 Liang Dong1†
Liang Dong1† Xinning Liu1†Bo Wu2†Chengwei Li1
Xinning Liu1†Bo Wu2†Chengwei Li1 Xiaomin Wei1Gulinuer Wumaier1Xiujuan Zhang1Jing Wang1
Xiaomin Wei1Gulinuer Wumaier1Xiujuan Zhang1Jing Wang1 Jingwen Xia1Yuanyuan Zhang1
Jingwen Xia1Yuanyuan Zhang1 Ruzetuoheti Yiminniyaze1
Ruzetuoheti Yiminniyaze1 Ning Zhu1Jing Li1Daibing Zhou1Youzhi Zhang1Shuanghui Li1Junzhu Lv1
Ning Zhu1Jing Li1Daibing Zhou1Youzhi Zhang1Shuanghui Li1Junzhu Lv1 Shengqing Li1*
Shengqing Li1*- 1Department of Pulmonary and Critical Care Medicine, Huashan Hospital, Fudan University, Shanghai, China
- 2Department of Lung Transplantation, Wuxi People’s Hospital, Wuxi, China
Background: Hypoxic pulmonary hypertension (HPH) is a challenging lung arterial disorder with remarkably high incidence and mortality, and so far patients have failed to benefit from therapeutics clinically available. Max interacting protein 1–0 (Mxi1-0) is one of the functional isoforms of Mxi1. Although it also binds to Max, Mxi1-0, unlike other Mxi1 isoforms, cannot antagonize the oncoprotein c-Myc because of its unique proline rich domain (PRD). While Mxi1-0 was reported to promote cell proliferation via largely uncharacterized mechanisms, it is unknown whether and how it plays a role in the pathogenesis of HPH.
Methods: GEO database was used to screen for genes involved in HPH development, and the candidate players were validated through examination of gene expression in clinical HPH specimens. The effect of candidate gene knockdown or overexpression on cultured pulmonary arterial cells, e.g., pulmonary arterial smooth muscle cells (PASMCs), was then investigated. The signal pathway(s) underlying the regulatory role of the candidate gene in HPH pathogenesis was probed, and the outcome of targeting the aforementioned signaling was evaluated using an HPH rat model.
Results: Mxi1 was significantly upregulated in the PASMCs of HPH patients. As the main effector isoform responding to hypoxia, Mxi1-0 functions in HPH to promote PASMCs proliferation. Mechanistically, Mxi1-0 improved the expression of the proto-oncogene c-Myc via activation of the MEK/ERK pathway. Consistently, both a MEK inhibitor, PD98059, and a c-Myc inhibitor, 10058F4, could counteract Mxi1-0-induced PASMCs proliferation. In addition, targeting the MEK/ERK signaling significantly suppressed the development of HPH in rats.
Conclusion: Mxi1-0 potentiates HPH pathogenesis through MEK/ERK/c-Myc-mediated proliferation of PASMCs, suggesting its applicability in targeted treatment and prognostic assessment of clinical HPH.
Introduction
Hypoxic pulmonary hypertension (HPH) due to lung diseases and/or chronic hypoxia is among the most common groups of pulmonary hypertension with high mortality (Badesch et al., 2010; Galiè et al., 2015). Although the two key pathological processes, vasoconstriction and pulmonary vascular remodeling, have been adequately investigated (Dunham-Snary et al., 2017; Woo et al., 2019; Young et al., 2019), the 3 years survival rate of HPH is significantly worse than of other categories of HPH (Hurdman et al., 2012). In advanced lung disease, regional hypoxic vasoconstriction improves the matching of perfusion and alveolar ventilation, but subsequently leads to the increase in pulmonary circulation pressure (Naeije and Dedobbeleer, 2013; Li et al., 2018), underlining the difficulty of HPH treatment despite the availability of novel drugs (Prins et al., 2017; Cassady and Reed, 2019). Therefore, there is an urgent need to develop novel therapeutics based on the identification of key players in the pathogenesis of HPH. In particular, although hypoxia-induced cell proliferation of vascular endothelial and smooth muscle cells is a hallmark of HPH, the underlying mechanisms remain largely elusive.
Max interacting protein 1–0 (Mxi1-0) is an alternative transcript of Mxi1 involved in the Myc-Max-Mad network (Zervos et al., 1993; Dugast-Darzacq et al., 2004). Mxi1-0 shares exons 2 to 6 with the originally cloned Mxi1 (here-in-after referred to as Mxi1-1). The common basic region helix-loop-helix/leucine zipper (bHLH/LZ) enables Mxi1-0 to bind to Max and regulate cell behavior (Hurlin and Huang, 2006). Compared with Mxi1-1, Mxi1-0 has an alternative first exon (exon 0), encoding a different SIN3 interaction domain (SID) (Engstrom et al., 2004), and is reported to promote proliferation of various types of cells, such as endothelial cells and neuroblastoma cells (Armstrong et al., 2013; Wu et al., 2017). However, it is uncharacterized whether and how Mxi1-0 plays a regulatory role in HPH-related over-proliferation of pulmonary arterial endothelial cells (PAECs) or pulmonary arterial smooth muscle cells (PASMCs).
In this study, we found in clinical specimens that Mxi1-0 was crucially involved in the development of HPH. Mxi1-0 but not Mxi1-1 improved the mitosis of PASMCs. Mechanistically, Mxi1-0 was transcriptionally activated by hypoxia and upregulates the proto-oncogene c-Myc via MEK/ERK signaling.
Materials and Methods
Sample Collection and Ethics Statement
6 chronic lung disease (CLD) samples with or without hypertension were obtained during lung transplantation, and three samples of donor lung tissues were taken from the lungs that were not transplanted. All experiments related to human samples were conducted in accordance with the Declaration of Helsinki and were approved by the ethics committee. All subjects provided written informed consent prior to participation in the study.
Hematoxylin and Eosin Staining
The fixed lungs were sliced in the mid-sagittal plane, embedded in paraffin, and cut into 5 μm thick sections with a microtome. Then, the sections were placed on glass slides, stained with hematoxylin and eosin (HE) staining for morphological analysis, and visualized under an Olympus BX41 microscope (Tokyo, Japan).
Immunohistochemistry and Immunofluorescence Staining
Paraffin-embedded lung tissue sections were deparaffinized in xylene and rehydrated in a graded ethanol series to PBS. Antigen retrieval was performed by pressure cooking in citrate buffer for 10 min. The sections were permeabilized by incubation with 0.3% Triton X-100 and blocked with 5% donkey serum albumin in a humidified chamber for 1h, and were immunostained with primary antibodies to Mxi1 (1:200, ab28740, Abcam Ltd., Cambridge, United Kingdom) or α-smooth muscle actin (α-SMA) (1:200, 48938, CST, Boston, United States). After overnight incubation, sections were washed and incubated with the respective secondary antibodies, donkey anti-rabbit IgG (1:1,000, Jackson Immuno, PA, United States), alexa 594 donkey anti-rabbit IgG (1:1,000, Jackson Immuno, PA, United States), or oralexa 488 donkey anti-mouse IgG (1:1,000, Jackson Immuno, PA, United States) for 1 h. For immunohistochemistry, sections were counterstained with hematoxylin and detected by incubation with the DAB substrate. For immunofluorescence staining, sections were counterstained with nuclear DAPI (1:1,000) and mounted with fluorescent mounting medium.
Cell Culture and Transfection
Primary human PASMCs were purchased from ScienCell, Inc (#3110) and cultured in smooth muscle cell medium (SMCM, ScienCell). Primary human PAECs were purchased from ScienCell, Inc (#3100) and cultured in endothelial cell medium (ECM, ScienCell). Cells were used at passages 4–7. In the cell-growth assay, PASMCs were exposed to normoxia, hypoxia, and hypoxia plus various antagonists as indicated. Cells in the normoxia group were maintained at 37°C in 21% O2, 74% N2, and 5% CO2 (Forma 370, Thermo, United States). Cells in the hypoxia groups were separately cultured at 1% O2, 94% N2, and 5% CO2 (Forma 3131, Thermo, United States). When cells reached 80% confluence, they were transfected with different siRNAs using Lipofectamine 3000 Transfection Reagent (Invitrogen). 6 h after transfection, cells were cultured in serum containing medium for a resting period of 24 h, followed by hypoxia exposure for different times. Mxi1-1 siRNA (target sequence: 5′-CGUCGCACAUGUUCC GGAACG-3′) and Mxi1-0 siRNA (target sequence: 5′-CAGCGAGAACUCGAUGGAGAATT-3′) were synthesized by GenePharma (Shanghai Gene Pharma Co.,). As a control, commercially available non-targeting siRNA (si-Control) was used.
