
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 12 April 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.652454
This article is part of the Research TopicCopy Number Variation in Rare DisordersView all 14 articles
 Julián Nevado1,2,3*†
Julián Nevado1,2,3*† Sixto García-Miñaúr1,2,3
Sixto García-Miñaúr1,2,3 María Palomares-Bralo1,2,3Elena Vallespín1,2,3Encarna Guillén-Navarro4Jordi Rosell5
María Palomares-Bralo1,2,3Elena Vallespín1,2,3Encarna Guillén-Navarro4Jordi Rosell5 Cristina Bel-Fenellós6,7María Ángeles Mori1,2,3Montserrat Milá8Miguel del Campo9
Cristina Bel-Fenellós6,7María Ángeles Mori1,2,3Montserrat Milá8Miguel del Campo9 Pilar Barrúz1Fernando Santos-Simarro1,2,3Gabriela Obregón10Carmen Orellana11
Pilar Barrúz1Fernando Santos-Simarro1,2,3Gabriela Obregón10Carmen Orellana11 Harry Pachajoa12Jair Antonio Tenorio1,2,3Enrique Galán13Juan C. Cigudosa14
Harry Pachajoa12Jair Antonio Tenorio1,2,3Enrique Galán13Juan C. Cigudosa14 Angélica Moresco10
Angélica Moresco10 César Saleme15Silvia Castillo16,17
César Saleme15Silvia Castillo16,17 Elisabeth Gabau18
Elisabeth Gabau18 Luis Pérez-Jurado2,19†
Luis Pérez-Jurado2,19† Ana Barcia20Maria Soledad Martín1Elena Mansilla1,2,3
Ana Barcia20Maria Soledad Martín1Elena Mansilla1,2,3 Isabel Vallcorba1,2,3
Isabel Vallcorba1,2,3 Pedro García-Murillo21
Pedro García-Murillo21 Franco Cammarata-Scalisi22
Franco Cammarata-Scalisi22 Natálya Gonçalves Pereira23Raquel Blanco-Lago24Mercedes Serrano25
Natálya Gonçalves Pereira23Raquel Blanco-Lago24Mercedes Serrano25 Juan Dario Ortigoza-Escobar25Blanca Gener26
Juan Dario Ortigoza-Escobar25Blanca Gener26 Verónica Adriana Seidel27Pilar Tirado28
Verónica Adriana Seidel27Pilar Tirado28 Pablo Lapunzina1,2,3† and Spanish PMS Working Group1Instituto de Genética Médica y Molecular (INGEMM)-IdiPAZ Hospital Universitario La Paz MadridSpain2CIBERER Centro de Investigación Biomédica en Red de Enfermedades Raras ISCIII MadridSpain3ITHACA-European Reference Network Hospital La Paz MadridSpain4Hospital Virgen de la Arrixaca MurciaSpain5Hospital Son Espases Palma de MallorcaSpain6Departamento de Investigación y Psicología en Educación Facultad de Educación UCM MadridSpain7CEE Estudio-3 Afanias MadridSpain8Hospital Clinic BarcelonaSpain9Hospital Vall D’Hebron BarcelonaSpain10Hospìtal Juan P. Garrahan Buenos AiresArgentina11Hospital La Fé ValenciaSpain12Universidad Icesi CaliColombia13Hospital Materno-Infantil Infanta Cristina BadajozSpain14NIM-Genetics Madrid AlcobendasSpain15Maternity Nuestra Señora de la Merced TucumánArgentina16Sección Genética Hospital Clínico Universidad de Chile SantiagoChile17Clínica Alemana SantiagoChile18Corporación Sanitaria Parc Taulí BarcelonaSpain19Servicio de Genética Instituto de Investigaciones Médicas Hospital del Mar (IMIM)/Universitat Pompeu Fabra BarcelonaSpain20Hospital Universitario Virgen del Rocío SevillaSpain21Unidad de Genética Hospital Virgen de la Salud ToledoSpain22Servicio de Pediatría Hospital Regional de Antofagasta AntofagastaChile23Clínica Reprodutiva NIDUS Juiz de ForaBrasil24Servicio de Neuropediatría Hospital Universitario Central de Asturias Oviedo (Asturias)Spain25Unidad de Neuropediatría Hospital San Joan de Deu BarcelonaSpain26Hospital Universitario de Cruces BilbaoSpain27Servicio de Genética Clínica Hospital Universitario Gregorio Marañon MadridSpain28Servicio de Neuropediatría Hospital Universitario La Paz MadridSpainPhelan-McDermid syndrome (PMS OMIM# 606232) results from either different rearrangements at the distal region of the long arm of chromosome 22 (22q13.3) or pathogenic sequence variants in the SHANK3 gene. SHANK3 codes for a structural protein that plays a central role in the formation of the postsynaptic terminals and the maintenance of synaptic structures. Clinically patients with PMS often present with global developmental delay absent or severely delayed speech neonatal hypotonia minor dysmorphic features and autism spectrum disorders (ASD) among other findings. Here we describe a cohort of 210 patients with genetically confirmed PMS. We observed multiple variant types including