
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 10 March 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.804202
This article is part of the Research Topic Advancing Our Understanding of the Genetic and Functional Basis of Skeletal Dysplasia View all 15 articles
 Liying Sun1†
Liying Sun1† Yingzhao Huang2,3,4†Sen Zhao2,3,4†Wenyao Zhong1†Jile Shi2,3,4Yang Guo1Junhui Zhao1Ge Xiong1Yuehan Yin1
Yingzhao Huang2,3,4†Sen Zhao2,3,4†Wenyao Zhong1†Jile Shi2,3,4Yang Guo1Junhui Zhao1Ge Xiong1Yuehan Yin1 Zefu Chen2,3,4
Zefu Chen2,3,4 Nan Zhang1
Nan Zhang1 Zongxuan Zhao1
Zongxuan Zhao1 Qingyang Li1Dan Chen1Yuchen Niu3,5
Qingyang Li1Dan Chen1Yuchen Niu3,5 Xiaoxin Li3,5Guixing Qiu2,3,4Zhihong Wu3,4,5
Xiaoxin Li3,5Guixing Qiu2,3,4Zhihong Wu3,4,5 Terry Jianguo Zhang2,3,4*Wen Tian1*
Terry Jianguo Zhang2,3,4*Wen Tian1* Nan Wu2,3,4*
Nan Wu2,3,4*Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder of connective tissue characterized by crumpled ears, arachnodactyly, camptodactyly, large joint contracture, and kyphoscoliosis. The nature course of CCA has not been well-described. We aim to decipher the genetic and phenotypic spectrum of CCA. The cohort was enrolled in Beijing Jishuitan Hospital and Peking Union Medical College Hospital, Beijing, China, based on Deciphering disorders Involving Scoliosis and COmorbidities (DISCO) study (http://www.discostudy.org/). Exome sequencing was performed on patients’ blood DNA. A recent published CCA scoring system was validated in our cohort. Seven novel variants and three previously reported FBN2 variants were identified through exome sequencing. Two variants outside of the neonatal region of FBN2 gene were found. The phenotypes were comparable between patients in our cohort and previous literature, with arachnodactyly, camptodactyly and large joints contractures found in almost all patients. All patients eligible for analysis were successfully classified into likely CCA based on the CCA scoring system. Furthermore, we found a double disease-causing heterozygous variant of FBN2 and ANKRD11 in a patient with blended phenotypes consisting of CCA and KBG syndrome. The identification of seven novel variants broadens the mutational and phenotypic spectrum of CCA and may provide implications for genetic counseling and clinical management.
Congenital contractural arachnodactyly (CCA), also known as Beals syndrome, is an autosomal dominantly inherited connective tissue disorder with an unknown prevalence (Putnam et al., 1995). The clinical manifestations of CCA primarily include arachnodactyly, congenital joint contractures, crumpled ears, kyphoscoliosis, chest deformity, dolichostenomelia, muscle hypoplasia, micrognathia and high arched palate (Guo et al., 2016; Meerschaut et al., 2019). CCA is caused by variants in fibrillin-2 (FBN2) gene. Fibrinllin-2 is an integral component of elastin fibers in extracellular matrix (ECM), which provides supporting structure for tissues and scaffolds for physiological processes (Olivieri et al., 2010). Fibrillin-2 mediates signaling molecules on cell surfaces, including transforming growth factor β(TGF-β), bone morphogenetic proteins (BMPs), integrins and controls ECM formation and remodeling. Pathogenic variants in FBN2 may weaken microfibril structure or disrupt binding capability, subsequently weaken the elastic fiber and perturbate ECM-mediated signaling, which leads to the anomalies of CCA (Ratnapriya et al., 2014).
Thus far, only 91 variants in FBN2 gene associated with CCA have been described, as listed in the Human Genome Mutation Database (HGMD). Most of these variants cluster in a hotspot region, which is known as neonatal region, spanning from exon 23 to exon 35 (Meerschaut et al., 2019), which encodes the calcium-binding epidermal growth factor-like (cbEGF) domains. However, a limited number of variants outside the neonatal region has also been reported (Callewaert et al., 2009).
Recently, a clinical scoring system for CCA was developed to classify patients into likely CCA or unlikely CCA, which was based on the presence or absence of the ten main clinical features of this disorder (Meerschaut et al., 2019). Developing a scoring system is particularly important due to the phenotype overlap between CCA and other connective tissue disorders like Marfan syndrome and type VI collagenopathies (Also named as Bethlem myopathy) (Bamshad et al., 1996). However, this scoring system has not been tested in independent cohort and the genotype-phenotype correlation of CCA is still elusive.