Immunocytofluorescence Assay
Human PASMCs grown on chamber slides were fixed with 4% paraformaldehyde, permeabilized by incubation with 0.3% Triton X-100 and blocked with 5% donkey serum albumin for 2 h. Then, slides were immunostained overnight with primary antibodies to Mxi1 (1:100, ab28740, Abcam Ltd., Cambridge, United Kingdom), followed by 1 h incubation with a secondary antibody, alexa 594 donkey anti-rabbit IgG (1:200, Jackson Immuno, PA, United States). After incubation, slides were counterstained with nuclear DAPI (1:1,000) and mounted with fluorescent mounting medium. Fluorescent images were taken with fluorescence microscope (Olympus, Japan).
Lentiviral Infection
The recombinant lentiviruses expressing Flag-tagged Mxi1-1 and HA-tagged Mxi1-0 were purchased from Genscript (Jiangsu, China). To prepare Mxi1-1 or Mxi1-0 overexpression viral particles, HEK293T cells were co-transfected with each viral vector and the packaging vectors (pMD2.0G and psPAX) using JetPEI purchased from Qbiogene (Montreal, Canada) following the manufacturer’s protocol. The medium was replaced 4 h after transfection, and cells were cultured for a further 36 h. Viral particles were harvested, filtered using a 0.45 µm syringe filter, and combined with 10 μg/ml polybrene (Sigma, MO, United States). PASMCs at 60% confluency were treated with these particles overnight. The culture medium was then replaced with fresh complete growth medium, and cells were cultured for a further 24 h and selected with puromycin (1 μg/ml). The selected cells were used in further experiments.
Quantitative Real-Time PCR
The mRNA level was determined by qRT-PCR. Total RNA was extracted from cells or tissues using Trizol reagent (Invitrogen, CA, United States) according to the manufacturer’s protocol. RNA was subsequently reverse transcribed into cDNA using ReverTra Ace qPCR RT Kit (TOYOBO, Japan). cDNA was amplified and detected using Hieff qPCR SYBR Green Master Mix (Yeasen, Shanghai, China). The PCR primers were as follows: 5′-GGACCTGACTGACTACCTCAT-3′ and 5′-CGTAGCACAGCTTCTCCTTAAT-3′ for β-actin, 5′-GAGGCTGCCGAGTTTTTGG-3′ and 5′-TCGGCATGGACGGGAAT-3′ for Mxi1-1, and 5′-GAGACCGACACACACTCCCATA-3′ and 5′-CGAAAAGCCGGCCTGACT-3′ for Mxi1-0. Fold change of RNA species was analyzed with the formula (2−ΔΔCt), and was normalized to β-actin expression.
Western Blotting
Total lysates of cells or tissues were homogenized in RIPA lysis buffer (Beyotime, Shanghai, China) supplemented with a protease inhibitor, phenylmethyl sulfonyl fluoride (1 mM). Equivalent amounts of protein were separated by SDS- polyacrylamide gels and transferred to 0.22 μM nitrocellulose membranes (Millipore, MA, United States). After blocking, the membranes were probed with one of the following primary antibodies overnight at 4°C: anti-Mxi1 (1:1,000, sc-130627, Santa Cruz, TX, United States), anti-MEK (1:1,000, 4,694, CST, Boston, United States), anti-phospho-MEK (1:1,000, 9,154, CST, Boston, United States), anti-ERK (1:1,000, 4,695, CST, Boston, United States), anti-phospho-ERK (1:1,000, 4,370, CST, Boston, United States) and anti-c-Myc (1:1,000, 13987, CST, Boston, United States). Then, membranes were incubated with secondary antibodies for 1 h at room temperature. Bound antibodies were detected using a Super ECL Detection Reagent (Yeasen, Shanghai, China) and imaged on a Tanon Western blotting detection system (Tanon, Shanghai, China).
Cell Viability Assay
Cells were seeded into 96-well plates at a density of 2000/well and incubated with either vehicles or inhibitors. After exposure to nomorxia or hypoxia, a total of 110 μL of DMEM containing the CCK-8 solution (Beyotime, Shanghai, China) [CCK8:DMEM (v/v) = 1:10] was added to each well, and cells were incubated for 4 h. Finally, cell viability was determined by measuring the absorbance at 450 nm using a multi-well spectrophotometer (Bio-RAD, CA, United States).
Cell Proliferation Assay
Proliferation of cells was detected with a BeyoClickEdU Cell Proliferation Kit with Alexa Fluor 647 (Beyotime, Shanghai, China) following the manufacturer’s protocol. Briefly, cells were seeded into 12-well plates at a density of 10000/well and incubated with siRNA or vehicle. After exposure to nomorxia or hypoxia for 48h, cells were treated with EdU (20 µm) for 3 h and subjected to fixing and permeabilization. Then, cells were exposed in click additive solution for 30 min followed by Hoechst staining for 10min, and finally observed with fluorescence microscope (Olympus, Japan).
Animal Models
Adult male Sprague-Dawley rats weighing 150–200 g were purchased from the Slake Company (Shanghai, China). All protocols and surgical procedures were approved by the Fudan University Veterinary Medicine Animal Care and Use Committee. Animals were randomly divided into five groups (n = 5/group): 1) normoxia (Nor), 2) chronic hypoxia (Hyp), 3) chronic hypoxia and treatment with DMSO (DMSO), 4) chronic hypoxia and treatment with 0.15 mg/kg/tiw PD98059 (PD0.15), and 5) chronic hypoxia and treatment with 0.3 mg/kg/tiw PD98059 (PD0.3). PD98059 was administered via intraperitoneal injection. Rats in the normoxia group were housed at ambient barometric pressure for 28 days (-718 mmHg, PO2 maintained in 150.6 mmHg). Rats in the hypoxia groups were housed in a hypobaric hypoxia chamber depressurized to 380 mmHg (PO2 in 79.6 mmHg) for 8 h/day for 28 days as previously described (Li et al., 2019). All animals were raised under a 12 h:12 h light-dark cycle and were freely supplied food and water. The room temperature was maintained at 25°C, and the bedding was changed once per week.
Echocardiography and Hemodynamic Analysis
After 28 days, rats were fasted overnight and initially anesthetized with isoflurane inhalation. Echocardiography was performed with Visual Sonics Vevo 2100 ultrasoundmachine and 12S rodent probe (GE Healthcare, CT, United States) to determine pulmonary artery acceleration time (PAAT) and tricuspid annulusplain systolic excursion (TAPSE). Data were analyzed with EchoPAC software (GE Healthcare, CT, United States). Then, haemodynamic analysis was performed as previously described (Xia et al., 2018). Briefly, anesthesia was given to rats with 20% ethylurethanm via injecting intraperitoneally (4 ml/kg). After intubation, right ventricular systolic pressure (RVSP) was recorded. To investigate right ventricular hypertrophy (RVH), the right ventricle (RV), the left ventricle plus septum (LV + S) and the body weight (BW) were weighed, and the RV/(LV + S) ratio and RV/BW ratio were determined.
Statistical Analysis
ImageJ software was used to scan the gray level of Western blotting images, and spss24.0 software was used for data statistical analysis. Statistical significance was assessed by comparing mean (±SD) values with Student’s t-test for independent groups. p < 0.05 was considered as statistically significant. The data were plotted with graphpad prism 8.0 software.
Results
Mxi1 Is Involved in the Clinical Development of Hypoxic Pulmonary Hypertension
To identify candidate players in HPH pathogenesis, we first screened for genes differentially expressed between normal and hypoxic PASMCs using the GEO database (Figure 1A, dataset: GSM1857126-1857131). As a result, we obtained 602 genes, of which 200 were up-regulated and 402 were down-regulated in hypoxia samples. GO and KEGG enrichment analyses were performed using DAVID 6.8 database. The GO results indicated that genes were significantly enriched in cell proliferation, aging and response to organic cyclic compound, mainly concentrated in biological processes related to proliferation (Figure 1B), whereas KEGG analysis demonstrated that genes were mainly enriched in growth and differentiation related pathways, such as Ras signaling pathway, PI3K-Akt signaling pathway, MAPK signaling pathway, FOXO signaling pathway, cell cycle and AMPK signaling pathway (Figure 1C).

