 18 fetuses diagnosed with LAI, the main intracardiac anomaly was the presence of bilateral left atrial appendages. Table 2 summarizes the distribution of visceral abnormalities among patients with left and right isomerism; it also summarizes the visceral abnormalities in patients with RAI. Most RAI patients presented with juxtaposition of the descending aorta and IVC on the same side, aspenia, and bilateral right bronchi. The main extracardiac anomalies in LAI were interrupted IVC, bilateral left bronchi (long), polysplenia, and arrhythmias.
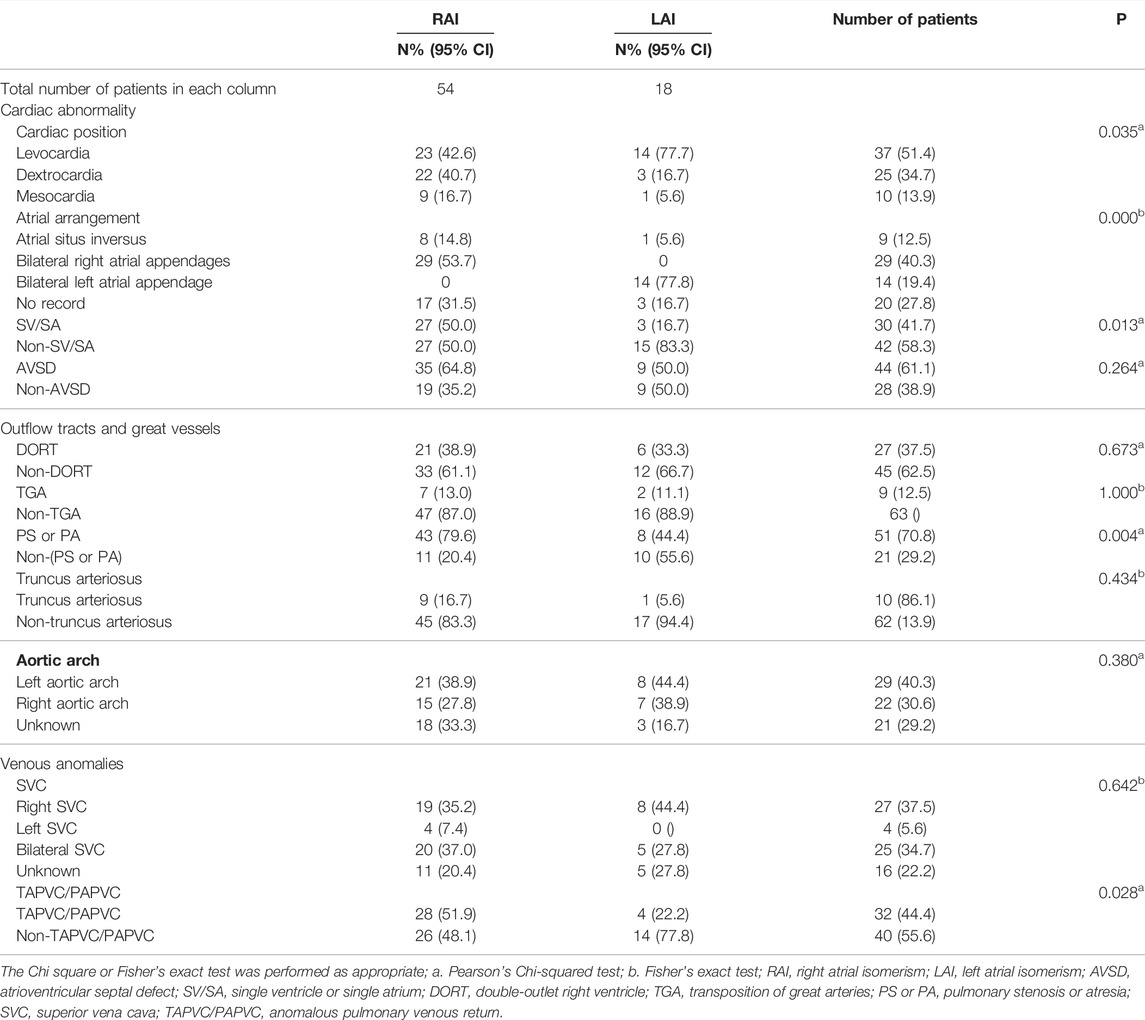
TABLE 1. Spectrum of cardiovascular abnormalities in right and left atrial isomerism patients.