In this study, we identified ten pathogenic FBN2 variants in 27 CCA patients from ten families, of which seven are novel variants. We provide their clinical manifestations and speculate variants’ impact on protein function based on variant location. Furthermore, we validated the clinical utility of a newly developed scoring system for CCA (Meerschaut et al., 2019).
We included ten families diagnosed with CCA and carrying pathogenic FBN2 variant from Beijing Jishuitan Hospital and Peking Union Medical College Hospital based on the Deciphering disorders Involving Scoliosis and COmorbidities (DISCO) study (http://www.discostudy.org/) (Tian et al., 2020; Sun et al., 2021). Informed consent was obtained from all patients or their guardians. This study was approved by the institutional review board at Beijing Jishuitan Hospital and Peking Union Medical College Hospital.
As a part of DISCO study, peripheral blood DNA from probands and available familial members were prepared into Illumina paired-end libraries and underwent whole-exome capture with the Agilent V5, followed by sequencing on the Illumina HiSeq 4,000 platform (Illumina, San Diego, CA, United States). In-house-developed Peking Union Medical college hospital Pipeline (PUMP) was used for variant calling and annotation (Zhao et al., 2020; Chen et al., 2021; Sun et al., 2021).
Variant-encoding amplicons were amplified by PCR from genomic DNA obtained from subjects and purified using an Axygen AP-GX-50 kit (lot no. 05915KE1) and conducted Sanger sequencing on an ABI3730XL instrument.
Rare variants with minor allele frequencies less than 0.01 in 1,000 Genomes (October 2013), Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org), the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org), and the in-house database of DISCO study (>8,394 exomes) were extracted. Besides variants in FBN2 gene, we also examined other variants according to American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015), The prediction of mutational effect was performed using Combined Annotation Dependent Depletion (CADD, https://cadd.gs.washington.edu/) (Rentzsch et al., 2019), Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/) (Ng and Henikoff, 2003), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2013).
We calculated the CCA clinical score of each affected individual based on their recorded phenotypes (Meerschaut et al., 2019). This scoring system was proposed to classify patients into likely CCA or unlikely CCA using phenotype-based scores. The scores were calculated based on the presence or absence of ten phenotypes. Four phenotypes were allotted for three points, including crumpled ears, arachnodactyly, camptodactyly and contractures of large joints. Two phenotypes were allotted for two points, including pectus deformity and dolichostenomelia. Four phenotypes were allotted for one point, including kyphoscoliosis, muscle hypoplasia, highly arched palate and micrognathia. A total score ≥7 and <7 indicated this patient was likely CCA or unlikely CCA, respectively. Only those with all the required phenotypes recorded were subjected for calculation.
This study included 10 unrelated families with a total of 27 cases affected with CCA (Figure 1). Eight of the families have more than one affected individual. Median age at admission was 4.95 years. Arachnodactyly (27/27, 100%), crumpled ears (26/27, 96.3%), camptodactyly (26/27, 96.3%), and muscle hypoplasia (22/26, 85%) were observed in almost all recruited cases (Table 1). More than half patients (16/25, 64%) presented with contracture of large joints, including elbow, wrist, knee, ankle, and shoulder. 54% patients (13/24) present with kyphosis or scoliosis; 29% patients (7/24) presented with pectus deformity, including four patients with pectus carinatum and three patients with pectus excavatum; 74% patients (17/23) presented with high arched palate and 71% patients (17/24) presented with micrognathia; 33% patients (9/27) presented with pes planus (Table1); Two patients presented with genu varus and no patient presented with dolichostenomelia. Besides characteristic features of CCA, one patient presented with cryptorchidism, global developmental delay, atrial septal defect, bulbous nose, and broad eyebrow.