FIGURE 1. Screening for hypoxic pulmonary hypertension (HPH)-related genes. (A) Heatmap of genes differentially expressed in normal and hypoxic pulmonary arterial smooth muscle cells (PASMCs) in GEO database (dataset: GSM1857126∼1857131). (B,C) GO (B) and KEGG (C) enrichment analyses for differentially expressed genes described in (A).
Among the most significantly changed genes in HPH is Mxi1, which is remarkably upregulated in HPH and established to participate in the regulation of cell division (Armstrong et al., 2013; Wu et al., 2017). We next investigated the expression of Mxi1 in clinical HPH samples. Specimens of nine patients, including 3 with CLD but not hypertension, 3 with HPH and three donors of normal lungs, were collected and examined (Figures 2A,B). Western blotting analysis indicated that Mxi1 was significantly higher in pulmonary of HPH tissues than in those of CLD patients or donors, suggesting a correlation between Mxi1 expression and HPH occurrence (Figure 2C). Immunohistochemical staining of the lung tissues of HPH patients demonstrated specific upregulation of Mxi1 in the medial layer, consisting mainly of PASMCs (Figure 2D). Consistently, immunofluorescent staining showed that Mxi1 co-localized with α-SMA (Figure 2E). Thus, Mxi1 expression is induced in PASMCs during the development of HPH.
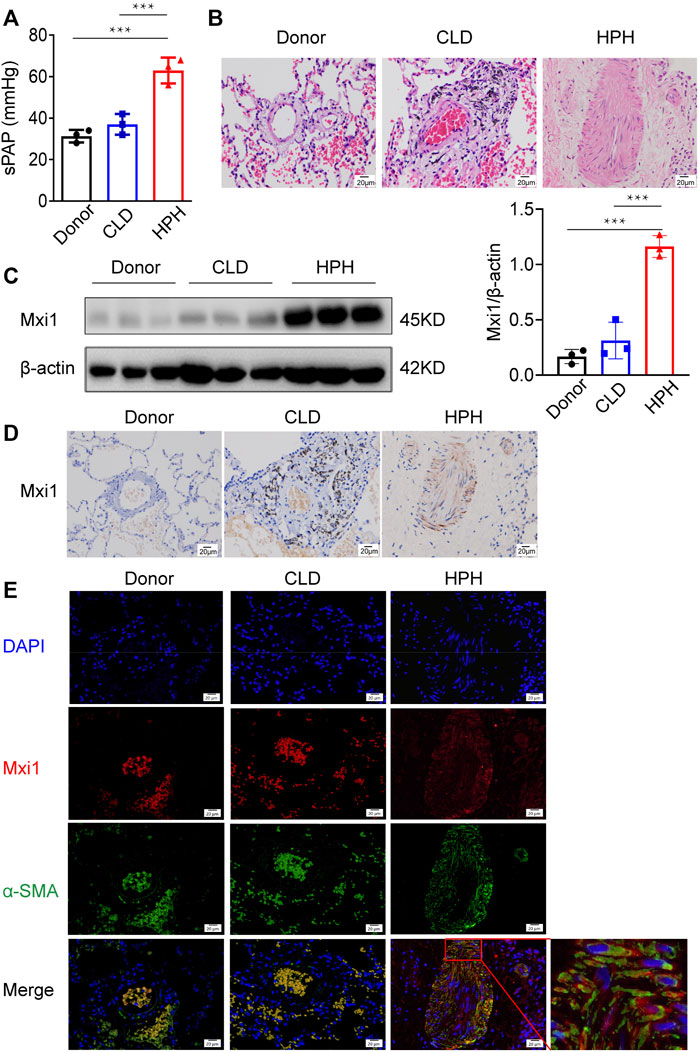
FIGURE 2. Mxi1 is upregulated in PASMCs of HPH patients. Lung samples of three donors, three chronic lung disease (CLD) patients and three hypoxic pulmonary hypertension (HPH) patients were collected and sectioned. (A) Hemodynamic analysis of patients before collection of samples. (B) Hematoxylin and eosin staining of paraffin-fixed lung sections was used for morphological analysis of pulmonary arteries. (C) The expression of Mxi1 was examined via Western blotting analysis, followed by quantification of the blots through densitometry and normalization to β-actin. (D) Representative paraffin lung tissue sections from donors, CLD, and HPH patients were subjected to immunohistochemical staining of Mxi1. (E) Representative sections in all groups were also subjected to immunofluorescence staining of Mxi1 and α-SMA with nuclei counterstained by DAPI (blue), and the image of HPH merge was zoomed. Scale bar, 20 μm. Data are shown as means ± SDs. For statistical significance, ***represents p < 0.001 compared with HPH patients.
Hypoxia Induces Expression of Mxi1-0 but Not Mxi1-1 in Pulmonary Arterial Smooth Muscle Cells
We next explored the expression of Mxi1 isoforms in pulmonary arterial cells when exposed to hypoxia. Western blotting analysis showed that Mxi1 was induced by hypoxia in a time-dependent manner and peaked when exposed to hypoxic conditions for 12 h (Figure 3A). Hypoxia significantly upregulated Mxi1 in PASMCs but not PAECs (Figure 3B). Consistent with the assays using clinical pulmonary specimens (Figure 2C), we detected a 45 kD protein using the pan-Mxi1 antibody (Figure 3B). In addition, to determine the major isoforms induced by hypoxia, we generated vectors for tagged Mxi1-0 and Mxi1-1, and ectopically overexpressed these proteins in PASMCs. Western blotting analyses using antibodies recognizing the tag sequence detected 2 proteins with distinct molecular weights (Figures 3C,D), suggesting that Mxi1-0 was the predominant isoform upregulated by hypoxia (Figure 3A). Unlike Mxi1-1 that was reported to exhibit a predominant nuclear localization (Engstrom et al., 2004; Erichsen et al., 2015), Mxi1-0 resided both in the cytoplasma and the nucleus (Figure 3E). These findings strongly suggest that Mxi1-0 is remarkably upregulated in PASMCs when exposed to hypoxic conditions.
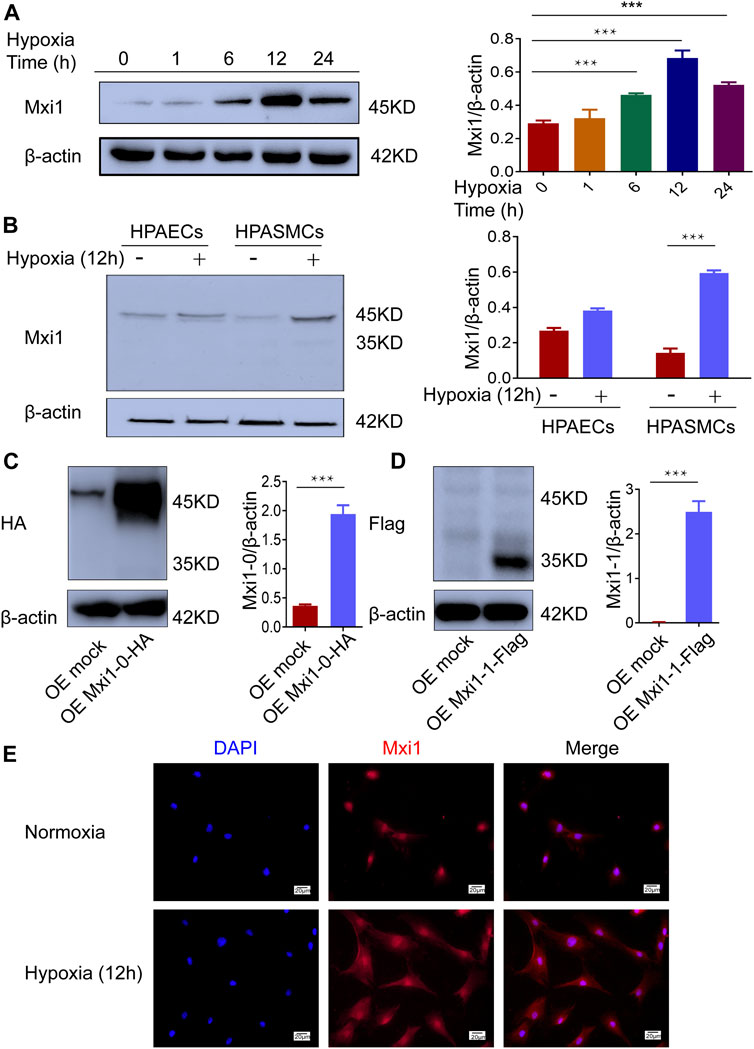
FIGURE 3. Hypoxia induces expression of Mxi1-0 but not Mxi1-1 in PASMCs. (A) PASMCs were exposed to hypoxia (1% O2) for indicated periods of time. Cell lysates were prepared and subjected to Western blotting assay. (B) Pulmonary arterial endothelial cells (PAECs) and PASMCs were exposed to normoxia (21% O2) or hypoxia for 12 h, and were subjected to Western blottinganalyses. (C,D) PASMCs were transfected with constructs for HA-tagged Mxi1-0 (C) or Flag-tagged Mxi1-1 (D), and were subjected to Western blotting analyses. (E) PASMCs were exposed to normoxia or hypoxia for 12 h, and were subjected to immunostaining with a Mxi1 antibody and counterstaining of the nuclei with DAPI. Scale bar, 20 μm. Data from three independent experiments are shown as means ± SDs. For statistical significance, ***represents p < 0.001 compared to normoxia or the mock-transfected group.