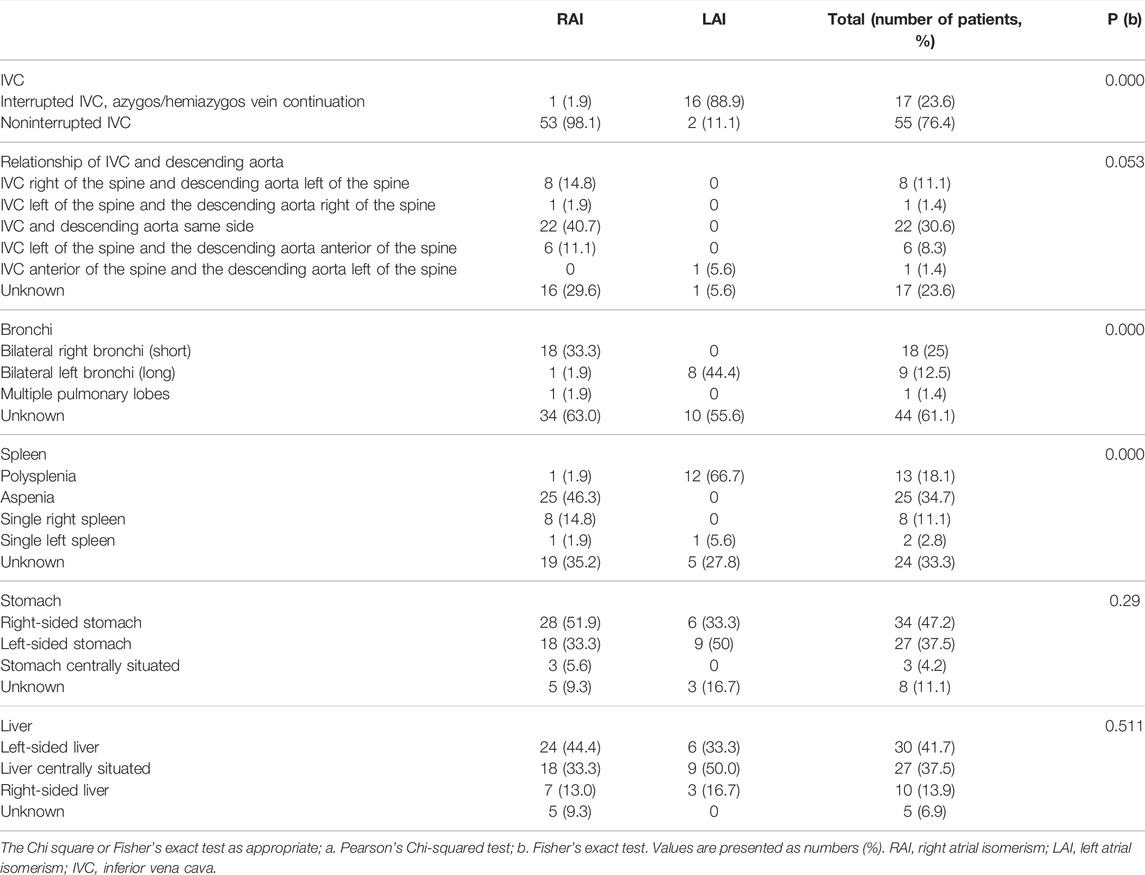
TABLE 2. Spectrum of visceral abnormalities in right and left atrial isomerism.
Among the 72 cases analyzed, eight had definitive pathogenic variants and three had likely pathogenic variants. The positive detection rate was approximately 15.3% (11/72). The frequencies of the genetic variants in patients with different presentations of heterotaxy are shown in Supplementary Table S1. The positive genetic detection rate for isomerism of the right atrial appendages was significantly higher than that for isomerism of the left atrial appendage (p = 0.04).
Among the 72 cases for whom CNV-seq analysis was performed, three (16.9%) had a chromosomal abnormality, including one (7.2%) with aneuploidy (21 chromosome trisomy) and two (9.6%) with a de novo pathogenic CNV (22q11.2 deletion). The three cases with such chromosomal abnormalities were prospectively excluded from the WES analysis.