a significant number of small deletions (<0.5 Mb 64/189) SHANK3 sequence variants (21 cases). We also detected multiple types of rearrangements among microdeletion cases including a significant number with post-zygotic mosaicism (9.0% 17/189) ring chromosome 22 (10.6% 20/189) unbalanced translocations (de novo or inherited 6.4%) and additional rearrangements at 22q13 (6.3% 12/189) as well as other copy number variations in other chromosomes unrelated to 22q deletions (14.8% 28/189). We compared the clinical and genetic characteristics among patients with different sizes of deletions and with SHANK3 variants. Our findings suggest that SHANK3 plays an important role in this syndrome but is probably not uniquely responsible for all the spectrum features in PMS. We emphasize that only an adequate combination of different molecular and cytogenetic approaches allows an accurate genetic diagnosis in PMS patients. Thus a diagnostic algorithm is proposed.
Pablo Lapunzina1,2,3† and Spanish PMS Working Group1Instituto de Genética Médica y Molecular (INGEMM)-IdiPAZ Hospital Universitario La Paz MadridSpain2CIBERER Centro de Investigación Biomédica en Red de Enfermedades Raras ISCIII MadridSpain3ITHACA-European Reference Network Hospital La Paz MadridSpain4Hospital Virgen de la Arrixaca MurciaSpain5Hospital Son Espases Palma de MallorcaSpain6Departamento de Investigación y Psicología en Educación Facultad de Educación UCM MadridSpain7CEE Estudio-3 Afanias MadridSpain8Hospital Clinic BarcelonaSpain9Hospital Vall D’Hebron BarcelonaSpain10Hospìtal Juan P. Garrahan Buenos AiresArgentina11Hospital La Fé ValenciaSpain12Universidad Icesi CaliColombia13Hospital Materno-Infantil Infanta Cristina BadajozSpain14NIM-Genetics Madrid AlcobendasSpain15Maternity Nuestra Señora de la Merced TucumánArgentina16Sección Genética Hospital Clínico Universidad de Chile SantiagoChile17Clínica Alemana SantiagoChile18Corporación Sanitaria Parc Taulí BarcelonaSpain19Servicio de Genética Instituto de Investigaciones Médicas Hospital del Mar (IMIM)/Universitat Pompeu Fabra BarcelonaSpain20Hospital Universitario Virgen del Rocío SevillaSpain21Unidad de Genética Hospital Virgen de la Salud ToledoSpain22Servicio de Pediatría Hospital Regional de Antofagasta AntofagastaChile23Clínica Reprodutiva NIDUS Juiz de ForaBrasil24Servicio de Neuropediatría Hospital Universitario Central de Asturias Oviedo (Asturias)Spain25Unidad de Neuropediatría Hospital San Joan de Deu BarcelonaSpain26Hospital Universitario de Cruces BilbaoSpain27Servicio de Genética Clínica Hospital Universitario Gregorio Marañon MadridSpain28Servicio de Neuropediatría Hospital Universitario La Paz MadridSpainPhelan-McDermid syndrome (PMS OMIM# 606232) results from either different rearrangements at the distal region of the long arm of chromosome 22 (22q13.3) or pathogenic sequence variants in the SHANK3 gene. SHANK3 codes for a structural protein that plays a central role in the formation of the postsynaptic terminals and the maintenance of synaptic structures. Clinically patients with PMS often present with global developmental delay absent or severely delayed speech neonatal hypotonia minor dysmorphic features and autism spectrum disorders (ASD) among other findings. Here we describe a cohort of 210 patients with genetically confirmed PMS. We observed multiple variant types including a significant number of small deletions (<0.5 Mb 64/189) SHANK3 sequence variants (21 cases). We also detected multiple types of rearrangements among microdeletion cases including a significant number with post-zygotic mosaicism (9.0% 17/189) ring chromosome 22 (10.6% 20/189) unbalanced translocations (de novo or inherited 6.4%) and additional rearrangements at 22q13 (6.3% 12/189) as well as other copy number variations in other chromosomes unrelated to 22q deletions (14.8% 28/189). We compared the clinical and genetic characteristics among patients with different sizes of deletions and with SHANK3 variants. Our findings suggest that SHANK3 plays an important role in this syndrome but is probably not uniquely responsible