Mxi1-0 but Not Mxi1-1 Mediates Hypoxia-Induced Pulmonary Arterial Smooth Muscle Cells Proliferation
Accumulating evidence has indicated that hypoxemia in the pulmonary vessels leads to increased PASMCs proliferation (Shah, 2012). To test whether Mxi1-0 plays a pathogenic role in the induction of this phenotype, we designed siRNAs that could specifically knockdown Mxi1-0 (siMxi1-0) or Mxi1-1 (siMxi1-1) (Figure 4A). While hypoxic treatment significantly promoted the growth of PASMCs, this was ablated by knockdown of Mxi1-0 but not that of Mxi1-1 (Figure4B). Conversely, overexpression of Mxi1-0 but not Mxi1-1 improved the growth rates of PASMCs exposed either to normoxia or hypoxia (Figure 4C). In line with these observations, we found that Mxi1-0 silencing in hypoxia-treated PASMCs reduced the ratios of EdU-positive cells, whereas Mxi1-0 overexpression enhanced these ratios in both normoxia- and hypoxia-exposed cells (Figure 4D), suggesting an essential role of Mxi1-0 in hypoxia-elicited mitosis of PASMCs. Collectively, these data indicate that Mxi1-0 but not Mxi1-1 drives the hyper-proliferative response in hypoxia-exposed PASMCs.
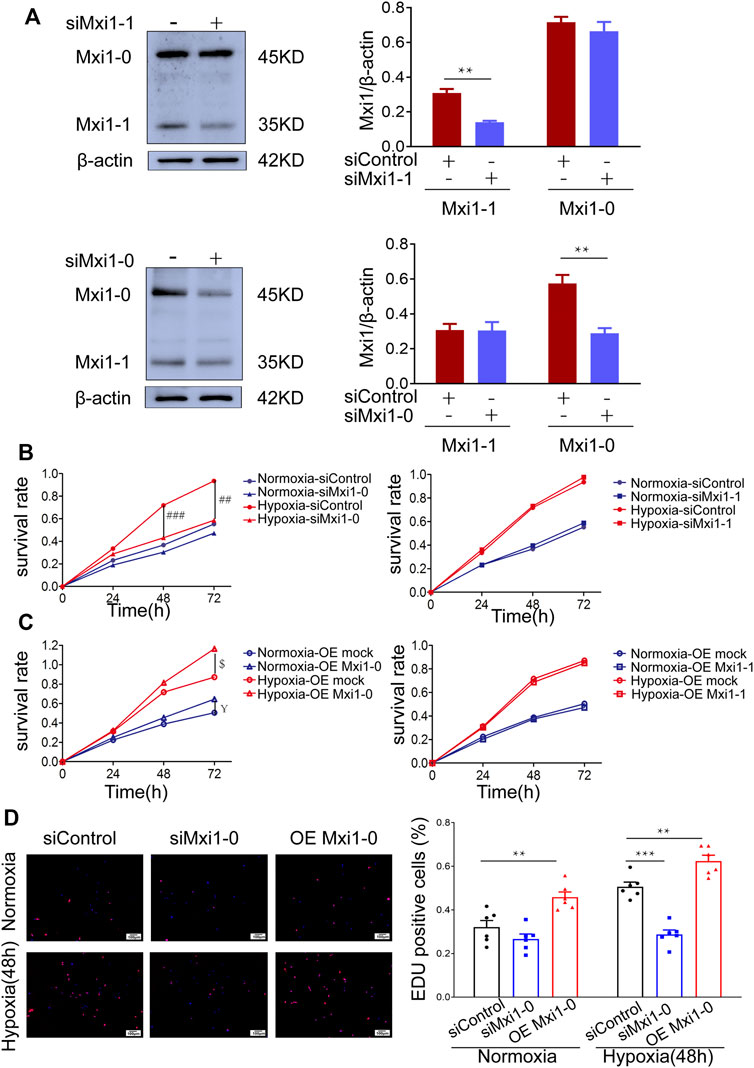
FIGURE 4. Mxi1-0 but not Mxi1-1 promotes PASMCs growth under hypoxic conditions. (A) PASMCs were transfected with siRNAs targeting either Mxi1-0 or Mxi1-1, and were subjected to Western blotting analyses. (B) PASMCs cultured in normoxic or hypoxic conditions were transfected with siRNAs targeting either Mxi1-0 or Mxi1-1, and CCK-8 assays were performed on indicated times. (C) PASMCs cultured in normoxic or hypoxic conditions were infected with recombinant lentiviruses to overexpress Mxi1-0 or Mxi1-1, and CCK-8 assays were performed on indicated times. (D) PASMCs were transfected with Mxi1-0-targeted siRNAs or infected with recombinant lentiviruses to overexpress Mxi1-0, and were cultured in normoxic or hypoxic conditions for 48 h. Cells were then subjected to immunofluorescence staining for EdU, followed by microscopy and calculation of the ratios of EdU-positive cells in three random fields. Scale bar, 100 μm. Data from three independent experiments are shown as means ± SDs. For statistical significance, *represents p < 0.05 compared to siControl, **represents p < 0.01 compared to siControl, ***represents p < 0.001 compared to siControl, ## represents p < 0.01 compared to Hypoxia-siControl, ### represents p < 0.001 compared to Hypoxia-siControl, $represents p < 0.05 compared to Hypoxia-OEmock, ¥represents p < 0.05 compared to Normoxia-OEmock.
Mxi1-0 Potentiates Pulmonary Arterial Smooth Muscle Cells Proliferation Via Upregulation of c-Myc
C-Myc is a proto-oncogene, which plays a key role in the regulation of cell growth and proliferation (Nesbit et al., 1999; Baluapuri et al., 2020). Previous studies have demonstrated that hypoxia leads to the high expression of c-Myc in the occurrence of pulmonary vascular diseases, and that Mxi1 is involved in the regulation of the transcription factor, c-Myc (Zervos et al., 1993; Voelkel et al., 2013). To mechanistically understand how Mxi1-0 might promote proliferation, we assessed the expression of c-Myc in hypoxic PASMCs further subjected to Mxi1-0 silencing or overexpression. As a result, hypoxic exposure increased c-Myc levels, which was further augmented by overexpression and counteracted by knockdown of Mxi1-0 (Figures 5A,B). By constrast, Mxi1-1 overexpression or knockdown failed to affect the expression of c-Myc in PASMCs cultured in normoxic or hypoxic conditions (Figures 5C,D), consistent with previous findings that Mxi1 forms a complex with Max to repress the transactivity rather than directly reduce the expression of c-Myc (Huang et al., 2018). We next probed whether Mxi1-0 promotes PASMCs growth via upregulation of c-Myc in hypoxic conditions. While hypoxia-induced proliferation of PASMCs was significantly impaired by a c-Myc inhibitor, 10058F4, this cannot be rescued by overexpression of Mxi1-0 (Figures 5E,F). These observations indicate that the pro-proliferative effect of Mxi1-0 is mediated by upregulation of c-Myc in hypoxic PASMCs.