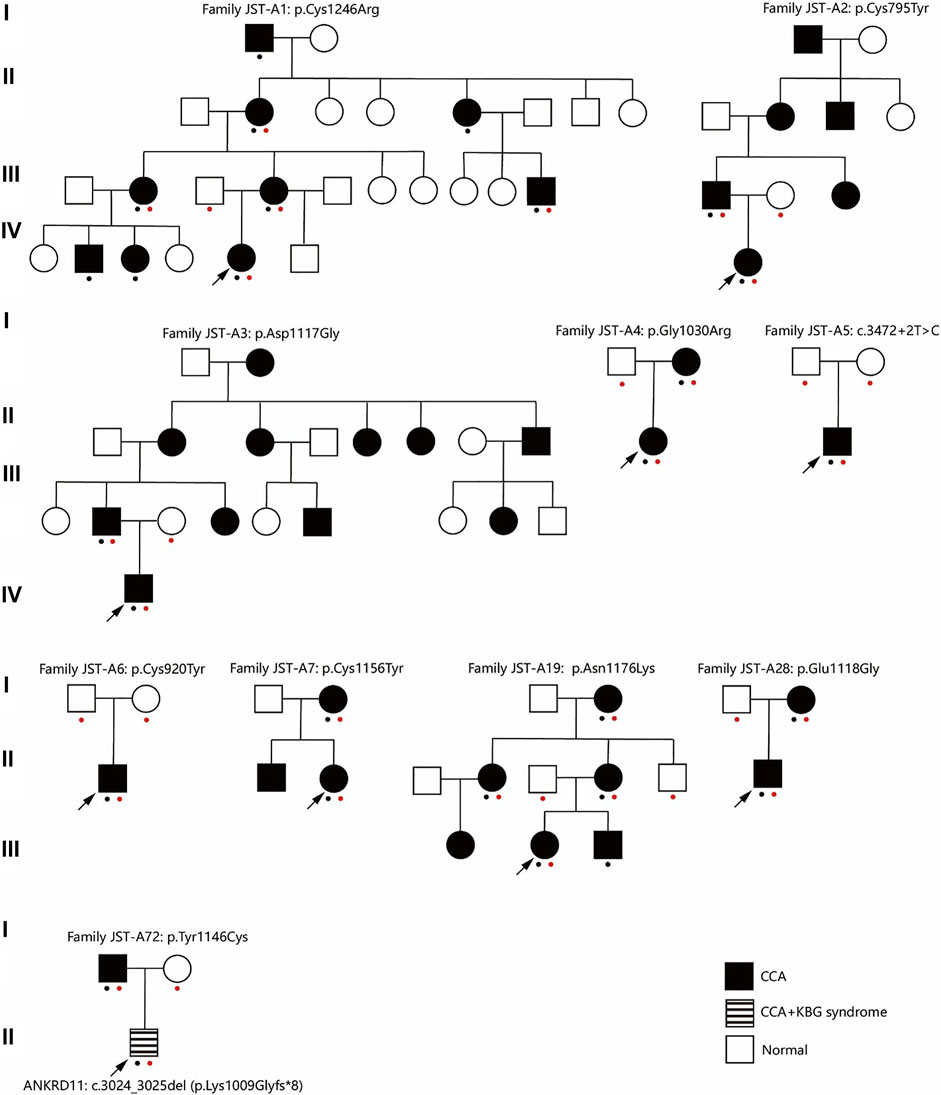
FIGURE 1. Pedigree of ten families. A red dot indicates this individual underwent genetic test (Sanger sequencing or exome sequencing). A black dot indicates this individual underwent phenotypes assessment. CCA, Congenital contractural arachnodactyly.
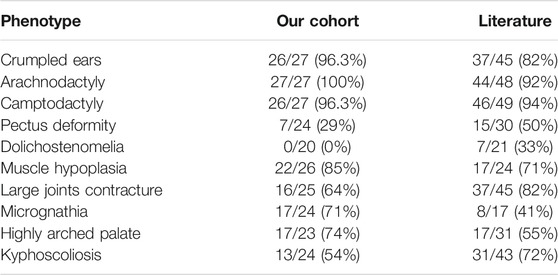
TABLE 1. Summary of patients’ phenotypes in our cohort and in the literature.
After ES and bioinformatic analyses, ten different FBN2 variants were identified from the ten families (Table 2). Two variants were validated to be de novo (Family JST-A5 and JST-A6) and eight variants were segregated with the phenotype. Of the ten variants, three have been previously reported, i.e. p.Tyr1146Cys, c.3472+2T>C and p.Asn1176Lys (Gupta et al., 2002; Callewaert et al., 2009). The remaining seven variants are all novel, i.e. p. Cys1246Arg, p. Glu1118Gly, p. Cys795Tyr, p. Asp1117Gly, p. Gly1030Arg, p. Cys920Tyr and p. Cys1156Tyr. All ten variants are evolutionary conserved and are absent from population controls.
