 Paola Granata1
Paola Granata1 Dario Cocciadiferro2
Dario Cocciadiferro2 Alessandra Zito1,3
Alessandra Zito1,3 Chiara Pessina1,3Alessandro Bassani1Fabio Zambonin4
Chiara Pessina1,3Alessandro Bassani1Fabio Zambonin4 Antonio Novelli2
Antonio Novelli2 Mauro Fasano5*
Mauro Fasano5* Rosario Casalone1*
Rosario Casalone1*- 1Cytogenetics and Medical Genetics Unit, Department of Services, ASST dei Sette Laghi, Varese, Italy
- 2Laboratory of Medical Genetics, Translational Cytogenomics Research Unit, Ospedale Pediatrico Bambino Gesù, Roma, Italy
- 3Department of Medicine and Surgery, University of Insubria, Varese, Italy
- 4Child Neuropsychiatry Unit, Department of Maternal and Child Health, ASST dei Sette Laghi, Varese, Italy
- 5Department of Science and High Technology and Center of Bioinformatics, University of Insubria, Busto Arsizio, Italy
The 16p13.11 microdeletion, whose prevalence in the general population is about 0.04%, is known in literature as a predisposition factor to neurodevelopmental disorders, being found in about 0.13% of patients with schizophrenia, in 0.5–0.6% of patient with epilepsy, cognitive impairment, autism spectrum disorder (ASD) and aggressiveness. The goal of this study was to identify a specific gene set pattern unique for the affected patients in comparison with other familial components. Due to the incomplete penetrance of this copy number variant (CNV), we studied by whole exome sequencing (WES), with particular regard of 850 SFARI genes, three families with an affected member carrier of inherited 16p13.11 and 16p13.11p12.3 microdeletion and one family with an affected member with a de novo 16p13.11 microdeletion. By combining a deductive approach together with personalized network models, we identified gene signatures potentially capable of explaining the clinical phenotype. Candidate variants in genes of interest were identified as possibly involved in determining the neurological phenotype of the four patients, such as compound heterozygosity in CECR2, variants in MTOR and RICTOR genes, compound heterozygous single nucleotide variants in the LRRK2 gene. Moreover, genes present in the microdeletion region were partially present as central nodes, with a focus on NDE1. No additional pathogenetic or uncertain CNVs were found in all four patients. No significant variants were detected in genes included in the microdeletion in patients 1, 2 and 3, excluding the finding of unmasked recessive variants. In conclusion, WES is a fundamental tool in the genetic investigation of patients having a predisposing variant, which is not sufficient to define the clinical phenotype. Moreover, the analysis of WES data using Systems medicine tools, such as personalized network models, led to the prioritization of genes on a high throughput scale and to discover variants in genes that were not prioritized at first.
Introduction
The microdeletion 16p13.11 (chr16:15.48-16.32 Mb, GRCh37/hg19) is a well known genomic rearrangement previously reported as predisposing to neurodevelopmental disorders (Ullmann et al., 2007; Hannes et al., 2009; Law et al., 2009; de Kovel et al., 2010; Heinzen et al., 2010; Mefford et al., 2010; Alkuraya et al., 2011; Balasubramanian et al., 2011; Ingason et al., 2011; Nagamani et al., 2011; Paciorkowski et al., 2013; Tropeano et al., 2013; Tropeano et al., 2014). Genes mapped in the deleted region include NDE1, the strongest candidate gene for neurodevelopmental phenotype and microcephaly, expressed in brain, which protein plays an essential role in microtubule organization, mitosis and neuronal migration. The region also includes NTAN1, involved in protein degradation and related to altered behavior, and MYH11, coding for the major contractile protein in smooth muscle cells [GeneCards (Stelzer et al., 2016)].
The microdeletion is found in about 0.13% of patients with schizophrenia (Ingason et al., 2011), in 0.5–0.6% of patients with epilepsy (de Kovel et al., 2010; Heinzen et al., 2010; Ingason et al., 2011) and it has been associated with a wide spectrum of multiple congenital anomalies such as facial dysmorphisms and different cognitive impairment, autism spectrum disorder (ASD), and aggressiveness (www.rarechromo.org). However, since the prevalence of the variation in the general population is about 0.04% (1:2,300–2,500 individuals) (Paciorkowski et al., 2013; Tropeano et al., 2014) and the variation is frequently present in unaffected parents and relatives of affected subjects, the pathological significance of 16p13.11 microdeletion remains uncertain. The association between abnormal phenotype and 16p13.11 microdeletion could be a mere coincidence. From a meta analysis by Rosenfeld and coworkers, the del16p13.11 frequency in postnatal array comparative genomic hybridization (aCGH) cases is 0.15%, while the frequency in controls is 0.05%. The de novo occurence in cases has a frequency of 21.7%, with a penetrance estimate of 13.1% (95% CI) (Rosenfeld et al., 2013). Indeed, the incomplete penetrance frequently observed in del16p13.11 families may be due to variable phenotypic expressivity, phenomenon of unmasking of a recessive variant, variants in genes outside the deletion, different extension of the microdeletion or a combination of all these events.
In order to identify the presence and significance of a possible second hit mutational event or other pathological variants in genes outside the deleted region, we performed whole exome sequencing (WES) in four del16p13.11 patients and their unaffected parents and siblings with the same microdeletion. The WES results were analyzed with two distinct approaches, in parallel. First, the classical deductive approach led to the identification of potentially relevant variants. Next, the inductive approach that characterizes Systems medicine allowed us to discover new variants in genes that were not prioritized at first. Altogether, results contributed to better define the clinical relevance of del16p13.11 and to shed light on the mechanisms of incomplete penetrance and of phenotypic heterogeneity.
Materials and Methods
Patients
Four patients with 16p13.11 microdeletion were referred to the Medical Genetics Unit of “ASST dei Sette Laghi” Hospital (Varese, Italy) for genetic investigation and counselling, with medical indication of developmental disorders, learning delay, intellectual disability, with or without congenital dysmorphic features. This study was based on results obtained for diagnostic purpose. A written informed consent was provided by the parents and relatives of the patients included in this study to perform aCGH and WES and to use the data for research.
Patient 1
Male patient, born at term without neonatal or prenatal distress, with weight = 3.70 kg, length = 52 cm, CC = 35 cm and APGAR = 9/10. He showed normal development, normal achievement of development milestones and no motor abnormalities; at the age of 8 he showed growth deficiency and dysmorphisms including low-set auricles, slow down eyelid rim, ogival palate with dental malocclusion, lower thick and down slated lip. The patient at this age developed difficulties in fine motor skills, showed low I.Q. (WISC-IV I.Q. 86), impulsiveness, spatial visualization difficulties, reading and understanding difficulties, dyscalculia, dysgraphia, spelling skills <5th percentile, mathematical difficulties and memory deficit, TCM rapidity <10th percentile, minimal atrioventricular valve insufficiency. At the age of 12, the patient showed hypotonia, persistent dysmorphism, epicanthus, scoliosis, winged shoulder blades, unilateral cryptorchidism, thinness, anxiety. No epilepsy was detected. The karyotype was 46,XY.
Patient 2
Male patient, born to term with caesarean section after normal pregnancy, with weight = 3.32 kg, length = 49 cm and CC = 34 cm. At the age of 5 he showed medium degree intellectual deficit (WISC-IV I.Q. 40), hypotonia, developmental and severe language delay, aggressivity and obsessive crises. He showed facial dysmorphisms (such as carp mouth), obesity, hyperphagia, but no hypogenitalism. Audiometric tests, MRI and EEG were normal. No epilepsy was detected. The karyotype was 46,XY and the methylation test for Prader-Willi/Angelman syndrome showed normal biparental methylation pattern.
Patient 3
Female patient, born at the 38th week of gestation with caesarean section, with weight = 3.42 kg, length = 49 cm, CC = 34 cm and no malformations. At the age of 8 she presented dysorthography (ICD-10 F81.1) and dysgraphia in evolution (ICD-10 F81.8) with associated expressive language disorder (ICD-10 F80.1); the intellectual level was at its lower limit (WISC-IV I.Q. 81). Psychomotor development was normal. The patient showed normal height (80th percentile) and no dysmorphisms. No epilepsy was detected (normal EEG). The karyotype was 46,XX.