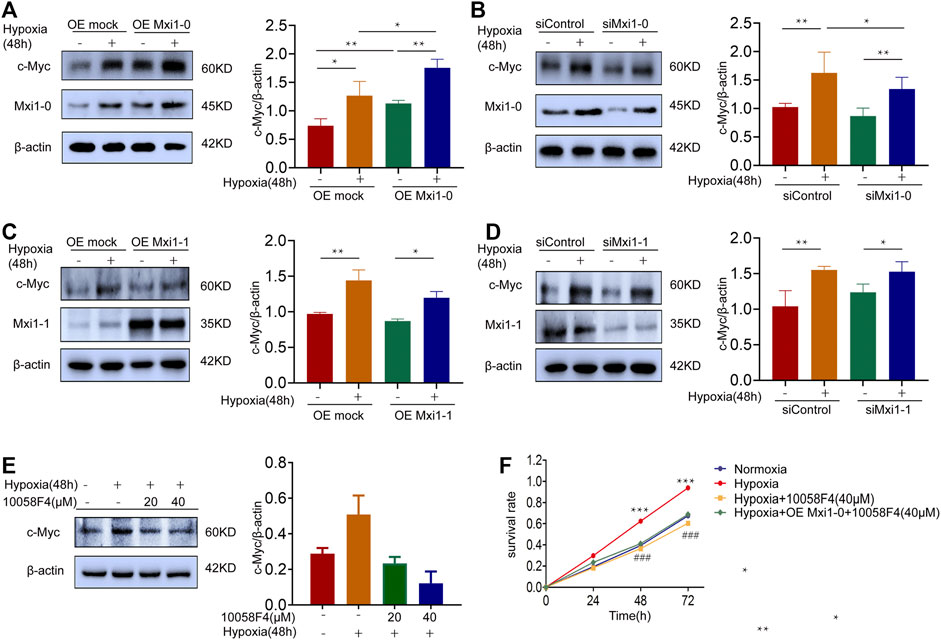
FIGURE 5. Mxi1-0 upregulates c-Myc production in hypoxic PASMCs. (A) PASMCs were infected with recombinant lentiviruses to overexpress Mxi1-0, and were cultured under normoxic or hypoxic conditions for 48 h. Cells were then subjected to Western blotting analysis. (B) PASMCs were transfected with Mxi1-0-targeted siRNAs, and were cultured under normoxic or hypoxic conditions for 48 h. Cells were then subjected to Western blotting analysis. (C) PASMCs were infected with recombinant lentiviruses to overexpress Mxi1-1, and were cultured under normoxic or hypoxic conditions for 48 h. Cells were then subjected to Western blotting analysis. (D) PASMCs were transfected with Mxi1-1-targeted siRNAs, and were cultured under normoxic or hypoxic conditions for 48 h. Cells were then subjected to Western blotting analysis. (E) PASMCs were cultured under normoxic or hypoxic conditions for 48 h and treated with indicated doses of 10058F4. Cells were then subjected to Western blotting analysis. (F) PASMCs were cultured under normoxic or hypoxic conditions, treated with 10058F4, and infected with control or Mxi1-0-overexpressing lentiviruses. Cells were then subjected to CCK-8 assays on indicated times. Data from three independent experiments are shown as means ± SDs. For statistical significance, *represents p < 0.05 compared between two groups, **represents p < 0.01 between two groups, ***represents p < 0.001 compared between Hypoxia and Normoxia, ### represents p < 0.001 compared between Hypoxia and Hypoxia + 10058F4 (40 μM).
Mxi1-0 Upregulates c-Myc Through MEK/ERK Signaling in Hypoxia-Exposed Pulmonary Arterial Smooth Muscle Cells
Previous studies suggested that the pathobiology of HPH was associated with the activation of MEK/ERK signaling, which was documented to promote c-Myc expression (Dang et al., 2006). In addition, bioinformatic analysis in the early stage of this study showed that the function of Mxi1-0 was enriched in Ras/MAPK signaling pathway. Hence, we examine whether Mxi1-0 upregulates c-Myc via MEK/ERK signaling in hypoxia-treated PASMCs. Indeed, hypoxia induced phosphorylation of MEK and ERK, which was inhibited by knockdown of Mxi1-0 but not Mxi1-1 (Figures 6A,B). Hypoxia-induced upregulation of c-Myc was counteracted by PD98059, a classical MEK antagonist, in a dose-dependent manner (Figure 6C). Similar to the inhibition of c-Myc, antagonizing MEK/ERK signaling abrogated hypoxia-induced hypoxia-elicited overgrowth of PASMCs, which was not rescured by overexpression of Mxi1-0 (Figure 6D). These data suggest that Mxi1-0 enhanced the proliferation of PASMCs through MEK/ERK-mediated upregulation of c-Myc.
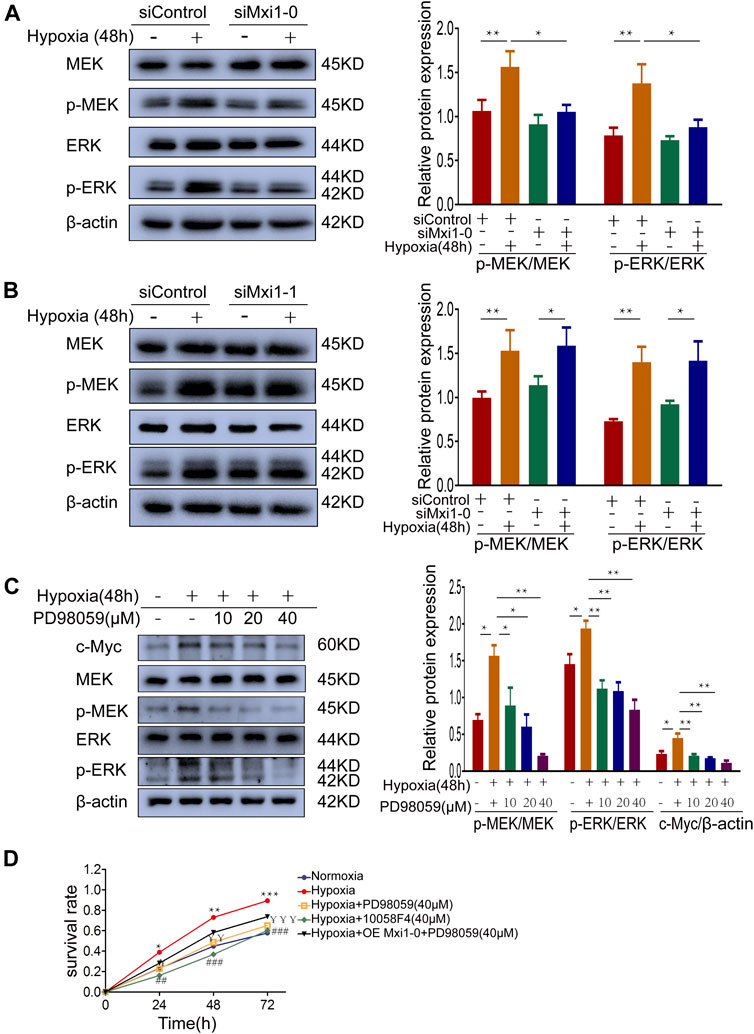
FIGURE 6. Mxi1-0 upregulates c-Myc through MEK/ERK signaling in hypoxic PASMCs. (A,B) PASMCs tranfected with siRNAs targeting Mxi1-0 (A) or Mxi1-1 (B) were cultured under normoxic or hypoxic conditions for 48 h. Western blotting assay was performed with the indicated antibodies. All the phospho-protein levels were measured by densitometry and normalized to that of β-actin. (C) PASMCs were cultured under normoxic or hypoxic conditions for 48 h and treated with different doses of PD98059, and the levels of indicated proteins were determined by Western blotting analysis and quantified by densitometry. (D) PASMCs were cultured under normoxic or hypoxic conditions, treated with PD98059 or 10058F4, and infected with control or Mxi1-0-overexpressing lentiviruses. Cells were then subjected to CCK-8 assays on indicated times. Data from three independent experiments are shown as means ± SDs. For statistical significance, *represents p < 0.05 compared between two groups or to Normoxia (D), **represents p < 0.01 compared between two groups or to Normoxia (D), ¥represents p < 0.05 compared between Hypoxia and Hypoxia + PD98059 (40 μM), ¥¥represents p < 0.01 compared between Hypoxia and Hypoxia + PD98059 (40 μM),¥¥¥represents p < 0.001 compared between Hypoxia and Hypoxia + PD98059 (40 μM), ## represents p < 0.01 compared between Hypoxia and Hypoxia+10058F4 (40 μM), ###represents p < 0.001 compared between Hypoxia and Hypoxia + 10058F4 (40 μM).