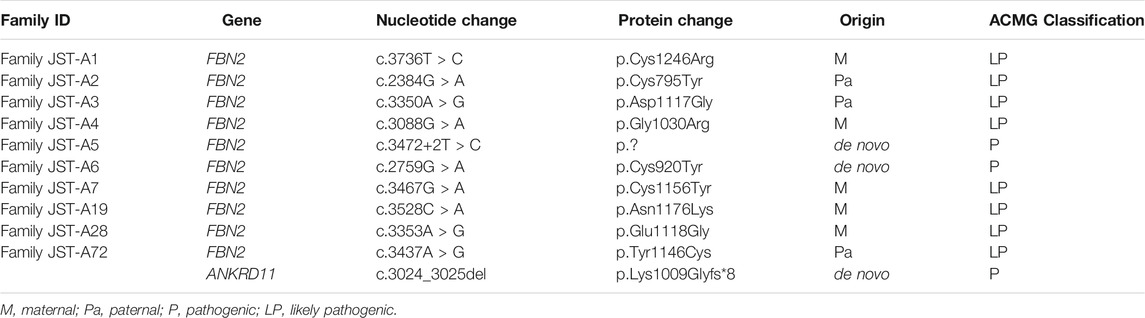
TABLE 2. Summary of identified FBN2 and ANKRD11 variants.
Eight out of ten variants were found in the neonatal region (exon 24-32) of the FBN2 gene. Among them, six variants alter the highly conserved cbEGF-like domain (p.Tyr1146Cys, p. Asp1117Gly, p. Glu1118Gly, p. Cys1156Tyr, p. Asn1176Lys, and p. Cys1246Arg) (Figure 2). One variant alters the TB domain (p.Gly1030Arg). One de novo splicing variant was identified (c.3472+2T > C), which has been verified to leading to an exon 26 skip (Gupta et al., 2002). Two variants were found outside the neonatal region of the FBN2 gene (p.Cys795Tyr and p. Cys920Tyr). The p. Cys920Tyr variant would affect the second hybrid domain and the p. Cys795Tyr would affect the cbEGF7 domain, respectively.
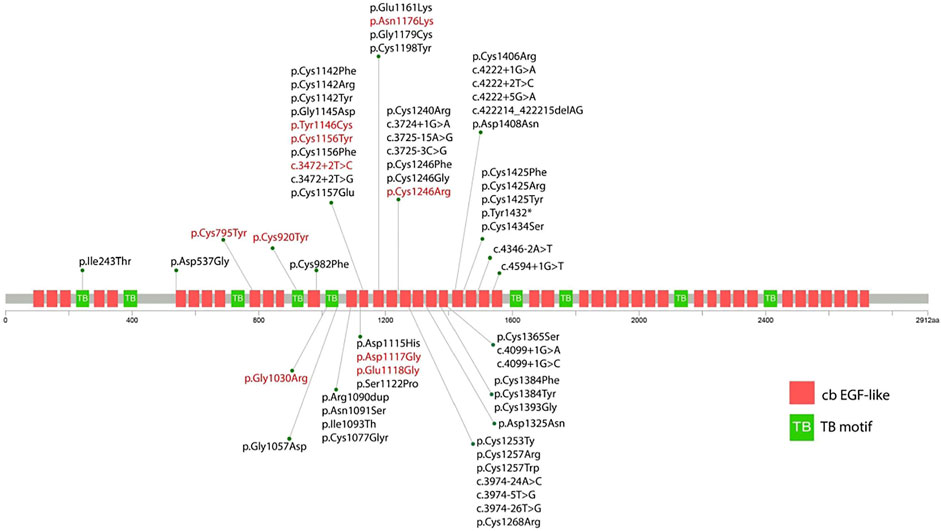
FIGURE 2. Schematic diagram showing the position of the identified variants in relation to the structural domains of the fibrillin-2 protein. The red variant indicates the variant reported in this study. The black variant indicates the variant reported in previous literature.