Patient 4
Male patient, born at the 42nd week of gestation with weight = 3.66 kg, length = 55, CC = 36, APGAR = 9/10. He showed normal psychomotor development and no dysmorphic features. The intellectual level was low (WISC-IV I.Q. 80). At the age of 5 he revealed severe global developmental and language delay and stereotypies. The diagnosis of non-syndromic ASD was assessed. No epilepsy was detected (normal EEG). The karyotype was 46,XY.
Array CGH
Array CGH was performed for all the components of the four families after DNA extraction (QIAmp DNA blood Maxi Kit, Qiagen, Hilden, Germany) from patients and relatives peripheral blood cells. CytoSure ISCA V2 4x180K platform with a backbone resolution of 1 probe/25 Kb and 1 probe/19 Kb in critical regions, human genome reference GRCh37/hg19 and sex matched normal human DNA pool (Kreatech, Amsterdam, Netherlands) as control were used. InnoScan 710 Microarray Scanner (Innopsys, Carbonne, France) and Mapix (Innopsys, Carbonne, France) were used to detect and analyze fluorescence levels, respectively. Results were interpreted using Cytosure Interpret Software (Oxford Gene Technology, Begbroke, United Kingdom). QC metrics: SD < 1.0 and DLR spread <0.3 were required. The significance of CNVs (copy number variants) was evaluated according to American College of Medical Genetics (ACMG) Joint Consensus (Riggs et al., 2012).
Whole Exome Sequencing
WES was performed on all the members of the four families using the Twist Human Core Exome Kit (Twist Bioscience, San Francisco, United States) according to the manufacturer’s protocol and sequenced with the Illumina NovaSeq 6000 platform. The BaseSpace pipeline (Illumina, San Diego, United States) and the TGex software (LifeMap Sciences, Alameda, United States) were used for the variant calling and annotation, respectively. Sequencing data were aligned to the GRCh37/hg19 human reference genome. Variants with a coverage lower than 10×, genotype quality (GQ) < 15, and gnomAD minor allele frequency (MAF) > 5% were excluded. WES results were interpreted according to ACMG guidelines 2015 (Richards et al., 2015).
Analyzed Genes
WES was applied to investigate: 850 neurodevelopmental genes from SFARI database, genes showing pathogenetic or likely pathogenetic variants following the American College of Medical Genetics and Genomics (ACMG) classification (Richards et al., 2015), genes included in the 16p13.11 and 16p1.11p12.3 microdeletions (NOMO1, PDXDC1, NTAN1, RRN3, MPV17L, MARF1, NDE1, MYH11, CEP20, ABCC1, ABCC6, NOMO3, XYLT1) and genes involved with NDE1 in cargo transport along the axon (i.e., PAFAH1B1, DCTN1, DCTN2, DCTN3, DCTN4, DCTN5, DCTN6, PIK3C3, and PPP1R10) (Kuijpers et al., 2016; Monda and Cheeseman, 2018). After the topological analysis of personalized networks, additional genes were filtered as it follows (second WES filtering). Patient 1: PTPN1, GRB2, ESR1, KIF4, MTFMT, MEOX2, TRAF2, EGFR, LLGL1, TNRC6A, HTT, NRP1. Patient 2: ESR1, MAPKAP1, LRRK2, RICTOR, RPTOR, LAMTOR1, LAMTOR5, MASP2, C4B_2, HTT, MYC, MLST8, CAV1. Patient 3: KMD1A, KMD3A, HPN, LRRK2, XRCC5, PARK7, ESR1, DAXX, DDX5, GRB2, ARID5A, FOXH1, PTPN1, CD33, PKN1, C1QC, SNW1, CALR. Patient 4: ESR1, LLGL1, HTT, KSR1, EWSR1, PIBF1, ATP2B4.
Criteria for Sequence Variants Selection in the Deductive Approach
All synonymous variants were excluded. In patient 1 (who inherited 16p13.11 microdeletion), and in patients 2 and 3 (with inherited 16p13.11p12.3 microdeletion) we selected hemizygous, recessive homozygous and compound heterozygous variants absent in healthy siblings carrying the 16p13.11 and 16p13.11p12.3 microdeletions, and heterozygous variants inherited from the non-carrier parent that were absent in both the healthy sibling and healthy parent carrier of the microdeletion. In patient 4, with de novo 16p13.11 microdeletion, all hemizygous, homozygous, compound heterozygous, and all heterozygous variants inherited from both healthy parents were selected. Variants showing a number of homozygotes higher than one in the general population were excluded (gnomAD v2.1.1 and v3.1), while all selected variants with a number of homozygotes equal to one or zero, or not annotated in the dataset, were included. The prediction of the effect on the protein structure and functionality of a single base variant was calculated using the PHRED quality score [CADD, Combined Annotation Dependent Depletion, genome build GRCh37 v1.4 (Rentzsch et al., 2019)]; the frameshift variants and the insertions or deletions involving two or more nucleotides were considered as deleterious. The hemizygous, homozygous, compound heterozygous or de novo heterozygous variants with PHRED <20.00 were considered, while the inherited heterozygous variants showing PHRED <20.00 were excluded.
The haploinsufficiency (HI) score (ClinGen Dosage Sensitivity Curation Page) and the gnomAD pLI value were considered for all genes showing variants selected as above. The haploinsufficiency of genes was expressed by HI score = 1 to 3, whereas the intolerance of the genes to variants causing loss of function (LoF) was expressed by gnomAD pLI value >0.9 (ClinGen Dosage Sensitivity Map). We have focused our attention on haploinsufficient genes or on genes showing high intolerance to variants causing LoF, since they might be more sensible to deleterious variants. All information about gene function and expression was taken from GeneCards and from GTEx Portal (Stelzer et al., 2016; The GTEx Consortium, 2020); the correlation of genes with Autosomal Recessive (AR)/Autosomal Dominant (AD)/X Linked disease was taken from OMIM database.
Protein-Protein Interaction Networks
Cytoscape 3.8.2 was used to generate networks (Su et al., 2014). The public database IntAct was queried through Cytoscape using the PSICQUIC standard (the Proteomics Standard Initiative Common QUery InterfaCe). Identities of all genes showing at least one variant, as obtained by aCGH and WES, were used to generate a network encompassing all gene products and their first interactors. The network was filtered for human proteins to remove homology inferences. All self-loops and duplicated edges were removed. The topological analysis was performed using the NetworkAnalyzer tool in the Cytoscape environment. Nodes were prioritized by betweenness centrality as already reported (Zito et al., 2021). Most central nodes not included in the first WES filtering were checked for additional variants (second WES filtering; see Analyzed Genes). PHRED scores were annotated to nodes corresponding to altered genes. Arbitrary, out-of-range values were associated to deleted genes for color-coding purpose.
Over-Representation Analysis
Over-representation analysis (ORA) was performed using WebGestalt (2019 release) by setting “geneontology_Biological_Process_noRedundant” as the enrichment database and “human genome, protein coding” as the reference database (Liao et al., 2019). p-values were calculated with the exact Fisher test and corrected for multiple testing according to the Benjamini-Hochberg method. The obtained GO (Gene Ontology) term with the lowest p-value was considered to check for the presence of variants in the enriched genes.
Results
Array CGH
Array CGH performed on DNA from four patients and their healthy parents and siblings showed a microdeletion in the short arm of chromosome 16, overlapping 16p13.11 region, in all patients, with different size. The characteristics of the microdeletions are described in Table 1. The microdeletion was inherited from an unaffected parent in Patient 1 (maternal inheritance), in Patient 2 (paternal inheritance) and in Patient 3 (maternal inheritance), while it was de novo in Patient 4. The protein-coding genes (according to OMIM, excluding RNA genes and pseudogenes) deleted in each patient are reported in Table 2. The genes MPV17L, MARF1, NDE1, MYH11, CEP20, ABCC1, ABCC6 were present in an overlapping common region. No other CNVs classified as pathogenic, likely pathogenic or VOUS (variant of uncertain significance) were found in all four patients.
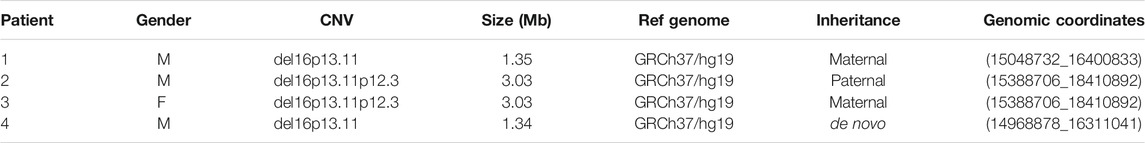
TABLE 1. Array CGH results.