We further explore the function of MEK/ERK signaling in HPH pathogenesis using rat models (Figure 7A). Sustained exposure of animals to hypoxia is sufficient to induce pulmonary hypertension as determined by hemodynamic (echocardiography and right heart catheter) and morphological analysis, e.g., decreased PAAT and TAPSE (Figure 7B). Treatment of rats with PD98059 significantly relieved hypertension as revealed by restored PAAT and TAPSE (Figure 7B). In addition, PD98059 treatment dramatically decreased RVSP and ameliorated RVH indices [including RV/BW ratio and RV/(LV + S) ratio], both of which are major characters of HPH (Figure 7C). HE staining revealed that PD98059 significantly improved hypoxia-induced pulmonary artery remodeling by reducing the population of PASMCs (Figure 7D). These data demonstrated that inhibition of MEK/ERK signaling protects rats against chronic hypoxia-induced HPH.
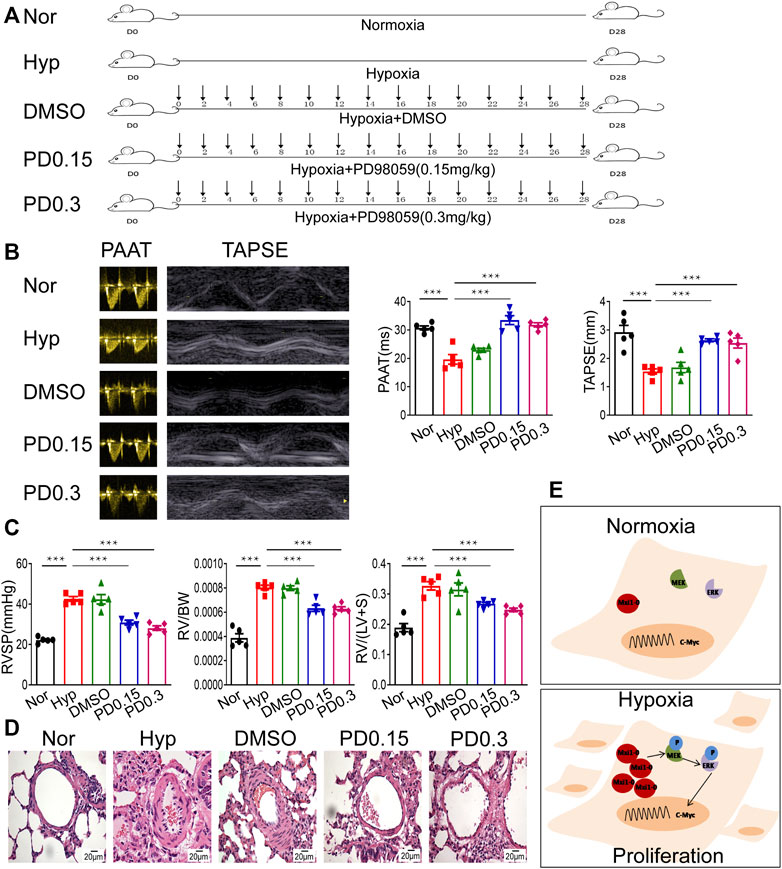
FIGURE 7. Inhibition of MEK/ERK signaling protects rats against HPH. (A) A rat model of hypoxic pulmonary hypertension (HPH) was generated (n = 5 animals per group). Rats exposed to chronic hypoxia were treated with vehicle (DMSO) or 0.15 mg/kg or 0.3 mg/kg PD98059. (B) Rats in all groups were subjected to echocardiography and measurement of PAATand TAPSE. (C) After intubation for rats described in (A), RVSP was recorded, and the right ventricular hypertrophy ratio of RV/BW and RV/LV + S were calculated. (D) Hematoxylin and eosin staining of paraffin-fixed lung sections prepared from rats described in (A) was performed for morphological analysis of the pulmonary arteries. (E) A diagram showing that hypoxia-induced Mxi1-0 promotes PASMCs proliferation via MEK/ERK/c-Myc signaling in the context of HPH. Scale bar, 20 μm. Data are shown as means ± SDs. For statistical significance, ***represents p < 0.001 compared to hypoxia. PAAT, pulmonary artery acceleration time; TAPSE, tricuspid annulusplain systolic excursion; RVSP, right ventricular systolic pressure; RV, right ventricle; BW, body weight; LV + S, left ventricle plus septum.
Discussion
HPH is a serious pulmonary disorder with systemic complications that aggravate its clinical consequences. To date, no effective treatment for this life-threatening disease has advanced, which is at least partially ascribed to the lack of appropriate biomarkers (Maron et al., 2016; Thompson and Lawrie, 2017). In the present study, we found that Mxi1 was overexpressed in the pulmonary arteries of HPH patients. Immunohistochemical staining showed that Mxi1 was expressed abundantly in the medial layer mainly composed of PASMCs, which was further validated by immunofluorescence indicating that Mxi1 overlapped with α-SMA, a specific biomarker of smooth muscle cells. Although Mxi1 has been extensively studied in the context of carcinogenesis and well documented as a tumor suppressor (Zervos et al., 1993; Huang et al., 2018), this is probably the first study to determine that it is also an important player in HPH, reminiscent of multiple shared pathomechanisms between pulmonary hypertension and cancer (Negi et al., 2021).
Protein isoforms are generated from the same gene due to transcription from different promoter, alternative splicing or varied translation initiation sites (Yap and Makeyev, 2016). Although these isoforms are structurally and functionally similar, it is also common that they play distinct roles or are competitively involved in a physiological or pathological process (Li et al., 2016). The Mxi1 gene is located on chromosome 10q24-q25 (Wechsler et al., 1994), and encodes proteins of three different isoforms, Mxi1-0, Mxi1-1 and Mxi1 WR, of which Mxi1 WR is considered to have no biological function due to lack of SID. Although it is highly homologous with Mxi1-1, Mxi1-0 has an additional N-terminal sequence consisting of 92 amino acids (Engstrom et al., 2004). This novel sequence has a proline rich domain (PRD), which is responsible for the cytoplasmic localization of Mxi1-0 (Armstrong et al., 2013). In the study, we established that Mxi1-0, but not Mxi1-1, was highly expressed in PASMCs of HPH patients. Mxi1-0, but not Mxi1-1, was induced by hypoxia and plays an essential role in the proliferation of PASMCs. These observations were in contrast to Mxi1-1, which was reported to suppress cell growth through binding to Max and impairing the transcriptional activity of c-Myc (Wechsler et al., 1997; Manni et al., 2002). The unique PRD in Mxi1-0 recruits specific protein chaperones may explain why Mxi1-0 has different cellular functions from Mxi1-1 (Dugast-Darzacq et al., 2004; Hurlin and Huang, 2006). A recent study showed that the deletion of PRD converts Mxi1-0 into a potent suppressor of c-Myc, which is considered to be a key mediator of HPASMCs proliferation (Dugast-Darzacq et al., 2007; Zhang et al., 2019). Other studies suggested that although Mxi1-0 could bind to Max protein, it failed to inhibit c-Myc-dependent transcription but might promote the transcription of the proto-oncogene c-Myc (Engstrom et al., 2004; Boult et al., 2008). Nonetheless, the detailed mechanisms underlying the role of Mxi1-0 in the division of PASMCs remain to be further dissected, e.g., whether Mxi1-0 and Mxi1-1 play opposite roles in the regulation of Max-Myc interaction or whether Mxi1-0 participates in the cellular machineries responsible for degradation of c-Myc.