Through interpretation of the ES data, we identified one patient with dual molecular diagnosis. Patient DISCO-JST A72 was a 5 months old boy born to nonconsanguineous northern Han parents. His father presented with typical features of CCA (crumpled ears, camptodactyly, arachnodactyly and dolichostenomelia), while his mother is healthy. At 5 months old, he presented with crumpled ears, camptodactyly, arachnodactyly, global developmental delay, short stature, cryptorchidism, intellectual disability, global developmental delay, atrial septal defect (3.5 mm), bulbous nose, and broad eyebrow. ES of this family reviewed a de novo heterozygous ANKRD11 variant c.3024_3025del (p.Lys1009Glyfs*8) in addition to the heterozygous FBN2 variant c.3437A > G (p.Tyr1146Cys) inherited from his affected father. The ANKRD11 frameshift variant is previously unreported and predicted to result in protein truncation or nonsense medicated decay. Loss-of-function of ANKRD11 gene is a known disease-causing mechanism (Kim et al., 2020). ANKRD11 is the only known causative gene of KBG syndrome, which is characterized by macrodontia, craniofacial findings, short stature, multiple skeletal anomalies including vertebrae and limbs, neurologic involvement including global developmental delay, seizures, and intellectual disability (Goldenberg et al., 2016). The clinical diagnostic criteria of KBG syndrome included four features macrodontia of upper central permanent incisors, characteristic facial anomalies, hand anomalies, neurologic involvement, significantly delayed bone age, costo-vertebral anomalies, postnatal short stature, and a first degree relative with KBG. For this patient, we observed a remarkable blended phenotype. For global developmental delay, cryptorchidism, intellectual disability, atrial septal defect (3.5 mm), bulbous nose, and broad eyebrow, we considered these phenotypes to be part of KBG syndrome; while crumpled ears, camptodactyly and arachnodactyly are characteristic features of CCA.
We calculated the diagnostic score for CCA in our FBN2-positive CCA patients. Totally, 16 patients with sufficient phenotype data were eligible for analysis. Figure 3 gives an overview of the distribution of the total scores calculated for each CCA patient. The minimal, median and maximum score is 8, 14 and 17. All patients’ CCA score is ≥ 7, which indicates all patients are successfully classified into likely CCA by the CCA scoring system. For the rest of 11 patients with insufficient clinical data, the clinical score of these patients are still ≥7.
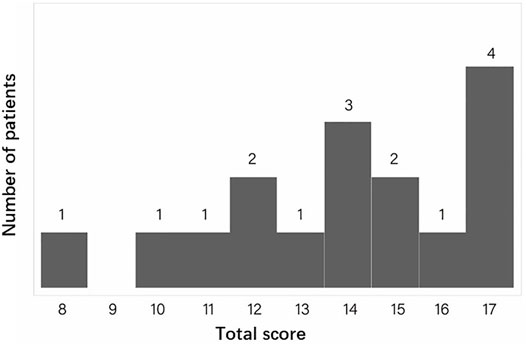
FIGURE 3. The distribution of the total CCA clinical scores for all eligible patients. The X ray indicates the total score. The Y ray indicates the number of patients.
In this study, we investigated 10 families with 27 patients diagnosed with CCA, based on their clinical and molecular profiles. We identified seven novel variants and three previously reported variants in FBN2 gene. Additionally, we reported an individual with dual molecular diagnosis of CCA and KBG syndrome.
FBN2 encodes a 2912-amino acids extracellular matrix protein related to the elasticity of the tissue, which includes nine TB motifs, and 46 cbEGF domains (Davis and Summers, 2012). Each EGF-like domain contains six conserved cysteine residues to support its native folding. Six conserved cysteine residues form three disulfide bridges to maintain protein stability (Davis and Summers, 2012). In the present study, we found that c.2384G > A (p.Cys795Tyr), c.2759G > A (p.Cys920Tyr), c.3437A > G (p.Tyr1146Cys), c.3467G > A (p.Cys1156Tyr), and c.3736T > C (p.Cys1246Arg) alter or produce cysteine residues in the cbEGF domain, which would potentially disrupt the disulfide bond and therefore impair the nature folding of fibrillin-2. This is considered as the major mechanism underlying the pathogenesis of CCA (Guo et al., 2016; Xu et al., 2020).
Nine out of ten variants reported in this study are missense variants, which is consistent with previous findings that nearly all currently reported human FBN2 pathogenic variants lead to single amino acids substitutions or in-frame exon deletions or duplications. This observation strongly suggests that gain-of-function is the key mechanism underlying the pathogenesis of CCA. This might explain why fbn2 null mice does not phenocopy human CCA (Shi et al., 2013; Sengle et al., 2015).
In our cohort, we detected only two pathogenic variant [c.2384G > A (p.Cys795Tyr) and c.2759G > A (p.Cys920Tyr)] located outside the neonatal region. There are only three variants outside the neonatal region have been previously reported to lead to CCA (Liu et al., 2019; Renner et al., 2019). Totally, only ∼8% CCA was caused by variants outside the neonatal region, which proves from another side of the substantial contribution of the neonatal region to the CCA phenotype. The pathogenicity of these two variants were supported by in silico predictions of pathogenicity, the absence of this variant from population controls, and segregation analysis.