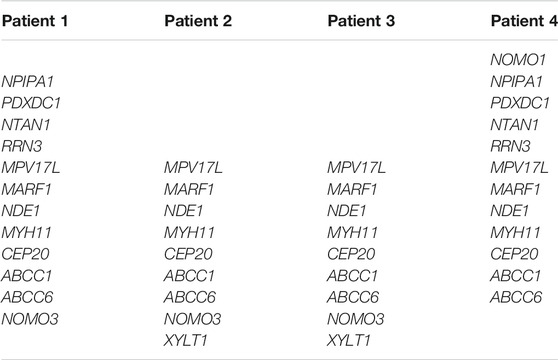
TABLE 2. Genes encompassed by the 16p13.11 and 16p13.11p12.3 microdeletions in the four patients.
Identification of Sequence Variants by Deductive Approach
WES analysis was performed on four trios. The sequencing data are reported in Table 3 and visualized in genealogical trees (Figure 1). In order to identify variants in genes included in the 16p13.11 deletion, as previously reported in some patients with microcephaly (Tan et al., 2017), all the deleted genes of each patient were sequenced. As a result, the heterozygous variant c.3901C>T in ABCC1 was detected in Patient 4, whereas no variants were found in genes involved with NDE1 in cargo transport along the axon (see 2.4 Analyzed Genes). Patient 1 only showed the heterozygous paternal frame-shift variant c.1148delC in CNGB3 gene. Patient 2 showed a compound heterozygosity in CECR2 and three maternal heterozygous variants in USP45, DNAH3 and UNC80. Concerning CECR2, the paternally inherited variant was c.1395+3A>G in an intron splice site region, whereas the maternal variant was the missense variant c.1322C>A. The maternally inherited heterozygous variants were the frame shift variant c.2239dupT in USP45, the intron splice site donor variant c.2099+1G>A in DNAH3 and the missense variant c.6775C>T in UNC80 gene. In Patient 3, paternal heterozygous missense variants c.32534C>T in TTN, c.959C>T in MET, c.2275G>A in DOCK8 and c.1063C>T in MYO9B were detected. Patient 4 displayed paternal missense heterozygous variants in four genes: c.8531G>C in SRCAP, c.1138G>A in DMPK, c.860C>T in NSMCE3, c.274C>T and c.163C>A in MCM6. In another transcript, the variant c.1131+7G>A in an intron splice site region of DMPK was also observed. The maternal missense heterozygous variants are c.2486G>A in PRR12, c.182G>A in SLC1A1, c.1385G>A in GRID1, c.1331G>A in CPEB4, c.2917T>G in ANKS1B, c.466T>A in EIF3G, c.5345C>T in PATJ, c.12284G>A in HECTD4, c.472C>T in CNTNAP4 and c.3901C>T in ABCC1, a gene encompassed by the 16p13.11 microdeletion.
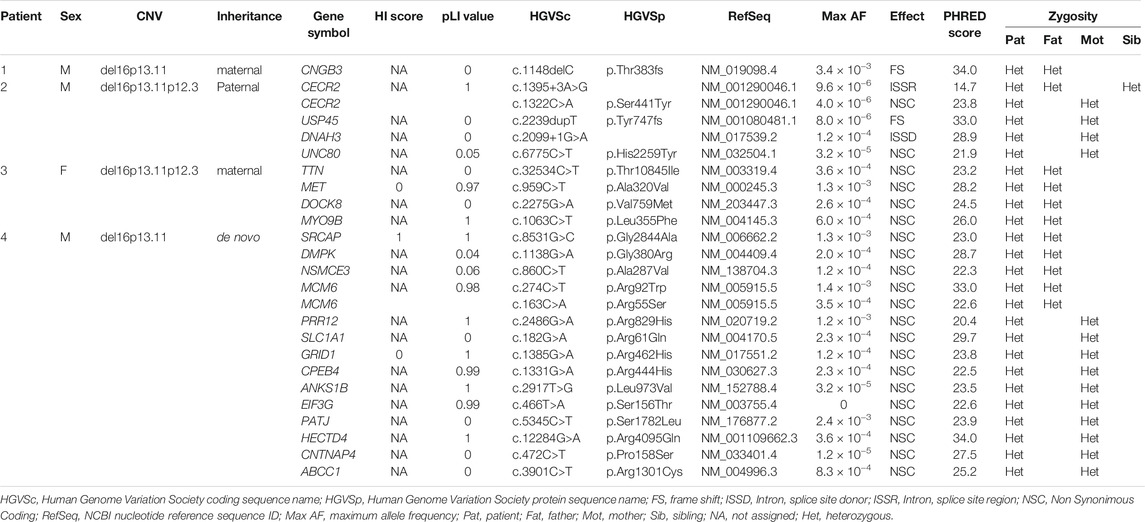
TABLE 3. Variants selected with deductive approach from WES data in the four patients.
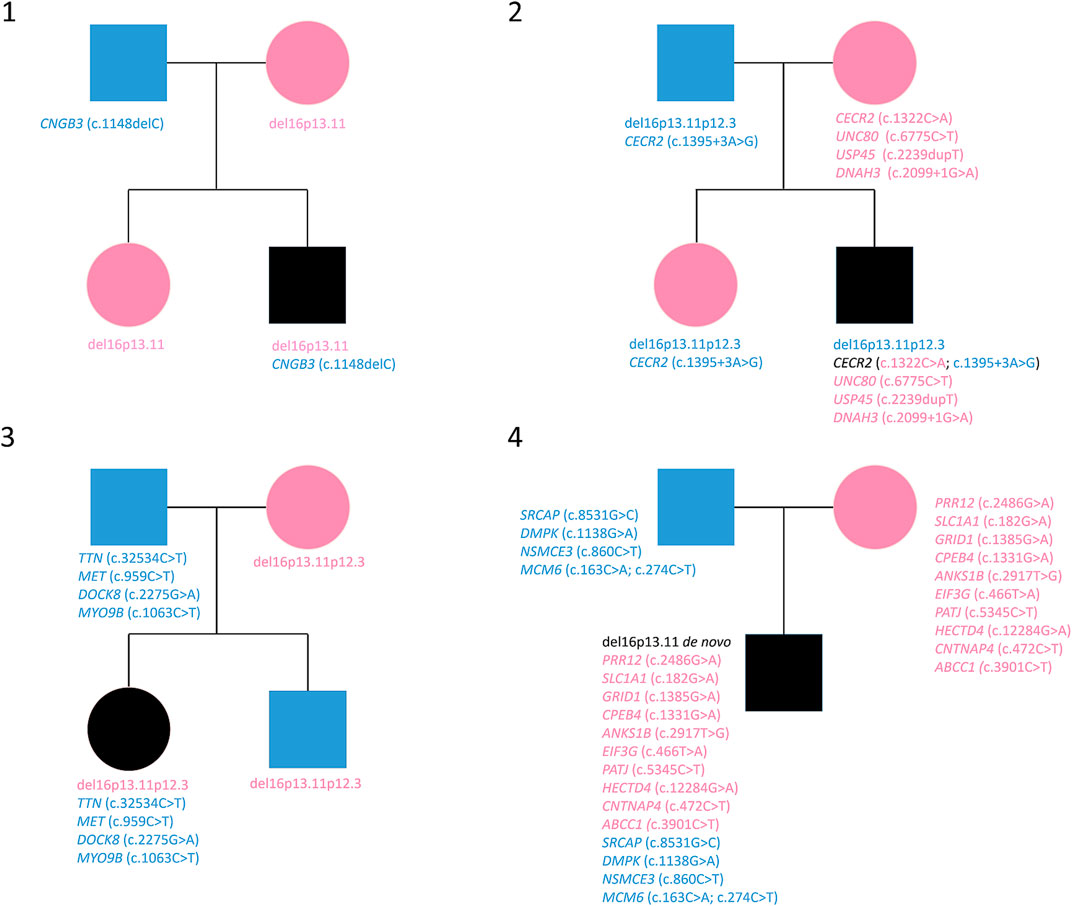
FIGURE 1. Variants reported in genealogical trees of the four families. Cyan: paternal inheritance. Magenta: maternal inheritance.