Of the 69 patients for whom WES analysis was performed, 11.6% (8/69) had a positive result, indicating an increased yield of 11.1% (8/72) and an overall diagnostic yield of 15.3% (11/72) in the entire cohort (Table 1). The genotype–phenotype information of the eight cases is shown in Table 3. The ratio of recessive genotypes was 8.3% (6/72), whereas dominant mutations accounted for 4.2% (3/72) of heterotaxy patients. De novo mutations accounted for 2.8% (2/72) of patients.
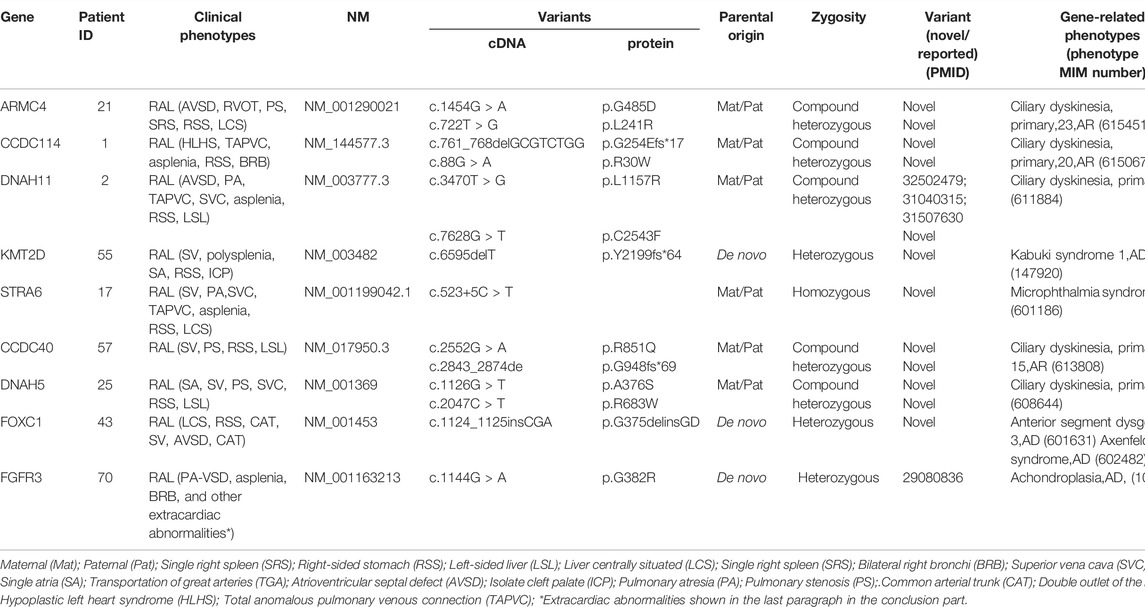
TABLE 3. Genotype–phenotype information of patients diagnosed with pathogenic variants, likely pathogenic variants, or variants of uncertain significance.
In our cohort, the largest contribution of pathogenic diagnostic genetic variant-related phenotypes was primary ciliary dyskinesia (PCD). We found evidence of recessive mutations (compound heterozygous alleles) in CCDC40, CCDC114, DNAH5, DNAH11, and ARMC4. All these genes are known to cause PCD (MIM 242650). The remaining likely pathogenic and candidate variants were identified in genes that have not been previously reported to be associated with heterotaxy, including FOXC1, KMT2D, and FGFR3. These genes have been reported to be related to heart development, except for FGFR3. The patient with the mutation in FGFR3 had extracardiac phenotypes of disorders of the cervical vertebrae (C4–7), lymph node cysts of the neck, abnormal appearance of the right ear, defects of the right radius, defects of the right thumb, single umbilical arteries, abnormal running of the portal vein, asplenia, and a double right main bronchus.
To the best of our knowledge, this is the first cohort-based study assessing the contribution of genetic variants to heterotaxy in a fetal population. Most importantly, we identified genetic abnormalities in 11 (15.3%) fetuses. These abnormalities represented aneuploidies, CNVs, and diagnostic genetic variants in 1.4, 2.8, and 11.1% of the cases, respectively. WES combined with CNV-seq, rather than just CNV-seq or targeted panels, which only provide partial information about the genome, led to a dramatic rise in the detection rate of genetic variants associated with heterotaxy in our prenatal cohort. This finding supports the notion that WES is an effective tool for detecting the genetic causes of heterotaxy as it can detect both chromosomal abnormalities and novel candidate genes.