MAPK signaling pathway is involved in the regulation of various biological functions of cells (Uehling and Harris, 2015). The activation of this pathway licenses the expression of a large cohort of genes regulating cell proliferation, differentiation and vascular development (Lei et al., 2015). MEK/ERK signaling is a classical MAPK signal transduction pathway and serves an important regulator of pulmonary hypertension (Preston et al., 2006). Consistently, we found here that HPH-related genes were significantly enriched in MAPK signaling pathway and ERK related biological processes. Mxi1-0, as a novel regulator of HPH, potentiates PASMC proliferation through MEK/ERK-dependent upregulation of c-Myc (Figure 7E). These findings are in agreement with previous studies showing that Mxi1-0 activates MEK/ERK signaling and improved the proliferation of human umbilical vein endothelial cells (HUVECs), and that Mxi1-0 underlies hypoxia-induced vascular endothelial growth factor production by hepatic carcinoma cells (Hu et al., 2017; Wu et al., 2017). Although it is still unknown how Mxi1-0 activates MEK/ERK signaling in PASMCs, we found that inhibition of this canonical pathway significantly repressed the development of pulmonary hypertension in rats exposed to chronic hypoxia.
Collectively, we report a hitherto unrecognized crucial role of Mxi1-0 in HPH, thereby providing rationale for the applicability of Mxi1-0 and downstream signaling as candidate targets for clinical treatment and potential biomarkers for prognostic assessment of HPH.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Huashan institutional review board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Fudan University Veterinary Medicine Animal Care and Use Committee.
Author Contributions
SQL designed the experiment and prepared the initial manuscript. LD, XL, and BW conducted experimental operation. CL, GW, and JW cultivated the cells. XW, JX, and XZ were responsible for experimental calculations. YYZ, RY, NZ, JL, DZ, YZZ, SHL, and JZL were involved in the experimental analysis. LD, XL, and BW contributed equally to this manuscript. SQL was a Correspondent.
Funding
This work was supported by the National Natural Science Foundation (Nos. 81670045 and 81970048) and by the General Project of Shanghai Municipal Health Commission (No.201840296).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for the financial support by the National Natural Science Foundation and by the General project of Shanghai Municipal Health Commission.
References
Armstrong, M. B., Mody, R. J., Ellis, D. C., Hill, A. B., Erichsen, D. A., and Wechsler, D. S. (2013). N-myc Differentially Regulates Expression of MXI1 Isoforms in Neuroblastoma. Neoplasia 15 (12), 1363–1370. PubMed PMID: 24403858; PubMed Central PMCID: PMC3884527. doi:10.1593/neo.11606
Badesch, D. B., Raskob, G. E., Elliott, C. G., Krichman, A. M., Farber, H. W., Frost, A. E., et al. (2010). Pulmonary Arterial Hypertension. Chest 137 (2), 376–387. PubMed PMID: 19837821. doi:10.1378/chest.09-1140
Baluapuri, A., Wolf, E., and Eilers, M. (2020). Target Gene-independent Functions of MYC Oncoproteins. Nat. Rev. Mol. Cel Biol 21 (5), 255–267. PubMed PMID: 32071436; PubMed Central PMCID: PMC7611238. doi:10.1038/s41580-020-0215-2
Boult, J. K. R., Tanière, P., Hallissey, M. T., Campbell, M. J., and Tselepis, C. (2008). Oesophageal Adenocarcinoma Is Associated with a Deregulation in the MYC/MAX/MAD Network. Br. J. Cancer 98 (12), 1985–1992. PubMed PMID: 18493233; PubMed Central PMCID: PMC2441969. doi:10.1038/sj.bjc.6604398
Cassady, S. J., and Reed, R. M. (2019). Pulmonary Hypertension in COPD: A Case Study and Review of the Literature. Medicina 55 (8), 432. PubMed PMID: 31382489; PubMed Central PMCID: PMC6723523. doi:10.3390/medicina55080432
Dang, C. V., O’Donnell, K. A., Zeller, K. I., Nguyen, T., Osthus, R. C., and Li, F. (2006). The C-Myc Target Gene Network. Semin. Cancer Biol. 16 (4), 253–264. PubMed PMID: 16904903. doi:10.1016/j.semcancer.2006.07.014
Dugast-Darzacq, C., Grange, T., and Schreiber-Agus, N. B. (2007). Differential Effects of Mxi1-Srα and Mxi1-Srβ in Myc Antagonism. FEBS J. 274 (17), 4643–4653. PubMed PMID: 17697116. doi:10.1111/j.1742-4658.2007.05992.x
Dugast-Darzacq, C., Pirity, M., Blanck, J. K., Scherl, A., and Schreiber-Agus, N. (2004). Mxi1-SRα: a Novel Mxi1 Isoform with Enhanced Transcriptional Repression Potential. Oncogene 23 (55), 8887–8899. PubMed PMID: 15467743. doi:10.1038/sj.onc.1208107
Dunham-Snary, K. J., Wu, D., Sykes, E. A., Thakrar, A., Parlow, L. R. G., Mewburn, J. D., et al. (2017). Hypoxic Pulmonary Vasoconstriction. Chest 151 (1), 181–192. PubMed PMID: 27645688; PubMed Central PMCID: PMC5310129. doi:10.1016/j.chest.2016.09.001
Engstrom, L. D., Youkilis, A. S., Gorelick, J. L., Zheng, D., Ackley, V., Petroff, C. A., et al. (2004). Mxi1-0, an Alternatively Transcribed Mxi1 Isoform, Is Overexpressed in Glioblastomas. Neoplasia 6 (5), 660–673. PubMed PMID: 15548375; PubMed Central PMCID: PMC1531670. doi:10.1593/neo.04244
Erichsen, D. A., Armstrong, M. B., and Wechsler, D. S. (2015). Mxi1 and Mxi1-0 Antagonize N-Myc Function and Independently Mediate Apoptosis in Neuroblastoma. Translational Oncol. 8 (1), 65–74. PubMed PMID: 25749179; PubMed Central PMCID: PMC4350643. doi:10.1016/j.tranon.2015.01.002
Galiè, N., Humbert, M., Vachiery, J.-L., Gibbs, S., Lang, I., Torbicki, A., et al. (2015. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Respir. J. 46 (4), 903–975. PubMed PMID: 26318161. doi:10.1183/13993003.01032-2015
Hu, Z., Dong, N., Lu, D., Jiang, X., Xu, J., Wu, Z., et al. (2017). A Positive Feedback Loop between ROS and Mxi1-0 Promotes Hypoxia-Induced VEGF Expression in Human Hepatocellular Carcinoma Cells. Cell Signal. 31, 79–86. PubMed PMID: 28065785. doi:10.1016/j.cellsig.2017.01.007
Huang, Y., Hu, K., Zhang, S., Dong, X., Yin, Z., Meng, R., et al. (2018). S6K1 Phosphorylation-dependent Degradation of Mxi1 by β-Trcp Ubiquitin Ligase Promotes Myc Activation and Radioresistance in Lung Cancer. Theranostics 8 (5), 1286–1300. PubMed PMID: 29507620; PubMed Central PMCID: PMC5835936. doi:10.7150/thno.22552
Hurdman, J., Condliffe, R., Elliot, C. A., Davies, C., Hill, C., Wild, J. M., et al. (2012). ASPIRE Registry: Assessing the Spectrum of Pulmonary Hypertension Identified at a REferral centre. Eur. Respir. J. 39 (4), 945–955. PubMed PMID: 21885399. doi:10.1183/09031936.00078411
Hurlin, P. J., and Huang, J. (2006). The MAX-interacting Transcription Factor Network. Semin. Cancer Biol. 16 (4), 265–274. PubMed PMID: 16908182. doi:10.1016/j.semcancer.2006.07.009