Topological Analysis of Personalized Protein-Protein Interaction Networks
Starting from the complete list of altered variants, a PPI network for each patient was built and analyzed to obtain betweenness centrality values for each node. Briefly, a gene signature is obtained for each patient that includes all variants without any a priori filtering. Then, edges are placed according to interaction evidence in public databases. Topological analysis is performed to quantify centrality parameters. Eventually, the network is compared to the GO database in order to find overlapping genes in the network and in the GO gene sets (Figure 2). Figure 3 shows subnetworks encompassing the 50 nodes with highest betweenness centrality for each patient. Genes that showed variants or belonged to the microdeletion region were identified with distinct colors (see caption). By merging the four lists of 50 genes each, a non-redundant list of 145 genes was obtained, with distinct centrality values for the same gene in distinct patients (Supplementary Table S1). Five nodes (i.e., NDE1, MARF1, ESR1, CTTNBP2, and TTN) were present in all four subnetworks, whereas MYH11 and HTT were present in three out of four models (Figure 4). Several nodes showed high centrality, although they did not show variants or they showed variants filtered out after WES analysis (green nodes). Therefore, WES results were filtered again including the “large green” nodes, i.e., nodes that were not included in the prioritization step and that showed high betweenness centrality. According to this second filtering, new variants with potential pathogenicity were discovered (Table 4).
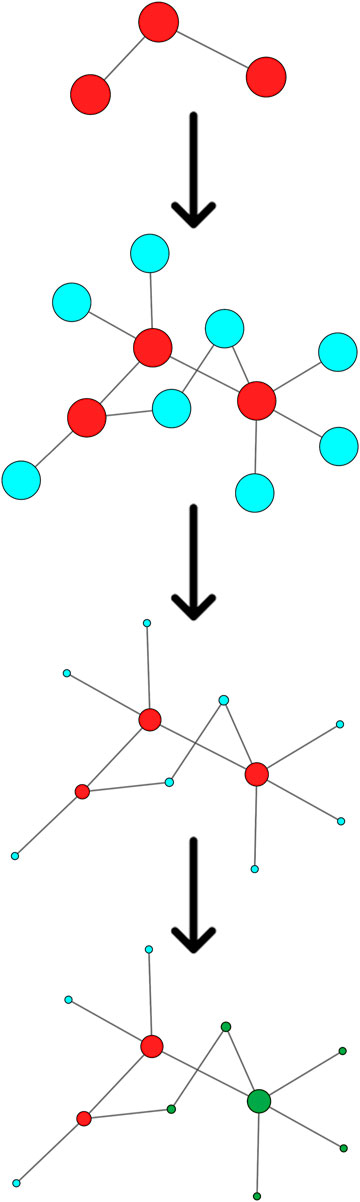
FIGURE 2. Schematic flowchart of the Systems biology approach. From top to bottom: A gene set (red nodes) is obtained for each patient. Interactors (blue nodes) are obtained from public databases. Centrality is calculated by topological analysis. Nodes overlapping with a GO gene set (green nodes) are identified.
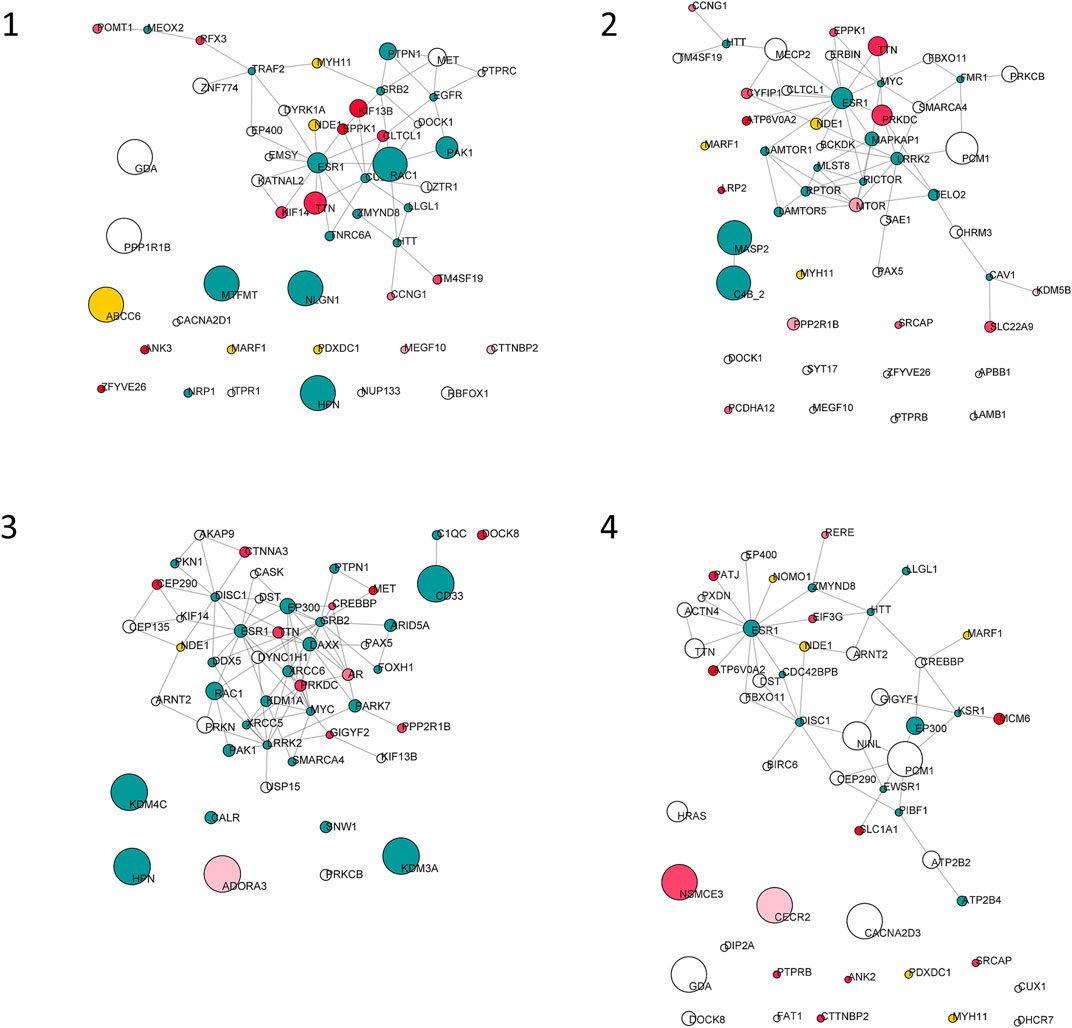
FIGURE 3. Top 50 nodes based on betweenness centrality. Node size is proportional to betweenness centrality. Node color is mapped on PHRED score as follows: white, score from 0 to 15; red shades, score from 15 to 20; red, score higher than 20; yellow, genes of the del16p13.11 microdeletion; green, unmapped.
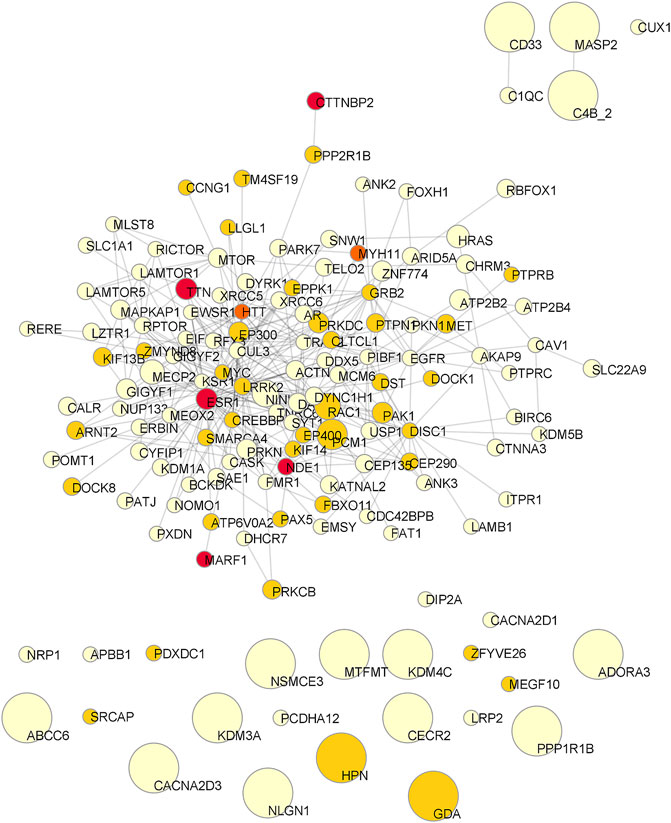
FIGURE 4. Consensus network of the Top50 genes of the four patients. Node size is proportional to the mean betweenness centrality in original networks. Color is associated to the number of observations (pale yellow: one patient; pale orange: two patients; dark orange: three patients; red: all patients).