We found one case with a blended phenotype consisting of CCA and KBG syndrome. Dual diagnoses in Mendelian disorders with complex phenotypes are being increasingly recognized. Coexisting diseases result in blended clinical phenotypes and poses challenge in diagnosis and management (Posey et al., 2017). One study have found 4.9% of cases suspected of having Mendelian disorders had multiple molecular diagnoses (Posey et al., 2017). Therefore, when phenotypes cannot be completely explained by one detected variant, additional genetic and clinical assessment should be considered. Interestingly, patients with CCA typically presented with tall stature, while KBG syndrome usually resulted in postnatal short stature. In this case, the patient has remarkable short stature.
Review of the phenotype data of CCA probands from literature and our cohort reveals a basically comparable phenotype. For example, external ear malformations like crumpled ears, which are a major characteristic of CCA, were found in 96.3% and 82% patients in our cohort or in the literature, respectively. The high prevalence of crumpled ears in CCA patients suggest it is particularly important in the differential diagnosis of connective tissue disorder. Interestingly, we found no patients in our cohort presented with dolichostenomelia. While in previous reports, 7/21 (33%) patients presented with dolichostenomelia (Fisher’s exact test, p = 0.0005). Additionally, we found no patients in our cohort presented with cardiovascular anomalies like aortic root dilation. Our findings suggest phenotype heterogeneity of CCA may exists in different populations.
Genotype-phenotype correlation analysis in this study and previous studies revealed no significant associations. Recently, mutational burden has been recognized has a key contributor to the molecular diagnosis of some patients with arthrogryposis through accumulation of multiple deleterious variants (Pehlivan et al., 2019). Multilocus variants might also contribute to the genotype-phenotype correlation and intrafamilial variability in CCA. Fibrillins-2 polymerize extracellularly and form microfibrils with many proteins, e.g., latent transforming growth factor beta binding proteins (LTBPs) and microfibril-associated proteins (MFAPs). The complex binding interactions between these molecules indicate variants in different genes could modify CCA phenotype. Further analysis of large samples would possibly provide insights into the genotype-phenotype correlation in CCA.
The newly developed clinical scoring system for CCA successfully classified all patients in this study into likely CCA, even in patients with insufficient clinical data, indicating the excellent sensitivity of this scoring system. However, we didn’t test the specificity of this scoring system due to lack of comprehensive clinical data of possibly misdiagnosed syndromes like Marfan syndrome. Marfan syndrome usually presented with cardiovascular, skeletal and ophthalmological manifestations (Bitterman and Sponseller, 2017). The overlapping phenotypes mainly include scoliosis, pectus deformity and cardiovascular deformity. In this scoring system, these overlapping phenotypes like kyphoscoliosis and pectus deformity were allotted for three points in total. While large joints contractures, camptodactyly, ear malformation, and arachnodactyly are usually absent in Marfan syndrome, which were allotted for three points each. Thus, we can speculate this scoring system would likely diagnose a Marfan syndrome patient into “unlikely CCA”. Nevertheless, more data are needed to validate the specificity of this scoring system.
Intrafamilial heterogeneity has been noted in some families with CCA. This phenomenon has also been observed in our cohort and showed by the CCA clinical score. In the family JST-A1 with eight patients eligible for analysis, the highest, lowest and median CCA clinical score was 17, 8 and 13, respectively. The intrafamilial variation in this large family was moderate and all patients were classified into “likely CCA”.
In conclusion, we report seven novel and three previously reported FBN2 mutations in 27 patients from ten families with CCA. Our report enriches the mutational spectrum of FBN2 and validate the novel CCA scoring system.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: <https://data.mendeley.com/drafts/cxgcj3s8t3, Mendeley.
The studies involving human participants were reviewed and approved by the ethics committee at Beijing Jishuitan Hospital (201808-09), and Peking Union Medical College Hospital (JS-098). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Conceptualization: WT, ZW, TJZ and NW. Cohort enrollment: LS, YG, ZZ, QL, YY, NZ, DC, GX, WZ and YH. Funding acquisition: NW, ZW, WT and TJZ. Experiments: ZC, XL, YN, JS and YH. Genetic data analysis: YH, LS, SZ, WZ, ZY, XL and NW. Bioinformatic analysis: SZ, and ZC. Writing—review and editing: TJZ, GQ, NW and ZW. Data interpretation: WT, ZW, TJZ and NW. Writing original draft: YH, LS, SZ, WZ, YG, GX, WT and NW.