for all the spectrum features in PMS. We emphasize that only an adequate combination of different molecular and cytogenetic approaches allows an accurate genetic diagnosis in PMS patients. Thus a diagnostic algorithm is proposed.In the past 15–20 years, the increasing use of genome-wide telomere screening by fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA, Schouten et al., 2002), and more recently chromosome microarrays (CMA) has provided evidence of the presence of subtle abnormalities involving telomeres in around 5% (range, 2%–30%) of patients with intellectual disability (ID) (Anderlid et al., 2002a; Shao et al., 2008). In the evaluation of ID patients, deletion of 22q13.3, also known as Phelan-McDermid syndrome (PMS; OMIM#:606232), is one of the most common subtelomeric deletions after 1p36.3 deletion syndrome (Heilstedt et al., 2003; Delahaye et al., 2009). PMS usually results from either the loss of genetic material at the distal region of the long arm of chromosome 22 (including SHANK3) or pathogenic sequence variants in SHANK3.
SHANK3 plays a central role in forming the postsynaptic environment, integrating the protein network of glutamate receptors at postsynaptic density and the maintenance of synaptic structures (Boeckers, 2006; Durand et al., 2007). Deletion sizes vary considerably among PMS individuals, ranging from intragenic deletions in the SHANK3 gene (∼13 Kb) to around 9 Mb (Bonaglia et al., 2011; Phelan et al., 2018). The deletion occurs with similar frequency in male and female. SHANK3 haploinsufficiency is proposed to be responsible for the major neurological features of the 22q13 deletion syndrome (Bonaglia M. C. et al., 2001; Anderlid et al., 2002b; Wilson et al., 2003; Durand et al., 2007; Phelan et al., 2018) and recently has also been shown to be involved in additional clinical features of the syndrome in humans (De Rubeis et al., 2018) and mice (Sauer et al., 2019). However, interstitial deletions disrupting the 22q13.3 band, not including SHANK3 (Wilson et al., 2008; Disciglio et al., 2014; Ha et al., 2017), are also reported. The clinical features in these patients overlap those of PMS, raising debate about whether they can be diagnosed as having PMS.
Although many PMS patients have been diagnosed worldwide, most of the individuals included in previous genotype-phenotype analyses had microdeletions (Cusmano-Ozog et al., 2007; Dhar et al., 2010; Sarasua et al., 2011; Soorya et al., 2013; Sarasua et al., 2014a,b; Tabet et al., 2017; Samogy-Costa et al., 2019). Indeed, the proportion of patients with SHANK3 variants in previous data is 3%–25% (Phelan et al., 2018; De Rubeis et al., 2018; and ClinVar, Varsome, LOVD databases) or 8.6% in the PMS International Registry (among genetically confirmed cases; Kolevzon et al., 2019). Thus, PMS seems to be underdiagnosed, and its exact prevalence in is unknown.
Here, we describe the clinical and molecular data of one of the largest cohorts of patients with confirmed genetic diagnosis of PMS, most of them with microdeletions (189/210, 90%) and 21 with SHANK3 sequence variants (10%). High-resolution CMA, cytogenetic, and MLPA techniques were necessary to delineate the size and gene content of the deletions and to identify additional rearrangements. Exome and/or target panel sequencing analysis of SHANK3 were preferentially applied for SHANK3 sequence variant analysis.
Between 2008 and 2020, 242 patients with confirmed PMS, mostly nonrelated (except for four individuals from two families), were recruited for this study in collaboration with the Spanish PMS Association and the Argentinean PMS Group. Twenty-eight of these had incomplete clinical or molecular data and were not included in this study. Three were excluded because they carried deletions at 22q13.33 nearby to SHANK3 but not including this gene, and one had an intragenic SHANK3 duplication and was also excluded because, at this time, we are not able to confirm that the duplication is in tandem and disrupts SHANK3. Thus, 210 individuals constituted the final cohort (Supplementary Figure S1).