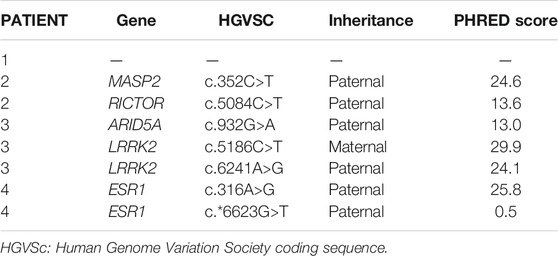
TABLE 4. New single nucleotide variants emerged from topological analysis of the four personalized networks.
Over-Representation Analysis of Personalized Protein-Protein Interaction Networks
The four subnetworks (Figure 3) were analyzed to identify functionally enriched gene ontologies (GOs). Table 5 reports, for each patient, GO terms whose enrichment is statistically significant in the over-representation analysis and the panel of genes that are present in both the subnetwork and in the geneset with the lowest p-value; in each panel the mutated genes and the genes included in the 16p13 microdeletion are highlighted. The analysis of the 50 nodes showing top betweenness centrality values in Patient 1 led to the identification of the following significantly enriched GOs: microtubule polymerization or depolymerization; actomyosin structure organization; phagocytosis; regulation of protein serine/threonine kinase activity; positive regulation of cell adhesion. Concerning Patient 2, the following GOs were highlighted: TOR signaling; peptidyl-serine modification; regulation of anatomical structure size; epithelial cell proliferation; covalent chromatin modification; process utilizing autophagic mechanism; muscle tissue development; protein polymerization; synaptic vesicle cycle. Patient 3 also displayed several significantly enriched GOs, i.e., intracellular receptor signaling pathway; intrinsic apoptotic signaling pathway; covalent chromatin modification; morphogenesis of a branching structure; interaction with symbiont; regulation of DNA-binding transcription factor activity; regulation of intracellular transport; regulation of chromosome organization. On the other hand, the analysis of the network of Patient 4 showed a single enriched pathway (establishment or maintenance of cell polarity). Concerning Patients 1 and 4, the most significantly enriched GOs comprised the NDE1 gene, contained in the deleted region.
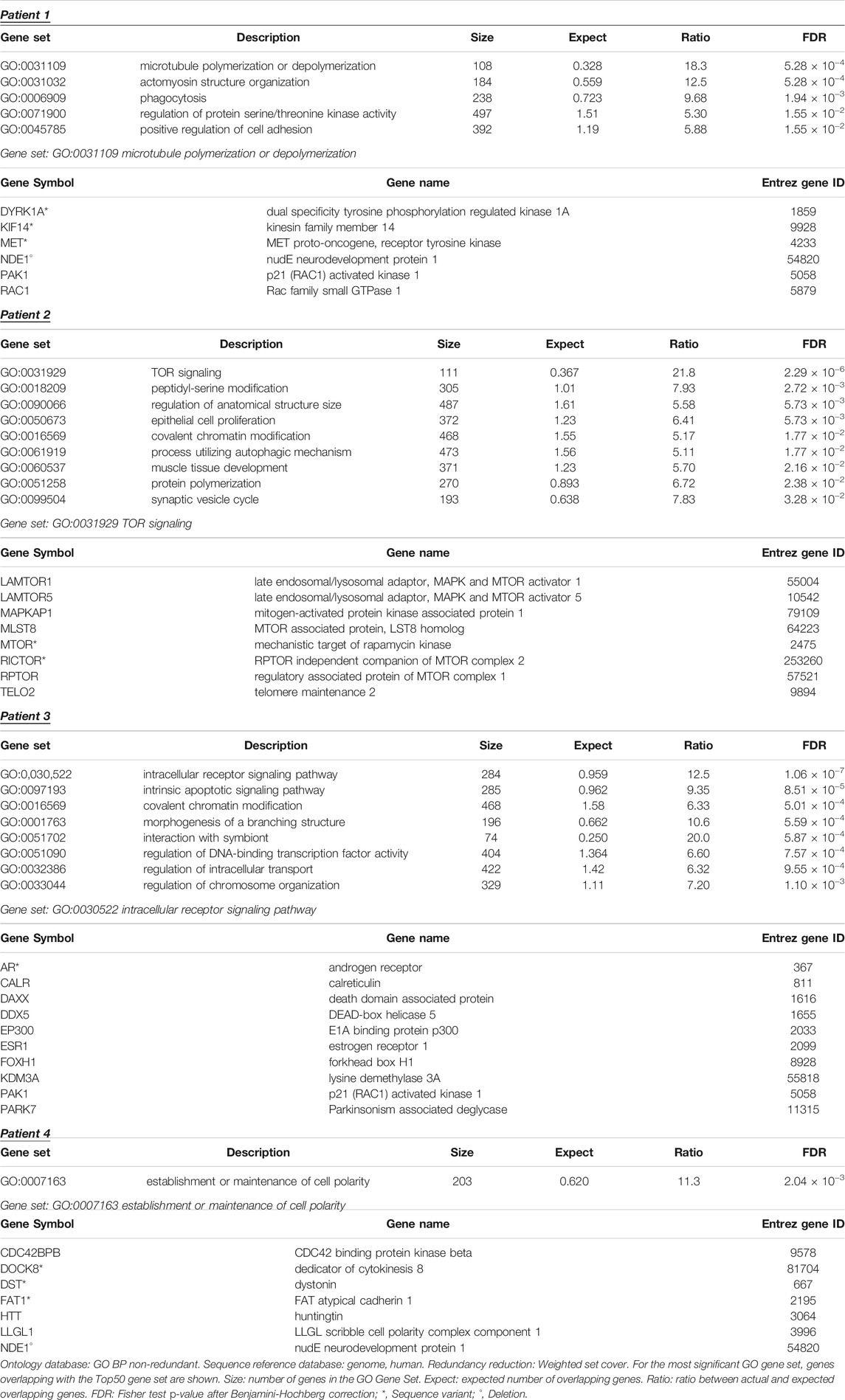
TABLE 5. Over-representation analysis of the Top50 genes for each patient.
Discussion
The 16p13.11 microdeletion has been reported in several cases in association with epilepsy, multiple congenital anomalies and cognitive impairment, but it has been also observed in normal subjects, so that a non-clear pathological significance can be attributed to this CNV (Paciorkowski et al., 2013; Tropeano et al., 2014). The variability of the phenotypic and clinical presentation of the 16p13.11 microdeletion in affected subjects may be due to incomplete penetrance, variable expressivity, unmasking of a recessive variant, different size of the microdeletion, variants in genes not involved in the deletion, or a combination of these conditions.
In order to identify the presence of a possible second hit mutational event or other pathological variants in genes outside the deletion, and their significance, we performed WES in four del16p13.11 patients and their unaffected relatives carrying the microdeletion. We did not find variants in genes included in the 16p deleted region in Patients 1, 2 and 3, so we excluded that their phenotype was due to the unmasking of a recessive variant. In Patient 4, with a de novo 16p13.11 microdeletion, we identified a maternal heterozygous variant in the ABCC1 gene. ABCC1 (ATP binding cassette subfamily C member 1) is a molecular transporter with ATPase activity, not expressed in brain, mediating the export of organic anions and drugs from the cytoplasm and the exchange of various molecules across extra- and intra-cellular membranes. Moreover, it was shown to be involved in multi-drug resistance (Nasr et al., 2020). The ABCC1 gene is related to autosomal dominant deafness-77 (DFNA77, OMIM #618915), a phenotype not observed in the patient and his mother, and did not show haploinsufficiency or LoF intolerance. For all these reasons, this variant does not seem to be an unmasked recessive variant with a pathological significance.
The selected variants obtained from WES highlighted a gene with compound heterozygosity in Patient 2 and heterozygous variants inherited from the parent without the 16p13.11 microdeletion in Patients 1, 2, 3. Additionally, heterozygous variants inherited from both parents were found in Patient 4. No variants were found in genes acting in axonal cargo transfer together with NDE1, the most important candidate gene for the neurodevelopmental phenotype encompassed by the 16p13.11 microdeletion, thus this function does not appear to be compromised.