Importantly, the WES results showed a spectrum of gene variants involved in the occurrence of heterotaxy, highlighting the need for implementing the use of this technique in a clinical setting to facilitate perinatal decision making and management when conventional tests such as karyotype testing or microarray analyses are inconclusive. Nearly half of the diagnostic genetic mutations identified in prenatal heterotaxy patients were associated with PCD, which is a recessive heterogeneous disorder of motile cilia that presents with chronic otosinopulmonary disease and organ lateral defects in 50% of the cases and with heterotaxy in 10% of the cases (Knowles et al., 2013). Moreover, a previous study reported that patients with heterotaxy and PCD had a higher prevalence of respiratory symptoms compared to patients with heterotaxy without PCD (Shapiro et al., 2014). In addition, another study has shown that patients with PCD and mutations in CCDC40 have a more severe disease course with an earlier onset and a higher prevalence of neonatal respiratory distress than PCD patients without such mutations (Davis et al., 2015). Hence, we recommend the use of WES in combination with standard diagnostic testing in patients prenatally diagnosed with heterotaxy to identify the pathogenic genetic variants and predict severity of the disease after birth.
Point mutations in Nodal signaling pathway members, which are known to be associated with laterality defects, were not routinely identified in our cohort. This may be due to the limitations of the screening conditions used in the previous studies. Numerous mutations identified in Nodal by previous studies were classified as benign in our research, according to the ACMG guidelines (Mohapatra et al., 2009). Mutations in ZIC3 are also well-documented to be related to situs abnormalities. In our cohort, we did not find any pathogenic or likely pathogenic variants of ZIC3. This may be because ZIC3 mutations underlie only a minority (3–5%) of sporadic heterotaxy cases (Bellchambers and Ware, 2018). Expanding the size of the cohort used may contribute to test this hypothesis. Finally, in our study, many genes related to PCD encoding components of cilia or the assembly of motile cilia were identified. This result is consistent with previous research showing that ultrastructural dynein arm defects strongly correlate with the development of situs abnormalities (Knowles et al., 2016). Collectively, these data highlight how the use of different research techniques or cohorts reveals differences in the genetic spectrum of heterotaxy. These differences should be considered in the clinical setting when performing molecular diagnostics.
Two genes that had not been previously associated with heterotaxy but have been found to be involved in early cardiac development (KMT2D and FOXC1) were identified in this study. The sequence alteration in coding exon 31 of KMT2D is identified in one family: a heterozygous deletion of one nucleotide, c.6595delT, predicting a frameshift with a premature stop codon at position 2,262 of the protein p.(Y2199fs*64). The variant was not detected in both parents, confirming that the variant is de novo. This variant has not yet been reported in public databases, including the Genome Aggregation Database (gnomAD), the Exome Aggregation Consortium (Exac), 1000 Genomes Project (1KGP), and Exome Sequencing Project (ESP6500). KMT2D is a major cause for Kabuki syndrome (OMIM:147920), in which CHD is predominantly presented. Nearly 31–58% of the patient had CHD, including a single ventricle with a common atrium, VSD, ASD, ToF, coarctation of aorta, and patent ductus arteriosus (Niikawa et al., 1988; Digilio et al., 2001). For this reason, the mutation was considered as a pathogenic mutation. Another sequence alteration in coding exon 6 of FOXC1 is identified in one proband: a heterozygous insertion of three nucleotides, c.11241125insCGA, predicting that the amino acid residue aspartic acid was inserted after position 375, p.G375delinsGD (Table 3). This variant has not yet been reported in public databases, including gnomAD, Exac, 1KGP, and ESP6500. The variant was not detected in both parents, confirming that the variant is de novo. One FOXC1-related phenotype showed in OMIM was Axenfeld–Rieger syndrome, type 3, the features of which included hearing loss, CHD, dental anomalies, developmental delay, and a characteristic facial appearance, central nervous hypoplasia (Maclean et al., 2005). In conclusion, the mutation was considered as a likely pathogenic mutation.