Most of the DNA samples from these patients were extracted and analyzed at INGEMM (Madrid, Spain). A minority of them had been previously analyzed outside of our institution by high-resolution CMA or next generation sequencing (NGS). The patients’ clinical information was obtained from the referring physicians and/or their clinical geneticists and compiled in two questionnaires. Data were completed by reviewing medical records and parents’ interviews. Parents or guardians provided informed consent. The Institutional Review Board of Hospital Universitario La Paz approved the study (PI: 2735 HULP, Madrid. Spain).
Cytogenetic analyses were performed on GTG-banded metaphases at a resolution of about 550 bands according to standard laboratory protocols using Chromosome Kit P (Euroclone, Siziano PV, Italy). FISH was performed according to standard laboratory protocols using the subtelomeric 22q13 probe (D22S1056, Kreatech Biotechnology B.V, Amsterdam, Netherlands) or the DiGeorge/VCFS probe mixture (Vysis Inc., IL, United States), containing a control probe in ARSA that maps to the 22q13.3 region. In some cases, the probe N25/N85A3 (Cytocell, Cambridge, United Kingdom) within the SHANK3 locus was also used.
We used highly polymorphic short tandem repeats (D22S1169, D22S1149, D22S444, D22S1170, D22S295, and D22S1141) mapping within the SHANK3 gene and around it to evaluate parental segregation. The forward primers were synthesized and labeled with fluorescein-amidite (Sigma-Aldrich, St. Louis, MO, United States), whereas the reverse primers were not labeled (primer sequences are available upon request). The region amplified by these primers depended on the number of repeats. Capillary electrophoresis (Applied Biosystems Genetic Analyzer System 3130) was used to detect the length of the fragments (Thermo Fisher, CA, United States).
We used several MLPA-Salsa kits in this study (MRC-Holland, Amsterdam, Netherlands). For patients referred to rule out subtelomeric rearrangements in the first years of the study, MLPA kits P036 and P070 were used. DNA samples of all patients with 22q13 deletions were further characterized with the specific MLPA P188 and P339 probe mixes for PMS (MRC-Holland). Both kits contain 34 sequence probes on chromosome 22q13 and control ones for other chromosomes (12 and 9, respectively). The majority of the 22q13 probes (22/34) are in the 1 Mb terminal region of the long arm (P188) and include multiple probes within SHANK3 (P339). Data analyses were performed according to the protocols supplied by the providers defining relative probe signals by dividing each measured peak area by the sum of all peak areas of the control probes of that sample. Each peak’s relative probe area ratio was then compared to a DNA control sample (Promega, United Kingdom), using Coffalysser.net (MRC- Holland).
Different array platforms were used in this study: 1) a clinical 60K-array CGH (INGEMM, KaryoArray-®, Vallespin et al., 2013) in 72 of 189 patients; 2) a high-resolution customized- 60K aCGH (INGEMM custom design, not published) at 22q13.3 in 30 of 189 patients; 3) different custom or commercial CGH-microarrays with a variety of resolutions in 59 patients (Supplementary Figure S2A); 4) a genome-wide scan of 850,00 tag SNPs (Illumina Infinium CytoSNP-850K BeadChip) in 56 patients (Supplemental Data, Supplementary Figure 2B) at INGEMM; and 5) a genome-wide scan of 750,00 tag SNPs (Affymetrix, ThermoFisher Scientific, Waltham, MA, United States) in 11 patients. Arrays in 1–3 were analyzed with Cytogenomics software (Agilent Corporation; Santa Clara, CA, United States). Image data from 4 were analyzed using the Chromosome Viewer tool contained in the Genome Studio package (Illumina, San Diego, CA, United States). In Chromosome Viewer, gene call scores <0.15 at any locus were considered “no calls.” In addition, allele frequency analysis was applied for all SNPs. For the analysis of 5, the ChAS software (Affymetrix, Thermo-Fisher Scientific, Waltham, MA, United States) was used.
All genomic coordinates were established according to the 2009 human genome build 19 (GRCh37/NCBI build 37.1). Deletion coordinates were plotted using the University of California at Santa Cruz Genome Browser (http://genome.ucsc.edu/).