Following a Systems medicine approach, the complete set of variants was used to build personalized models for each patient, in which altered genes are connected together either directly or by common interactors. To identify genes that potentially play a key role in the pathogenetic mechanism, each network model was analyzed to identify most central nodes (and, in turn, genes). This analysis generated several important findings. First, some of the genes in the microdeletion are often present in the first 50 positions (top50 subset), when nodes are ranked by betweenness centrality (Zito et al., 2021). Moreover, deleted genes may display interactions with mutated genes. In particular, NDE1 was always observed in the Top50 subsets and interacted either directly or through common interactors with other mutated genes, suggesting its potential role in a common pathogenetic mechanism. The second aspect that emerged from the analysis of personalized network models was the presence of very central nodes that were not included in the first prioritization of variants. Indeed, we found that first interactors of altered genes that were observed to be highly central in networks were actually altered in their sequence. ESR1, which played a central role in all four models, appeared to be altered in Patient 4 only, with two paternally inherited single nucleotide variants (SNVs), one of them with a high PHRED score. ESR1 is known to mediate the effect of endocrine disruptors in impairing neurodevelopment (Xu et al., 2020). Another interesting finding was the identification for Patient 3 of two SNVs in compound heterozygosity in LRRK2, both showing high PHRED score. Actually, LRRK2 is a kinase with several activities and several variants were reported (OMIM *609007). The most common one is the G2019S (c.6055G>A), that is, responsible of an AD form of Parkinson’s disease (PARK8, OMIM #607060). Interestingly, variants of LRRK2 were also associated to cognitive development leading to intellectual disability and ASD (Labonne et al., 2020). Hereafter, variants observed in each patient are discussed.
In Patient 1, a male patient with maternal 16p13.11 microdeletion, dysmorphisms and mild intellectual disability, the selected heterozygous paternal variant is a frame-shift variant in CNGB3 (OMIM *605080), a gene encoding the beta subunit of a cyclic nucleotide-gated ion channel with a possible role in modulation of channel function in cone photoreceptors. This gene is not expressed in brain and is not related to AR or AD diseases, neither it shows haploinsufficiency or LoF intolerance. For all these reasons, although CNGB3 is an emerging SFARI gene with score = 3, we exclude correlation of the present variant with the patient’s phenotype. No additional variants were identified after the second WES filtering. Concerning the functional enrichment analysis, we observed a significant over-representation of microtubule polymerization and depolymerization. Among the proteins responsible for this significance, we found NDE1. The corresponding gene is included within the microdeletion region and the encoded protein is a member of the nuclear distribution E (NudE) family, with a role in microtubule organization and neurodevelopment (Monda and Cheeseman, 2018). Although NDE1 has been associated to microhydranencephaly and lissencephaly-4 (OMIM #605013 and #614019, respectively), our observation supports its role in neurodevelopmental disorders in the absence of cerebral malformations (Allach El Khattabi et al., 2020). Variants were found in other genes of the enriched ontology, i.e., DYRK1A (PHRED = 9.75), KIF14 (PHRED = 23.00) and MET (PHRED = 6.39). Therefore, we may hypothesize that the microtubule polymerization and depolymerization pathway is impaired in this patient and could be at the origin of the observed clinical phenotype. The role of 16p13.11 microdeletion in determining the complex phenotype of Patient 1 can be suggested by the involvement of NDE1 in microtubule functionality pathways and the CNV may be considered as a contributing cause variant.
Patient 2, a male patient with a paternal 16p13.11p12.3 microdeletion and with severe neuropsychiatric phenotype, showed a compound heterozygosity in CECR2 (Histone Acetyl-Lysine Reader; OMIM *607576) gene, composed by a paternal variant in an intron splice site region and by a maternal missense variant. This genetic condition is unique to the patient and is not present in other healthy members of the family with or without 16p13.11 microdeletion. CECR2, highly expressed in cerebellum, is involved in chromatin remodeling and may play a role in DNA damage response and in several developmental processes. CECR2 is a component of the CECR2-containing remodeling factor (CERF), involved in neurulation and postnatal brain development (Banting et al., 2005). No AD or AR diseases are known to be related to CECR2 in OMIM. The observed compound heterozygosity in CECR2 seems to be a possible cause of the patient’s phenotype. The maternal heterozygosities were a frame-shift variant in USP45 (OMIM *618439), an intron splice site donor region variant in DNAH3 (OMIM *603334) and a missense variant in UNC80 (OMIM *612636). None of these three genes were related to AD diseases or showed haploinsufficiency or LoF intolerance, nor they were among the top50 genes in the network model of Patient 2. By performing ORA, we found that TOR signaling was the most significant and most enriched GO term, with eight geneset nodes overlapping with the network model (see Table 5). Among these eight nodes, we found variants in MTOR (c.2805G>A, PHRED = 18.86) and RICTOR (c.5084C>T, PHRED = 13.60) genes. These findings support the role of autophagy in neurodevelopmental diseases (Marsh and Dragich, 2019; Lv et al., 2020) with a specific reference to mTORopathies (Karalis and Bateup, 2021) that lead to epilepsy, ASD and, present in this patient, intellectual disability, obesity and hyperphagia. Worthy of note, NDE1 was included in the top50 genes ranked by betweenness centrality, highlighting its role in the network topology. Therefore, we cannot exclude a possible pathogenic contribution of the microdeletion. On the other hand, it was not possible to significantly define its involvement in any biological pathway as described by GO.
Patient 3, a female patient with maternal 16p13.11p12.3 microdeletion with a mild neurocognitive phenotype, showed paternal missense heterozygous variants in TTN (OMIM *188840), MET (OMIM *164860), DOCK8 (OMIM *611432) and MYO9B (OMIM *602129). The genes showing heterozygous variants inherited from the healthy father and related to AD diseases are TTN, related to AD myopathies and cardiomyopathies and MET, related to AD susceptibility to Osteofibrous Dysplasia (OMIM #607278). No signs of these pathologies are present in both Patient 3 and his father. MYO9B is a susceptibility factor to Celiac disease (OMIM #609753) and showed gnomAD pLI = 1. The protein encoded by MET is a hepatocyte growth factor receptor (HGFR); during embryonic development MET plays a role in neuronal precursors; in adults participates in organ regeneration and tissue remodeling and promotes differentiation and proliferation of hematopoietic cells. There is no evidence of a hypothetically causative role of these variants in determining the patient’s neurocognitive phenotype. The second filtering of WES data allowed us to identify two compound heterozygous SNVs in the LRRK2 gene (c.1586C>T, PHRED = 29.9; c.6241A>G, PHRED = 24.1) and a paternal SNV in the ARID5A gene (c.932G>A, PHRED = 13.0). The over-representation analysis of the network model highlighted intracellular receptor signaling as the most significantly enriched pathway. Among the genes mapped on this GO term, AR showed two variants with different deleteriousness (c.636G>A, PHRED = 14.8, and c.1174C>T, PHRED = 20.5). As for Patient 2, NDE1 was included in the Top50 genes, highlighting its role in the network topology. Again, we cannot exclude a possible pathogenic contribution of the microdeletion. On the other hand, it was not possible to significantly define its involvement in any biological pathway as described by GO.
In Patient 4, a male patient with a de novo 16p13.11 microdeletion diagnosed with non-syndromic ASD, we pointed our attention to mutated genes showing dosage sensitivity or LoF intolerance. The patient showed paternal variants in the haploinsufficient gene SRCAP, the core catalytic component of the chromatin remodeling complex. Among LoF intolerant genes, we found MCM6 (OMIM *601806) with paternal heterozygosity and PRR12 (OMIM *616633), GRID1 (OMIM *610659), CPEB4 (OMIM *610607), ANKS1B (OMIM *607815), EIF3G (OMIM *603813) and HECTD4 (not found in OMIM) with maternal heterozygosity. SRCAP, with a missense variant, encodes the core catalytic component of the chromatin-remodeling SRCAP complex. The gene is related to AD Floating-Harbor syndrome (OMIM #136140) (Hood et al., 2012). MCM6, essential for the initiation of eukaryotic genome replication, is related to AD Lactase persistence/non-persistence (OMIM #223100) (Almazar et al., 2019) and has low expression in brain. PRR12 encodes a proline-rich protein nuclear factor associated to neurodevelopmental disorders and intellectual disability (Leduc et al., 2018). No AD diseases are related to this gene. GRID1 encodes a subunit of glutamate receptor channels mediating most of the fast excitatory synaptic transmission in the central nervous system and plays key roles in synaptic plasticity. No AD diseases are related to this gene. CPEB4 has low expression in brain and plays a role in promoting tumor growth and progression. No AD diseases are related to this gene. We cannot exclude a hypothetical damaging role of this gene, known to be associated with autism (SFARI score = 2), in some function of central nervous system. ANKS1B is expressed in all cerebral districts, with a possible role in normal brain development. No AD diseases are related to this gene. EIF3G shows low expression in brain and is required for initiation of protein translation. No AD diseases are related to this gene. HECTD4 encodes HECT Domain E3 ubiquitin-protein ligase; the gene is highly expressed at cerebellar level and is not related to AD diseases. No evidences of cited AD diseases are present in the patient and his parents. The second WES filtering highlighted two paternal SNVs in the ESR1 gene, one of them with a high PHRED score (c.316A>G, PHRED = 25.8). The over-representation analysis of the Top50 network model suggested a single enriched GO, i.e., establishment or maintenance of cell polarity. Among enriched genes, we found the microdeletion gene NDE1, together with genes showing variants with low PHRED scores (DOCK8, DST, FAT1).