This research was funded in part by the National Key Research and Development Program of China (2016YFC0901500), Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China (2016ZX310174-4), Beijing JST Research Funding (ZR-201907, ZR-201911 and 2019-YJ03), Beijing Jishuitan Hospital Nova Program (XKXX201818), National Natural Science Foundation of China (81822030, 82072391, 81672123, 81972037, 81772299, 81930068, 81772301, and 81972132), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2019PT320025), Beijing Natural Science Foundation (JQ20032 and 7191007), Tsinghua University-Peking Union Medical College Hospital Initiative Scientific Research Program, CAMS Initiative Fund for Medical Sciences (2016-I2M-3-003, 2016-I2M-2-006 and 2017-I2M-2-001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the families who participated in this research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.804202/full#supplementary-material
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting Functional Effect of Human Missense Mutations Using PolyPhen‐2. Curr. Protoc. Hum. Genet. 76. doi:10.1002/0471142905.hg0720s76
Bamshad, M., Jorde, L. B., and Carey, J. C. (1996). A Revised and Extended Classification of the Distal Arthrogryposes. Am. J. Med. Genet. 65, 277–281. doi:10.1002/(sici)1096-8628(19961111)65:4<277::aid-ajmg6>3.0.co;2-m
Bitterman, A. D., and Sponseller, P. D. (2017). Marfan Syndrome. J. Am. Acad. Orthopaedic Surgeons 25, 603–609. doi:10.5435/JAAOS-D-16-00143
Callewaert, B. L., Loeys, B. L., Ficcadenti, A., Vermeer, S., Landgren, M., Kroes, H. Y., et al. (2009). Comprehensive Clinical and Molecular Assessment of 32 Probands with Congenital Contractural Arachnodactyly: Report of 14 Novel Mutations and Review of the Literature. Hum. Mutat. 30, 334–341. doi:10.1002/humu.20854
Chen, N., Zhao, S., Jolly, A., Wang, L., Pan, H., Yuan, J., et al. (2021). Perturbations of Genes Essential for Müllerian Duct and Wölffian Duct Development in Mayer-Rokitansky-Küster-Hauser Syndrome. Am. J. Hum. Genet. 108, 337–345. doi:10.1016/j.ajhg.2020.12.014
Davis, M. R., and Summers, K. M. (2012). Structure and Function of the Mammalian Fibrillin Gene Family: Implications for Human Connective Tissue Diseases. Mol. Genet. Metab. 107, 635–647. doi:10.1016/j.ymgme.2012.07.023
Goldenberg, A., Riccardi, F., Tessier, A., Pfundt, R., Busa, T., Cacciagli, P., et al. (2016). Clinical and Molecular Findings in 39 Patients with KBG Syndrome Caused by Deletion or Mutation ofANKRD11. Am. J. Med. Genet. 170, 2847–2859. doi:10.1002/ajmg.a.37878
Guo, X., Song, C., Shi, Y., Li, H., Meng, W., Yuan, Q., et al. (2016). Whole Exome Sequencing Identifies a Novel Missense FBN2 Mutation Co-segregating in a Four-Generation Chinese Family with Congenital Contractural Arachnodactyly. BMC Med. Genet. 17, 91. doi:10.1186/s12881-016-0355-6
Gupta, P. A., Putnam, E. A., Carmical, S. G., Kaitila, I., Steinmann, B., Child, A., et al. (2002). Ten novelFBN2mutations in Congenital Contractural Arachnodactyly: Delineation of the Molecular Pathogenesis and Clinical Phenotype. Hum. Mutat. 19, 39–48. doi:10.1002/humu.10017
Kim, S. J., Yang, A., Park, J. S., Kwon, D. G., Lee, J.-S., Kwon, Y. S., et al. (2020). Two Novel Mutations of ANKRD11 Gene and Wide Clinical Spectrum in KBG Syndrome: Case Reports and Literature Review. Front. Genet. 11, 579805. doi:10.3389/fgene.2020.579805
Liu, H., Tsui, Y., Wang, J., Su, C., Zheng, R., Shao, Y., et al. (2019). Establishment of a Beals Syndrome Patient-Derived Human Induced Pluripotent Stem Cell Line HELPi001-A. Stem Cel Res. 40, 101535. doi:10.1016/j.scr.2019.101535
Meerschaut, I., De Coninck, S., Steyaert, W., Barnicoat, A., Bayat, A., Benedicenti, F., et al. (2020). A Clinical Scoring System for Congenital Contractural Arachnodactyly. Genet. Med. 22, 124–131. doi:10.1038/s41436-019-0609-8