These studies were performed either at INGEMM or outside of our institution, using different NGS approaches, all under the manufacturer’s guidelines: 1) exome sequencing by trio analysis using the Agilent SureSelect XT clinical research exome (Agilent Tech) and IDT Technologies (Coralville, IA, United States); 2) singleton exome sequencing CentoXome Gold®, and NOVAGENE (Agilent all exon V6) and MedExome, Q-Genomics (Barcelona, Spain); and 3) a customized gene panel of specific genes related to ID or/and autism (Agilent-based Technologies). Most samples (98%) were run in Illumina instruments (such as Nextseq500; Miseq, Hiseq 2000/4000; Illumina, San Diego, CA, United States). Classification of the variants follows ACMG/AMP criteria (Richards et al., 2015), using VarSome 10.2 as a web source.
We estimated an individual severity score in our cohort using different features taken from the questionnaires and weighed them by Human Ontology Phenotype (HPO) term frequencies on a numerical scale of core features of the syndrome. The GFAP was constructed as follows: items with a frequency between 0% and 20%, 1 point; between 20% and 35%, 2 points; between 35% and 70%, 5 points, and >70%, 10 points. Principal component analysis (PCA) was used to validate the GFAP construct, containing Kaiser-Meyer-Olkin’s measure and Barlett’s test.
Statistical analyses were performed with SPSS version 25 (IBM Corporation, Chicago, IL, United States). Descriptive analysis included mean ± SD for continuous variables and frequency tables for categorical variables (Table 1). The categorical variables were taken from our two questionnaires curated from medical records and were expressed as “1” (condition present at some point) or “0” (condition not present at any time). Correlation associations were calculated using Pearson’s linear correlation coefficient (continuous variables) or Spearman’s Rho and Kendall’s tau_b (categorical variables). Comparisons between two groups were performed either with Student’s t-test (for continuous variables) or chi-square tests (for categorical ones). For more than two groups, ANOVA (followed by Bonferroni’s or