Our results suggest that performing WES in presence of 16p13.11 microdeletion is necessary and that the present dual deductive and inductive approach, when applied to other families, will help to clarify the clinical contribution of 16p13.11 microdeletion, when the non-deleted alleles of the genes included in the microdeletion do not show variants.
Conclusion
In conclusion, we have analyzed WES data from four patients carrying the 16p13.11 microdeletion by both a deductive approach and a Systems medicine analysis based on network models. As a whole, we identified several variants potentially involved in the pathogenetic mechanism at the basis of their phenotype. In particular, no additional CNVs were found in the four patients. Accordingly, the low penetrance of the microdeletion and the variability of phenotypes cannot be explained by the presence of other CNVs. From WES analysis, no potential pathogenic variants were found in the non-deleted alleles of genes in 16p13.11 or 16p13.11p12.3 regions, thus excluding unmasked recessive conditions.
Concerning variants identified by WES, we did not identify relevant variants in Patient 1, with a mild phenotype. However, the microdeletion gene NDE1 was found to be one of the enriched genes in the microtubule polymerization or depolymerization pathway, together with DYRK1A, KIF14, and MET, potentially explaining the observed phenotype. We identified a compound heterozygosity in CECR2 in Patient 2, with a severe phenotype. Additionally, two SNVs were identified in MTOR and RICTOR genes, both present in the enriched TOR signaling ontology and central in the network model of Patient 2. We could therefore hypothesize a possible association among the clinical phenotype observed in Patient 2, its genotype and an mTORopathy. Patient 3, with a mild phenotype, showed heterozygous paternal variants in TTN, MET, DOCK8 (all three central genes in the network model of this patient) and MYO9B. Moreover, the network topological analysis led to the identification of compound heterozygous SNVs in the LRRK2 gene. Eventually, paternal heterozygous variants in SRCAP and MCM6 and maternal heterozygous variants in PRR12, GRID1, CPEB4, ANKS1B, EIF3G, and HECTD4 were found in Patient 4, diagnosed with non-syndromic ASD. Furthermore, ESR1, the most central gene in all network models, showed a SNV only in Patient 4, and the single significant GO included the microdeletion gene NDE1.
Concerning the role of 16p13.11 microdeletion in determining the neurodevelopmental phenotype of the four patients, we can conclude that the CNV can be considered a contributing cause variant in patients 1 and 4, since NDE1 is present in both Top50 networks and GO terms. Conversely, it may be considered a candidate factor in patients 2 and 3, since NDE1 is present with high centrality only in the Top50 networks, without being associated to the most significant enriched pathway.
Altogether, we have demonstrated that WES is a fundamental tool in the genetic investigation of patients having a predisposing variant, but this is not sufficient to define the clinical phenotype. It would be interesting to consider additional sequence variants in the gene set recently prioritized by Leblond et al. in the first filtering step (Leblond et al., 2021). Additionally, the analysis of WES data using Systems medicine tools, such as personalized network models, allowed us to prioritize genes on a high throughput scale and to discover variants in genes that were not prioritized at first. Indeed, the dual study approach, classical deductive on WES results and inductive by personalized network models, is a good strategy to clarify, in each patient, the clinical contribution of an uncertain or predisposing CNV. Additionally, uncovering novel enriched pathways associated to a specific phenotype may also provide new insights for personalized therapeutic strategies.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/eva/, European Variation Archive Project: PRJEB41629, Analyses: ERZ4208684 https://decipher.sanger.ac.uk/, 350680 (Patient 1), 414066 (Patient 2), 414067 (Patient 3), and 318359 (Patient 4).
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
Conceptualization: PG, AZ, MF, and RC. Data curation: PG, DC, AZ, and AN. Formal analysis: AZ and MF. Funding acquisition: PG and RC. Investigation: DC, AZ, CP, and AB. Methodology: PG, AZ, MF, and RC. Project administration: PG and RC. Resources: FZ and RC. Softwares: AZ and MF. Supervision: RC. Visualization: AZ and MF. Writing—original draft: PG, AZ, MF, and RC. Writing—review & editing: AN, MF, and RC.
Funding
This research was supported by “la gemma rara” ODV, Varese and in part by “Il Ponte del Sorriso” Foundation, Varese.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully acknowledge the kind cooperation and agreement of patients and their parents.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.798607/full#supplementary-material
References
Alkuraya, F. S., Cai, X., Emery, C., Mochida, G. H., Al-Dosari, M. S., Felie, J. M., et al. (2011). Human Mutations in NDE1 Cause Extreme Microcephaly with Lissencephaly. Am. J. Hum. Genet. 88 (5), 536–547. doi:10.1016/j.ajhg.2011.04.003
Allach El Khattabi, L., Heide, S., Caberg, J. H., Andrieux, J., Doco Fenzy, M., Vincent-Delorme, C., et al. (2020). 16p13.11 Microduplication in 45 New Patients: Refined Clinical Significance and Genotype-Phenotype Correlations. J. Med. Genet. 57 (5), 301–307. doi:10.1136/jmedgenet-2018-105389
Almazar, A. E., Chang, J. Y., Larson, J. J., Atkinson, E. J., Locke, G. R., Talley, N. J., et al. (2019). Comparison of Lactase Variant MCM6 -13910 C>T Testing and Self-Report of Dairy Sensitivity in Patients with Irritable Bowel Syndrome. J. Clin. Gastroenterol. 53 (6), e227–e231. doi:10.1097/mcg.0000000000001065
Balasubramanian, M., Smith, K., Mordekar, S. R., and Parker, M. J. (2011). Clinical Report: An Interstitial Deletion of 16p13.11 Detected by Array CGH in a Patient with Infantile Spasms. Eur. J. Med. Genet. 54 (3), 314–318. doi:10.1016/j.ejmg.2011.01.008
Banting, G. S., Barak, O., Ames, T. M., Burnham, A. C., Kardel, M. D., Cooch, N. S., et al. (2005). CECR2, a Protein Involved in Neurulation, Forms a Novel Chromatin Remodeling Complex with SNF2L. Hum. Mol. Genet. 14 (4), 513–524. doi:10.1093/hmg/ddi048
de Kovel, C. G. F., Trucks, H., Helbig, I., Mefford, H. C., Baker, C., Leu, C., et al. (2010). Recurrent Microdeletions at 15q11.2 and 16p13.11 Predispose to Idiopathic Generalized Epilepsies. Brain 133 (1), 23–32. doi:10.1093/brain/awp262
Hannes, F. D., Sharp, A. J., Mefford, H. C., de Ravel, T., Ruivenkamp, C. A., Breuning, M. H., et al. (2009). Recurrent Reciprocal Deletions and Duplications of 16p13.11: the Deletion Is a Risk Factor for MR/MCA while the Duplication May Be a Rare Benign Variant. J. Med. Genet. 46 (4), 223–232. doi:10.1136/jmg.2007.055202
Heinzen, E. L., Radtke, R. A., Urban, T. J., Cavalleri, G. L., Depondt, C., Need, A. C., et al. (2010). Rare Deletions at 16p13.11 Predispose to a Diverse Spectrum of Sporadic Epilepsy Syndromes. Am. J. Hum. Genet. 86 (5), 707–718. doi:10.1016/j.ajhg.2010.03.018