Additionally, we also identified FGFR3 as a candidate gene. FGFR3 is known to cause achondroplasia with rhizomelic shortening of the limbs, characteristic facies, exaggerated lumbar lordosis, a limitation of elbow extension, genu varum, and trident hand (Bellus et al., 1995). In our study, the fetus with a de novo FGFR3 mutation was diagnosed with RA (PA-VSD, right-sided aortic arch accompanied by a mirror branch, bilateral right bronchi, and the absence of the spleen) and achondroplasia (disordered arrangement of cervical vertebrae, missing right radius, abnormal appearance of the right ear, trident right hand, and a higher measurement volume of the lateral ventricle) (Nelson et al., 1988). However, FGFR3 has not been reported to be the genetic cause of heterotaxy to date and has rarely been shown to be related to cardiac abnormalities, except for one case of FGFR3-related craniosynostosis combined with a cleft mitral valve (Agochukwu et al., 2012). Functional experiments have shown that FGFR3 is highly expressed in primary cardiomyocytes (Touchberry et al., 2013) and regulates the intracellular calcium levels and cardiac contractility in cardiac hypertrophy, suggesting that it plays a role in cardiomyocytes or heart development.
Although prenatal diagnosis of heterotaxy has been well studied, the spectrum of underlying gene variants has rarely been reported. Our retrospective cohort study demonstrated the spectrum of cardiac and other associated anomalies in heterotaxy in a prenatal cohort and confirmed previous findings of fetal atrial isomerism. Our results showed a greater incidence of RAI than LAI, which is consistent with other studies in Asian cohorts (Yan et al., 2008). It has been previously reported that patients with RAI have more severe cardiac abnormalities compared with patients with LAI (Buca et al., 2018). We observed that the positive genetic detection rate is 3 times higher in RAI than in LAI, even though the difference is not significant for right and left isomerism (Supplementary Table S1). In the future, it is necessary to evaluate this in larger cohorts. Based on these results, we recommended the use of genetic testing, particularly for fetuses diagnosed with RAI. Additionally, the results of this study indicate that the genetic spectrum of fetal heterotaxy must be evaluated in larger studies.
This study had some limitations. First, the sample size of this study was relatively small. Second, subtle dysmorphic features cannot be determined using fetal ultrasound, and some phenotypes, particularly neurodevelopmental disorders, cannot be determined in the prenatal setting.
Another major limitation of our study is the deviation of our cohort. As a matter of fact, we work in a referral center which is famous for diagnosing fetal cardiac abnomalities, and the patients we see normally have complicate malformations which may be difficult for primary doctors to diagnose. Therefore, the abnormality of the fetus heart we see in our center is more complicated compared to other centers. For this reason, our cohort may not represent the clinical characteristic of whole prenatal heterotaxy patients neither the TOP rate under normal conditions.
If we calculate the termination of pregnancy rate in our cohort, we may be shocked by the extremely high rate. This may be partly explained by the deviation of our cohort we mentioned above, and more importantly, it may be caused by two reasons: first, in China, the decision to choose TOP is considered a private matter of the couple (Lou et al., 2018). Second, high-quality, targeted counseling following prenatal diagnosis of CHD was not available in China in previous years. In China, the TOP following prenatal diagnose was 97.4% in Hunan and 84.8% in Beijing (Yang et al., 2009; Xie et al., 2017; Li et al., 2018). For now, high-quality, targeted counseling following the prenatal diagnosis of CHDs was established and disseminated, and this condition may improve in the future.