T3-Dunnett post hoc tests) were run for continuous variables and z-tests between column proportions for categorical variables. Ward’s minimum variance method was the criterion used in hierarchical cluster analysis, and the number of clusters was selected using the Bayesian information criterion (BIC) or Akaike information criterion (AIC). A p-value lower than .05 was considered to indicate a statistically significant difference.
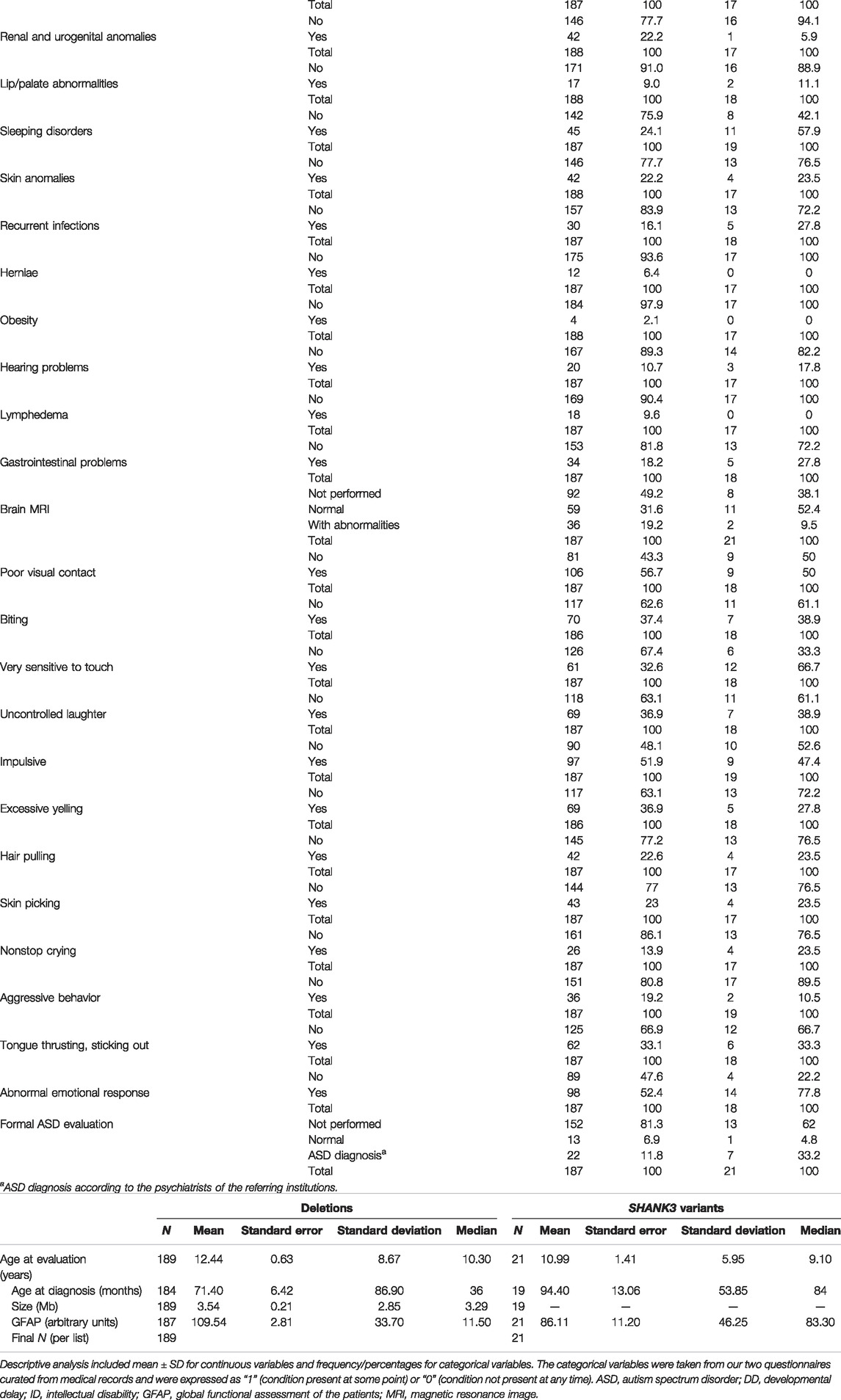
TABLE 1. Descriptive statistics and frequencies of variables used in the study of 22q13.3 microdeletions and SHANK3 variants.a) Categorical variables
Individuals (n = 210), all previously nonreported, are mostly from Spain, all over the country (n = 178), and from South America (n = 32), mainly from Argentina (Supplementary Figure S1). The female/male ratio, 1.12:1 (111/99), was similar to previous reports, and ages ranged from birth to 62 years. Descriptive statistics (for continuous variables) and frequencies (for categorical items) are shown in Table 1. The majority of individuals with PMS in our cohort are of pediatric age (between 0 and 16 years old, 146 patients; 69.5%). The mean age at diagnosis was around 6 years old for deletions (Table 1b) and around 8 years for the group with sequence variants in SHANK3. The mean age at evaluation were 12.44 ± 8.7 years and 10.99 ± 5.95 years for deletions and SHANK3 sequence variants, respectively (Table 1b).
The clinical features observed in this cohort by weighed-HPO terms are listed in Table 2a for 22q13.3 microdeletions. Table 2a also shows the frequencies of clinical features observed in other representative studies with deletion cases (Sarasua et al., 2014a; Tabet et al., 2017, Samogy-Costa et al., 2019). Table 2b shows the frequencies of clinical features observed in patients with SHANK3 variants, and data from De Rubeis et al. (2018) and other previously published cases (Gauthier et al., 2009; Boccuto et al., 2013; Leblond et al., 2014; O’Roak et al., 2014; Bramswig et al., 2015; Nemirovsky et al., 2015; Zhang et al., 2015; Holder & Quach 2016; Bowling et al., 2017; Lim et al., 2017; Yuen et al., 2017).