Hood, R. L., Lines, M. A., Nikkel, S. M., Schwartzentruber, J., Beaulieu, C., Nowaczyk, M. J. M., et al. (2012). Mutations in SRCAP, Encoding SNF2-Related CREBBP Activator Protein, Cause Floating-Harbor Syndrome. Am. J. Hum. Genet. 90 (2), 308–313. doi:10.1016/j.ajhg.2011.12.001
Ingason, A., Rujescu, D., Rujescu, D., Cichon, S., Sigurdsson, E., Sigmundsson, T., et al. (2011). Copy Number Variations of Chromosome 16p13.1 Region Associated with Schizophrenia. Mol. Psychiatry 16 (1), 17–25. doi:10.1038/mp.2009.101
Karalis, V., and Bateup, H. S. (2021). Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 43, 1–16. doi:10.1159/000515672
Kuijpers, M., van de Willige, D., Freal, A., Chazeau, A., Franker, M. A., Hofenk, J., et al. (2016). Dynein Regulator NDEL1 Controls Polarized Cargo Transport at the Axon Initial Segment. Neuron 89 (3), 461–471. doi:10.1016/j.neuron.2016.01.022
Labonne, J. D. J., Driessen, T. M., Harris, M. E., Kong, I.-K., Brakta, S., Theisen, J., et al. (2020). Comparative Genomic Mapping Implicates LRRK2 for Intellectual Disability and Autism at 12q12, and HDHD1, as Well as PNPLA4, for X-Linked Intellectual Disability at Xp22.31. J. Clin. Med. 9 (1), 274. doi:10.3390/jcm9010274
Law, L., Lau, T., Fung, T., Leung, T., Wang, C., and Choy, K. (2009). De Novo 16p13.11 Microdeletion Identified by High-Resolution Array CGH in a Fetus with Increased Nuchal Translucency. BJOG: Int. J. Obstet. Gynaecol. 116 (2), 339–343. doi:10.1111/j.1471-0528.2008.01948.x
Leblond, C. S., Le, T.-L., Malesys, S., Cliquet, F., Tabet, A.-C., Delorme, R., et al. (2021). Operative List of Genes Associated with Autism and Neurodevelopmental Disorders Based on Database Review. Mol. Cell Neurosci. 113, 103623. doi:10.1016/j.mcn.2021.103623
Leduc, M. S., Mcguire, M., Madan-Khetarpal, S., Ortiz, D., Hayflick, S., Keller, K., et al. (2018). De Novo apparent Loss-Of-Function Mutations in PRR12 in Three Patients with Intellectual Disability and Iris Abnormalities. Hum. Genet. 137 (3), 257–264. doi:10.1007/s00439-018-1877-0
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z., and Zhang, B. (2019). WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 47 (W1), W199–W205. doi:10.1093/nar/gkz401
Lv, M., and Ma, Q. (2020). “Autophagy in Neurodevelopmental Disorders,” in Autophagy: Biology and Diseases: Clinical Science. Editor W. Le (Singapore: Springer), 171–182. Advances in Experimental Medicine and Biology. doi:10.1007/978-981-15-4272-5_11
Marsh, D., and Dragich, J. M. (2019). Autophagy in Mammalian Neurodevelopment and Implications for Childhood Neurological Disorders. Neurosci. Lett. 697, 29–33. doi:10.1016/j.neulet.2018.04.017
Mefford, H. C., Muhle, H., Ostertag, P., von Spiczak, S., Buysse, K., Baker, C., et al. (2010). Genome-Wide Copy Number Variation in Epilepsy: Novel Susceptibility Loci in Idiopathic Generalized and Focal Epilepsies. Plos Genet. 6 (5), e1000962. doi:10.1371/journal.pgen.1000962
Monda, J. K., and Cheeseman, I. M. (2018). Nde1 Promotes Diverse Dynein Functions through Differential Interactions and Exhibits an Isoform-specific Proteasome Association. MBoC 29 (19), 2336–2345. doi:10.1091/mbc.e18-07-0418
Nagamani, S. C. S., Erez, A., Bader, P., Lalani, S. R., Scott, D. A., Scaglia, F., et al. (2011). Phenotypic Manifestations of Copy Number Variation in Chromosome 16p13.11. Eur. J. Hum. Genet. 19 (3), 280–286. doi:10.1038/ejhg.2010.184
Nasr, R., Lorendeau, D., Khonkarn, R., Dury, L., Pérès, B., Boumendjel, A., et al. (2020). Molecular Analysis of the Massive GSH Transport Mechanism Mediated by the Human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 10 (1), 7616. doi:10.1038/s41598-020-64400-x
Paciorkowski, A. R., Keppler‐Noreuil, K., Robinson, L., Sullivan, C., Sajan, S., Christian, S. L., et al. (2013). Deletion 16p13.11 Uncovers NDE1 Mutations on the Non‐deleted Homolog and Extends the Spectrum of Severe Microcephaly to Include Fetal Brain Disruption. Am. J. Med. Genet. 161 (7), 1523–1530. doi:10.1002/ajmg.a.35969
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 47 (D1), D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Jackson, L., Miller, D. T., and Van Vooren, S. (2012). Phenotypic Information in Genomic Variant Databases Enhances Clinical Care and Research: The International Standards for Cytogenomic Arrays Consortium Experience. Hum. Mutat. 33 (5), 787–796. doi:10.1002/humu.22052
Rosenfeld, J. A., Coe, B. P., Eichler, E. E., Cuckle, H., and Shaffer, L. G. (2013). Estimates of Penetrance for Recurrent Pathogenic Copy-Number Variations. Genet. Med. 15 (6), 478–481. doi:10.1038/gim.2012.164
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 54 (1), 1.30.1–1.30.33. doi:10.1002/cpbi.5
Su, G., Morris, J. H., Demchak, B., and Bader, G. D. (2014). Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinformatics 47 (1), 8.13.1–24. doi:10.1002/0471250953.bi0813s47
Tan, L., Bi, B., Zhao, P., Cai, X., Wan, C., Shao, J., et al. (2017). Severe Congenital Microcephaly with 16p13.11 Microdeletion Combined with NDE1 Mutation, a Case Report and Literature Review. BMC Med. Genet. 18 (1), 141. doi:10.1186/s12881-017-0501-9
The GTEx Consortium (2020). The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 369 (6509), 1318–1330. doi:10.1126/science.aaz1776
Tropeano, M., Ahn, J. W., Dobson, R. J. B., Breen, G., Rucker, J., Dixit, A., et al. (2013). Male-Biased Autosomal Effect of 16p13.11 Copy Number Variation in Neurodevelopmental Disorders. PLOS ONE 8 (4), e61365. doi:10.1371/journal.pone.0061365
Tropeano, M., Andrieux, J., and Collier, D. A. (2014). Clinical Utility Gene Card for: 16p13.11 Microdeletion Syndrome. Eur. J. Hum. Genet. 22 (5), 713. doi:10.1038/ejhg.2013.230
Ullmann, R., Turner, G., Kirchhoff, M., Chen, W., Tonge, B., Rosenberg, C., et al. (2007). Array CGH Identifies Reciprocal 16p13.1 Duplications and Deletions that Predispose to Autism And/or Mental Retardation. Hum. Mutat. 28 (7), 674–682. doi:10.1002/humu.20546
Xu, S., Zhang, H., Pao, P.-C., Lee, A., Wang, J., Suen Chan, Y., et al. (2020). Exposure to Phthalates Impaired Neurodevelopment through Estrogenic Effects and Induced DNA Damage in Neurons. Aquat. Toxicol. 222, 105469. doi:10.1016/j.aquatox.2020.105469
Keywords: 16p13.11 microdeletion, copy number variants (CNVs), whole exome sequencing (WES), neurodevelopmental disorders, protein-protein interactions (PPIs)
Citation: Granata P, Cocciadiferro D, Zito A, Pessina C, Bassani A, Zambonin F, Novelli A, Fasano M and Casalone R (2022) Whole Exome Sequencing in 16p13.11 Microdeletion Patients Reveals New Variants Through Deductive and Systems Medicine Approaches. Front. Genet. 13:798607. doi: 10.3389/fgene.2022.798607
Received: 08 November 2021; Accepted: 23 February 2022;
Published: 15 March 2022.
Edited by:
Guillaume Huguet, CHU Sainte-Justine, CanadaReviewed by:
Anne Claude Tabet, Hôpital Robert Debré, FrancePatrice Eydoux, University of British Columbia, Canada
Copyright © 2022 Granata, Cocciadiferro, Zito, Pessina, Bassani, Zambonin, Novelli, Fasano and Casalone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Fasano, bWF1cm8uZmFzYW5vQHVuaW5zdWJyaWEuaXQ=; Rosario Casalone, cm9zYXJpby5jYXNhbG9uZUBhc3N0LXNldHRlbGFnaGkuaXQ=