Clinical phenotyping and next-generation sequencing of a cohort of fetuses with heterotaxy revealed that 1. diagnostic genetic variants were the main genetic cause of fetal heterotaxy, 2. mutations related to PCD were common in the prenatal heterotaxy cohort, and 3. mutations in genes related to Nodal signaling were rare. In contrast to other studies based on animal models or human case reports, our study was conducted on a cohort of prenatal heterotaxy cases, which provided novel insights into the genetic etiology of heterotaxy syndrome. Our results raise the question of whether the insights obtained from animal models and isolated cases with heterotaxy can be extended to fetal patients, especially when determining the candidate genes to be included in sequencing panels. Our results also demonstrate that WES is a promising method for identifying diagnostic genetic variants and novel candidate genes in heterotaxy patients.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Medical Ethics Committee of Beijing Anzhen Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TY and YH designed the study. YF, XH, XZ, LS, XW, YZ, XG, JH and YG collected the study materials and samples. HS and TY performed sequencing experiments. YF, XH, XZ and XW collected and aggregated the data. TY and HS analyzed and interpreted the data. TY drafted the manuscript. All the authors have contributed to the manuscript and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (8217020250 and 82100322), the Beijing Lab for Cardiovascular Precision Medicine, Beijing, China (PXM 2020_014226_000054), and the Beijing Advanced Innovation Center for Big Data-based Precision Medicine (PXM 2021_014226_000026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.818241/full#supplementary-material
Agochukwu, N. B., Solomon, B. D., Doherty, E. S., and Muenke, M. (2012). Palatal and Oral Manifestations of Muenke Syndrome (FGFR3-Related Craniosynostosis). J. Craniofac. Surg. 23 (3), 664–668. doi:10.1097/scs.0b013e31824db8bb
Baban, A., Cantarutti, N., Adorisio, R., Lombardi, R., Calcagni, G., Piano Mortari, E., et al. (2018). Long-term Survival and Phenotypic Spectrum in Heterotaxy Syndrome: A 25-year Follow-Up Experience. Int. J. Cardiol. 268, 100–105. doi:10.1016/j.ijcard.2018.02.050
Bartz, P. J., Driscoll, D. J., Dearani, J. A., Puga, F. J., Danielson, G. K., O’Leary, P. W., et al. (2006). Early and Late Results of the Modified Fontan Operation for Heterotaxy Syndrome. J. Am. Coll. Cardiol. 48 (11), 2301–2305. doi:10.1016/j.jacc.2006.07.053
Bellchambers, H. M., and Ware, S. M. (2018). ZIC3 in Heterotaxy. Adv. Exp. Med. Biol. 1046, 301–327. doi:10.1007/978-981-10-7311-3_15
Bellus, G. A., Hefferon, T. W., Ortiz de Luna, R. I., Hecht, J. T., Horton, W. A., Machado, M., et al. (1995). Achondroplasia Is Defined by Recurrent G380R Mutations of FGFR3. Am. J. Hum. Genet. 56 (2), 368–373.
Buca, D. I. P., Khalil, A., Rizzo, G., Familiari, A., Di Giovanni, S., Liberati, M., et al. (2018). Outcome of Prenatally Diagnosed Fetal Heterotaxy: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 51 (3), 323–330. doi:10.1002/uog.17546
Carvalho, J. S., Allan, L. D., Chaoui, R., Copel, J. A., DeVore, G. R., Hecher, K., et al. (2013). ISUOG Practice Guidelines (Updated): Sonographic Screening Examination of the Fetal Heart. Ultrasound Obstet. Gynecol. 41 (3), 348–359. doi:10.1002/uog.12403
Cowan, J. R., Tariq, M., Shaw, C., Rao, M., Belmont, J. W., Lalani, S. R., et al. (2016). Copy Number Variation as a Genetic Basis for Heterotaxy and Heterotaxy-Spectrum Congenital Heart Defects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371 (1710). doi:10.1098/rstb.2015.0406
Davis, S. D., Ferkol, T. W., Rosenfeld, M., Lee, H.-S., Dell, S. D., Sagel, S. D., et al. (2015). Clinical Features of Childhood Primary Ciliary Dyskinesia by Genotype and Ultrastructural Phenotype. Am. J. Respir. Crit. Care Med. 191 (3), 316–324. doi:10.1164/rccm.201409-1672oc