Lei, Z., van Mil, A., Brandt, M. M., Grundmann, S., Hoefer, I., Smits, M., et al. (2015). MicroRNA-132/212 Family Enhances Arteriogenesis after Hindlimb Ischaemia through Modulation of the Ras-MAPK Pathway. J. Cel. Mol. Med. 19 (8), 1994–2005. PubMed PMID: 25945589; PubMed Central PMCID: PMC4549050. doi:10.1111/jcmm.12586
Li, D., Wang, B., Wang, H., and Liu, Q. (2018). Prognostic Significance of Pulmonary Hypertension in Patients with Cystic Fibrosis. Medicine 97 (7), e9708. PubMed PMID: 29443734; PubMed Central PMCID: PMC5839836. doi:10.1097/MD.0000000000009708
Li, W., Liu, C.-C., Kang, S., Li, J.-R., Tseng, Y.-T., and Zhou, X. J. (2016). Pushing the Annotation of Cellular Activities to a Higher Resolution: Predicting Functions at the Isoform Level. Methods 93, 110–118. PubMed PMID: 26238263. doi:10.1016/j.ymeth.2015.07.016
Li, Y., Yang, L., Dong, L., Yang, Z.-w., Zhang, J., Zhang, S.-l., et al. (2019). Crosstalk between the Akt/mTORC1 and NF-Κb Signaling Pathways Promotes Hypoxia-Induced Pulmonary Hypertension by Increasing DPP4 Expression in PASMCs. Acta Pharmacol. Sin 40 (10), 1322–1333. PubMed PMID: 31316183; PubMed Central PMCID: PMC6786428. doi:10.1038/s41401-019-0272-2
Manni, I., Tunici, P., Cirenei, N., Albarosa, R., Colombo, B. M., Roz, L., et al. (2002). Mxi1 Inhibits the Proliferation of U87 Glioma Cells through Down-Regulation of Cyclin B1 Gene Expression. Br. J. Cancer 86 (3), 477–484. PubMed PMID: 11875718; PubMed Central PMCID: PMC2375210. doi:10.1038/sj.bjc.6600065
Maron, B. A., Machado, R. F., and Shimoda, L. (2016). Pulmonary Vascular and Ventricular Dysfunction in the Susceptible Patient (2015 Grover Conference Series). Pulm. Circ. 6 (4), 426–438. PubMed PMID: 28090285; PubMed Central PMCID: PMC5210067. doi:10.1086/688315
Naeije, R., and Dedobbeleer, C. (2013). Pulmonary Hypertension and the Right Ventricle in Hypoxia. Exp. Physiol. 98 (8), 1247–1256. PubMed PMID: 23625956. doi:10.1113/expphysiol.2012.069112
Negi, V., Yang, J., Speyer, G., Pulgarin, A., Handen, A., Zhao, J., et al. (2021). Computational Repurposing of Therapeutic Small Molecules from Cancer to Pulmonary Hypertension. Sci. Adv. 7 (43), eabh3794. PubMed PMID: 34669463. doi:10.1126/sciadv.abh3794
Nesbit, C. E., Tersak, J. M., and Prochownik, E. V. (1999). MYC Oncogenes and Human Neoplastic Disease. Oncogene 18 (19), 3004–3016. PubMed PMID: 10378696. doi:10.1038/sj.onc.1202746
Preston, I. R., Hill, N. S., Warburton, R. R., and Fanburg, B. L. (2006). Role of 12-lipoxygenase in Hypoxia-Induced Rat Pulmonary Artery Smooth Muscle Cell Proliferation. Am. J. Physiology-Lung Cell Mol. Physiol. 290 (2), L367–L374. PubMed PMID: 16199435. doi:10.1152/ajplung.00114.2005
Prins, K. W., Duval, S., Markowitz, J., Pritzker, M., and Thenappan, T. (2017). Chronic Use of PAH‐specific Therapy in World Health Organization Group III Pulmonary Hypertension: a Systematic Review and Meta‐analysis. Pulm. Circ. 7 (1), 145–155. PubMed PMID: 28680574; PubMed Central PMCID: PMC5448533. doi:10.1086/690017
Shah, S. J. (2012). Pulmonary Hypertension. Jama 308 (13), 1366–1374. PubMed PMID: 23032553. doi:10.1001/jama.2012.12347
Thompson, A. A. R., and Lawrie, A. (2017). Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Molecular Medicine 23 (1), 31–45. PubMed PMID: 27989641. doi:10.1016/j.molmed.2016.11.005
Uehling, D. E., and Harris, P. A. (2015). Recent Progress on MAP Kinase Pathway Inhibitors. Bioorg. Med. Chem. Lett. 25 (19), 4047–4056. PubMed PMID: 26298497. doi:10.1016/j.bmcl.2015.07.093
Voelkel, N. F., Mizuno, S., and Bogaard, H. J. (2013). The Role of Hypoxia in Pulmonary Vascular Diseases: a Perspective. Am. J. Physiology-Lung Cell Mol. Physiol. 304 (7), L457–L465. PubMed PMID: 23377344. doi:10.1152/ajplung.00335.2012
Wechsler, D. S., Shelly, C. A., Petroff, C. A., and Dang, C. V. (1997). MXI1, a Putative Tumor Suppressor Gene, Suppresses Growth of Human Glioblastoma Cells. Cancer Res. 57 (21), 4905–4912. PubMed PMID: 9354456.
Wechsler, D. S., Hawkins, A. L., Li, X., Jabs, E. W., Griffin, C. A., and Dang, C. V. (1994). Localization of the Human Mxi1 Transcription Factor Gene (MXI1) to Chromosome 10q24-Q25. Genomics 21 (3), 669–672. PubMed PMID: 7959753. doi:10.1006/geno.1994.1336
Woo, K. V., Ornitz, D. M., and Singh, G. K. (2019). Diagnosis and Pathophysiological Mechanisms of Group 3 Hypoxia-Induced Pulmonary Hypertension. Curr. Treat. Options. Cardio Med. 21 (3), 16. PubMed PMID: 30903302. doi:10.1007/s11936-019-0718-3
Wu, W., Hu, Z., Wang, F., Gu, H., Jiang, X., Xu, J., et al. (2017). Mxi1-0 Regulates the Growth of Human Umbilical Vein Endothelial Cells through Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) and Interleukin-8 (IL-8)-dependent Pathways. PloS one 12 (6), e0178831. PubMed PMID: 28575053; PubMed Central PMCID: PMC5456372. doi:10.1371/journal.pone.0178831
Xia, J., Yang, L., Dong, L., Niu, M., Zhang, S., Yang, Z., et al. (2018). Cefminox, a Dual Agonist of Prostacyclin Receptor and Peroxisome Proliferator-Activated Receptor-Gamma Identified by Virtual Screening, Has Therapeutic Efficacy against Hypoxia-Induced Pulmonary Hypertension in Rats. Front. Pharmacol. 9, 134. PubMed PMID: 29527168; PubMed Central PMCID: PMC5829529. doi:10.3389/fphar.2018.00134
Yap, K., and Makeyev, E. V. (2016). Functional Impact of Splice Isoform Diversity in Individual Cells. Biochem. Soc. Trans. 44 (4), 1079–1085. PubMed PMID: 27528755; PubMed Central PMCID: PMC4984447. doi:10.1042/BST20160103
Young, J. M., Williams, D. R., and Thompson, A. A. R. (2019). Thin Air, Thick Vessels: Historical and Current Perspectives on Hypoxic Pulmonary Hypertension. Front. Med. 6, 93. PubMed PMID: 31119132; PubMed Central PMCID: PMC6504829. doi:10.3389/fmed.2019.00093
Zervos, A. S., Gyuris, J., and Brent, R. (1993). Mxi1, a Protein that Specifically Interacts with Max to Bind Myc-Max Recognition Sites. Cell 72 (2), 223–232. PubMed PMID: 8425219. doi:10.1016/0092-8674(93)90662-a
Keywords: hypoxic pulmonary hypertension (HPH), max interacting protein 1–0 (Mxi1-0), pulmonary arterial smooth muscle cells (PASMCs), cell proliferation, MEK/ERK, c-Myc
Citation: Dong L, Liu X, Wu B, Li C, Wei X, Wumaier G, Zhang X, Wang J, Xia J, Zhang Y, Yiminniyaze R, Zhu N, Li J, Zhou D, Zhang Y, Li S, Lv J and Li S (2022) Mxi1-0 Promotes Hypoxic Pulmonary Hypertension Via ERK/c-Myc-dependent Proliferation of Arterial Smooth Muscle Cells. Front. Genet. 13:810157. doi: 10.3389/fgene.2022.810157
Received: 06 November 2021; Accepted: 08 February 2022;
Published: 23 March 2022.
Edited by:
Andy T Y Lau, Shantou University, ChinaCopyright © 2022 Dong, Liu, Wu, Li, Wei, Wumaier, Zhang, Wang, Xia, Zhang, Yiminniyaze, Zhu, Li, Zhou, Zhang, Li, Lv and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengqing Li, c2hlbmdxaW5nbGlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work