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting Amino Acid Changes that Affect Protein Function. Nucleic Acids Res. 31, 3812–3814. doi:10.1093/nar/gkg509
Olivieri, J., Smaldone, S., and Ramirez, F. (2010). Fibrillin Assemblies: Extracellular Determinants of Tissue Formation and Fibrosis. Fibrogenesis Tissue Repair 3, 24. doi:10.1186/1755-1536-3-24
Pehlivan, D., Bayram, Y., Gunes, N., Coban Akdemir, Z., Shukla, A., Bierhals, T., et al. (2019). The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am. J. Hum. Genet. 105, 132–150. doi:10.1016/j.ajhg.2019.05.015
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 376, 21–31. doi:10.1056/NEJMoa1516767
Putnam, E. A., Zhang, H., Ramirez, F., and Milewicz, D. M. (1995). Fibrillin-2 (FBN2) Mutations Result in the Marfan-like Disorder, Congenital Contractural Arachnodactyly. Nat. Genet. 11, 456–458. doi:10.1038/ng1295-456
Ratnapriya, R., Zhan, X., Fariss, R. N., Branham, K. E., Zipprer, D., Chakarova, C. F., et al. (2014). Rare and Common Variants in Extracellular Matrix Gene Fibrillin 2 (FBN2) Are Associated with Macular Degeneration. Hum. Mol. Genet. 23, 5827–5837. doi:10.1093/hmg/ddu276
Renner, S., Schüler, H., Alawi, M., Kolbe, V., Rybczynski, M., Woitschach, R., et al. (2019). Next-generation Sequencing of 32 Genes Associated with Hereditary Aortopathies and Related Disorders of Connective Tissue in a Cohort of 199 Patients. Genet. Med. 21, 1832–1841. doi:10.1038/s41436-019-0435-z
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 47, D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sengle, G., Carlberg, V., Tufa, S. F., Charbonneau, N. L., Smaldone, S., Carlson, E. J., et al. (2015). Abnormal Activation of BMP Signaling Causes Myopathy in Fbn2 Null Mice. Plos Genet. 11, e1005340. doi:10.1371/journal.pgen.1005340
Shi, Y., Tu, Y., Mecham, R. P., and Bassnett, S. (2013). Ocular Phenotype ofFbn2-Null Mice. Invest. Ophthalmol. Vis. Sci. 54, 7163–7173. doi:10.1167/iovs.13-12687
Sun, L., Huang, Y., Zhao, S., Zhao, J., Yan, Z., Guo, Y., et al. (2021). Deciphering the Mutational Signature of Congenital Limb Malformations. Mol. Ther. - Nucleic Acids 24, 961–970. doi:10.1016/j.omtn.2021.04.012
Tian, W., Huang, Y., Sun, L., Guo, Y., Zhao, S., Lin, M., et al. (2020). Phenotypic and Genetic Spectrum of Isolated Macrodactyly: Somatic Mosaicism of PIK3CA and AKT1 Oncogenic Variants. Orphanet J. Rare Dis. 15, 288. doi:10.1186/s13023-020-01572-9
Xu, P., Li, R., Huang, S., Sun, M., Liu, J., Niu, Y., et al. (2020). A Novel Splicing Mutation in the FBN2 Gene in a Family with Congenital Contractural Arachnodactyly. Front. Genet. 11, 143. doi:10.3389/fgene.2020.00143
Keywords: FBN2 (fibrillin-2), congenital contractural arachnodactyly, arthrogryposis, novel variants, clinical genetics, musculo-skeletal diseases
Citation: Sun L, Huang Y, Zhao S, Zhong W, Shi J, Guo Y, Zhao J, Xiong G, Yin Y, Chen Z, Zhang N, Zhao Z, Li Q, Chen D, Niu Y, Li X, Qiu G, Wu Z, Zhang TJ, Tian W and Wu N (2022) Identification of Novel FBN2 Variants in a Cohort of Congenital Contractural Arachnodactyly. Front. Genet. 13:804202. doi: 10.3389/fgene.2022.804202
Received: 29 October 2021; Accepted: 08 February 2022;
Published: 10 March 2022.
Edited by:
Long Guo, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Bert Callewaert, Ghent University, BelgiumCopyright © 2022 Sun, Huang, Zhao, Zhong, Shi, Guo, Zhao, Xiong, Yin, Chen, Zhang, Zhao, Li, Chen, Niu, Li, Qiu, Wu, Zhang, Tian and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry Jianguo Zhang, amd6aGFuZ19wdW1jaEB5YWhvby5jb20=; Wen Tian, d2VudGlhbkpTVEAxNjMuY29t; Nan Wu, ZHIud3VuYW5AcHVtY2guY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.