Diderich, K. E. M., Romijn, K., Joosten, M., Govaerts, L. C. P., Polak, M., Bruggenwirth, H. T., et al. (2021). The Potential Diagnostic Yield of Whole Exome Sequencing in Pregnancies Complicated by Fetal Ultrasound Anomalies. Acta Obstet. Gynecol. Scand. 100 (6), 1106–1115. doi:10.1111/aogs.14053
Digilio, M. C., Marino, B., Toscano, A., Giannotti, A., and Dallapiccola, B. (2001). Congenital Heart Defects in Kabuki Syndrome. Am. J. Med. Genet. 100 (4), 269–274. doi:10.1002/ajmg.1265
Dykes, I. (2014). Left Right Patterning, Evolution and Cardiac Development. Jcdd 1 (1), 52–72. doi:10.3390/jcdd1010052
Fakhro, K. A., Choi, M., Ware, S. M., Belmont, J. W., Towbin, J. A., Lifton, R. P., et al. (2011). Rare Copy Number Variations in Congenital Heart Disease Patients Identify Unique Genes in Left-Right Patterning. Proc. Natl. Acad. Sci. U.S.A. 108 (7), 2915–2920. doi:10.1073/pnas.1019645108
Grimes, D. T., and Burdine, R. D. (2017). Left-Right Patterning: Breaking Symmetry to Asymmetric Morphogenesis. Trends Genet. 33 (9), 616–628. doi:10.1016/j.tig.2017.06.004
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F., and South, S. T. (2011). American College of Medical Genetics Standards and Guidelines for Interpretation and Reporting of Postnatal Constitutional Copy Number Variants. Genet. Med. 13 (7), 680–685. doi:10.1097/gim.0b013e3182217a3a
Kim, S.-J., Kim, W.-H., Lim, H.-G., and Lee, J.-Y. (2008). Outcome of 200 Patients after an Extracardiac Fontan Procedure. J. Thorac. Cardiovasc. Surg. 136 (1), 108–116. doi:10.1016/j.jtcvs.2007.12.032
Knowles, M. R., Daniels, L. A., Davis, S. D., Zariwala, M. A., and Leigh, M. W. (2013). Primary Ciliary Dyskinesia. Recent Advances in Diagnostics, Genetics, and Characterization of Clinical Disease. Am. J. Respir. Crit. Care Med. 188 (8), 913–922. doi:10.1164/rccm.201301-0059ci
Knowles, M. R., Zariwala, M., and Leigh, M. (2016). Primary Ciliary Dyskinesia. Clin. Chest Med. 37 (3), 449–461. doi:10.1016/j.ccm.2016.04.008
Li, A. H., Hanchard, N. A., Azamian, M., D’Alessandro, L. C. A., Coban-Akdemir, Z., Lopez, K. N., et al. (2019). Genetic Architecture of Laterality Defects Revealed by Whole Exome Sequencing. Eur. J. Hum. Genet. 27 (4), 563–573. doi:10.1038/s41431-018-0307-z
Li, M. M., Datto, M., Duncavage, E. J., Kulkarni, S., Lindeman, N. I., Roy, S., et al. (2017). Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 19 (1), 4–23. doi:10.1016/j.jmoldx.2016.10.002
Li, Y., Rongming, X., and Qiuju, L. (2018). The Analysis of 860 Congenital Heart Disease Cases in Beijing, China. Women Child. Health China 33 (03), 588–591.
Lin, A. E., Ticho, B. S., Houde, K., Westgate, M.-N., and Holmes, L. B. (2000). Heterotaxy: Associated Conditions and Hospital-Based Prevalence in Newborns. Genet. Med. 2 (3), 157–172. doi:10.1097/00125817-200005000-00002
Liu, S., Song, L., Cram, D. S., Xiong, L., Wang, K., Wu, R., et al. (2015). Traditional Karyotypingvscopy Number Variation Sequencing for Detection of Chromosomal Abnormalities Associated with Spontaneous Miscarriage. Ultrasound Obstet. Gynecol. 46 (4), 472–477. doi:10.1002/uog.14849
Lord, J., McMullan, D. J., Eberhardt, R. Y., Rinck, G., Hamilton, S. J., Quinlan-Jones, E., et al. (2019). Prenatal Exome Sequencing Analysis in Fetal Structural Anomalies Detected by Ultrasonography (PAGE): a Cohort Study. Lancet 393 (10173), 747–757. doi:10.1016/S0140-6736(18)31940-8
Lou, S., Carstensen, K., Petersen, O. B., Nielsen, C. P., Hvidman, L., Lanther, M. R., et al. (2018). Termination of Pregnancy Following a Prenatal Diagnosis of Down Syndrome: A Qualitative Study of the Decision-Making Process of Pregnant Couples. Acta Obstet. Gynecol. Scand. 97 (10), 1228–1236. doi:10.1111/aogs.13386
Ma, L., Selamet Tierney, E. S., Lee, T., Lanzano, P., and Chung, W. K. (2012). Mutations in ZIC3 and ACVR2B Are a Common Cause of Heterotaxy and Associated Cardiovascular Anomalies. Cardiol. Young 22 (2), 194–201. doi:10.1017/s1047951111001181
Maclean, K., Smith, J., Heaps, L. S., Chia, N., Williams, R., Peters, G. B., et al. (2005). Axenfeld-Rieger Malformation and Distinctive Facial Features: Clues to a Recognizable 6p25 Microdeletion Syndrome. Am. J. Med. Genet. 132A (4), 381–385. doi:10.1002/ajmg.a.30274
Mohapatra, B., Casey, B., Li, H., Ho-Dawson, T., Smith, L., Fernbach, S. D., et al. (2009). Identification and Functional Characterization of NODAL Rare Variants in Heterotaxy and Isolated Cardiovascular Malformations. Hum. Mol. Genet. 18 (5), 861–871. doi:10.1093/hmg/ddn411
Nelson, F. W., Hecht, J. T., Horton, W. A., Butler, I. J., Goldie, W. D., and Miner, M. (1988). Neurological Basis of Respiratory Complications in Achondroplasia. Ann. Neurol. 24 (1), 89–93. doi:10.1002/ana.410240117
Niikawa, N., Kuroki, Y., Kajii, T., Matsuura, N., Ishikiriyama, S., Tonoki, H., et al. (1988). Kabuki Make-Up (Niikawa-Kuroki) Syndrome: a Study of 62 Patients. Am. J. Med. Genet. 31 (3), 565–589. doi:10.1002/ajmg.1320310312
Pelliccia, J. L., Jindal, G. A., and Burdine, R. D. (2017). Gdf3 Is Required for Robust Nodal Signaling during Germ Layer Formation and Left-Right Patterning. Elife 6. doi:10.7554/eLife.28635
Primo, A. E. L., Brock Zacharias, R. S., de Moraes, A. D., da Silva Filho, L. V. R. F., Trujillo, D. R., Yoshida, R. A. M., et al. (2020). Primary Ciliary Dyskinesia as a Cause of Repeating Atelectasis in the Neonatal Period. Am. J. Case Rep. 21, e921949. doi:10.12659/AJCR.921949
Reller, M. D., Strickland, M. J., Riehle-Colarusso, T., Mahle, W. T., and Correa, A. (2008). Prevalence of Congenital Heart Defects in Metropolitan Atlanta, 1998-2005. J. Pediatr. 153 (6), 807–813. doi:10.1016/j.jpeds.2008.05.059
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2021). Correction: Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 23 (11), 2230. doi:10.1038/s41436-021-01150-9
Shapiro, A. J., Davis, S. D., Ferkol, T., Dell, S. D., Rosenfeld, M., Olivier, K. N., et al. (2014). Laterality Defects Other Than Situs Inversus Totalis in Primary Ciliary Dyskinesia. Chest 146 (5), 1176–1186. doi:10.1378/chest.13-1704
Sutherland, M. J., and Ware, S. M. (2009). Disorders of Left-Right Asymmetry: Heterotaxy and Situs Inversus. Am. J. Med. Genet. 151C (4), 307–317. doi:10.1002/ajmg.c.30228
Touchberry, C. D., Green, T. M., Tchikrizov, V., Mannix, J. E., Mao, T. F., Carney, B. W., et al. (2013). FGF23 Is a Novel Regulator of Intracellular Calcium and Cardiac Contractility in Addition to Cardiac Hypertrophy. Am. J. Physiology-Endocrinology Metab. 304 (8), E863–E873. doi:10.1152/ajpendo.00596.2012
Xie, D., Wang, H., Liu, Z., Fang, J., Yang, T., Zhou, S., et al. (2017). Perinatal Outcomes and Congenital Heart Defect Prognosis in 53313 Non-selected Perinatal Infants. PLoS One 12 (6), e0177229. doi:10.1371/journal.pone.0177229
Yan, Y. L., Tan, K. B., and Yeo, G. S. (2008). Right Atrial Isomerism: Preponderance in Asian Fetuses. Using the Stomach-Distance Ratio as a Possible Diagnostic Tool for Prediction of Right Atrial Isomerism. Ann. Acad. Med. Singap 37 (11), 906–912.
Keywords: congenital heart, heterotaxy syndrome, whole exome, CNV (copy number variant), echocardiagraphy
Citation: Yi T, Sun H, Fu Y, Hao X, Sun L, Zhang Y, Han J, Gu X, Liu X, Guo Y, Wang X, Zhou X, Zhang S, Yang Q, Fan J and He Y (2022) Genetic and Clinical Features of Heterotaxy in a Prenatal Cohort. Front. Genet. 13:818241. doi: 10.3389/fgene.2022.818241
Received: 19 November 2021; Accepted: 09 March 2022;
Published: 19 April 2022.
Edited by:
Wenhao Zhou, Fudan University, ChinaReviewed by:
Zirui Dong, The Chinese University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Yi, Sun, Fu, Hao, Sun, Zhang, Han, Gu, Liu, Guo, Wang, Zhou, Zhang, Yang, Fan and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yihua He, aGV5aWh1YWVjaG9AaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.