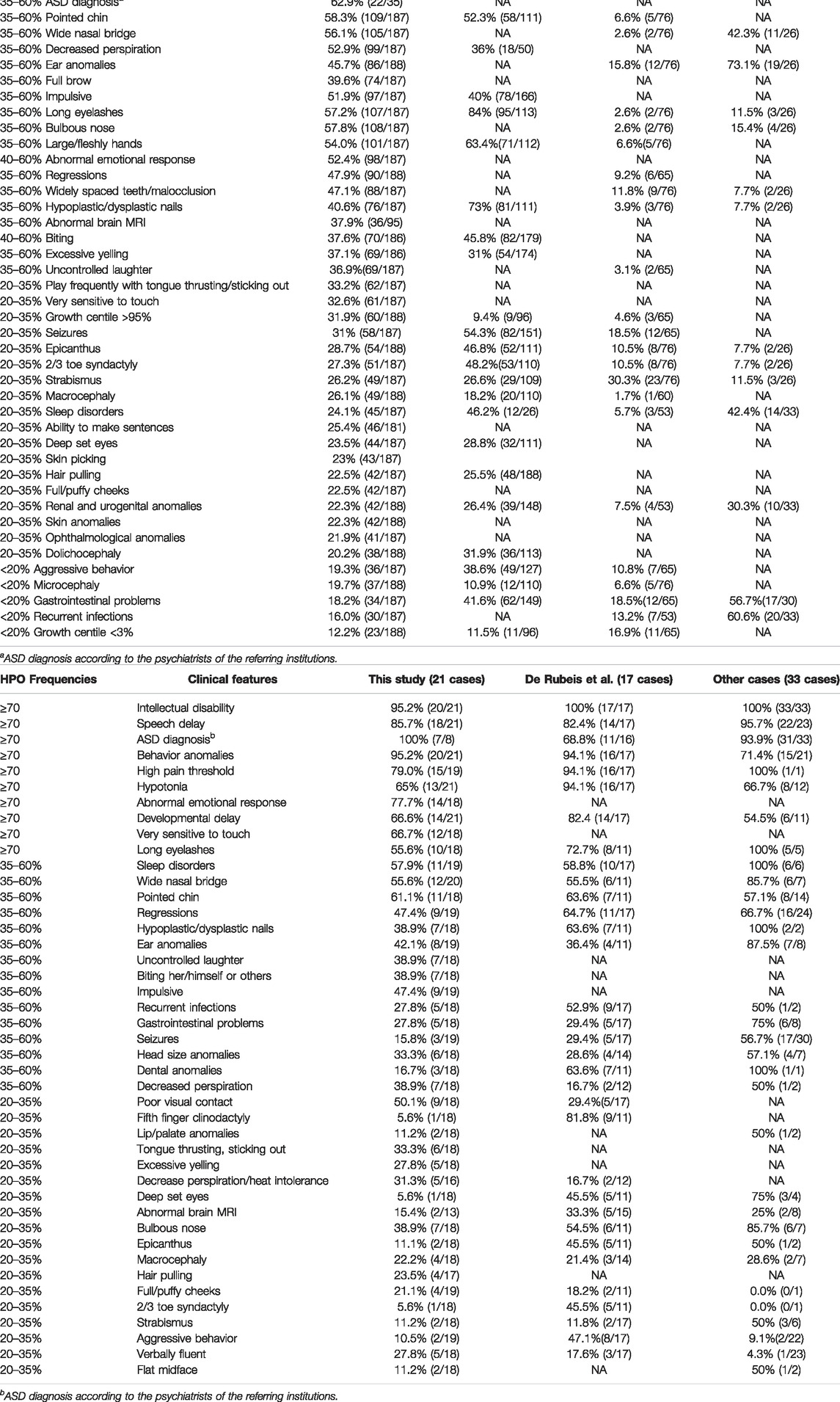
TABLE 2. Frequency of clinical features observed in this cohort.a) Microdeletions at 22q13.3
Figure 1 shows that facial features are neither typical nor specific for PMS. Patients presented a high degree of facial variability even among individuals with similar deletion size. Significant facial differences can be observed when comparing bigger deletions (>5 Mb) with either small deletions (≤0.5 Mb) or sequence variants in SHANK3 (Figure 1). Facial features such as bulbous nose, pointed chin, ear anomalies, full eyebrows, long eyelashes, and wide nasal bridge were observed in around 35%–80% of the individuals (Table 2a). These facial features, together with hypotonia, high pain threshold, developmental delay, speech delay, ID, behavior abnormalities, large/fleshly hands, hypoplastic/dysplastic nails, decreased perspiration, and ASD, should be considered as core features of this syndrome (at least in patients with microdeletions; Table 2a). On the other hand, patients with variants in SHANK3 seemed to have fewer dysmorphic features than patients with microdeletions (Figure 1 and Table 2b).

FIGURE 1. Facial views of individuals with PMS with 22q13.3 deletions or SHANK3 sequence variants.
Interestingly, many of these core features seem to be inter-related among them. Significant positive correlations were observed when Kendall’s tau_b analysis was performed between categorical variables (Supplementary Table S1). An example with three of these categorical variables is schematized in Figure 2.
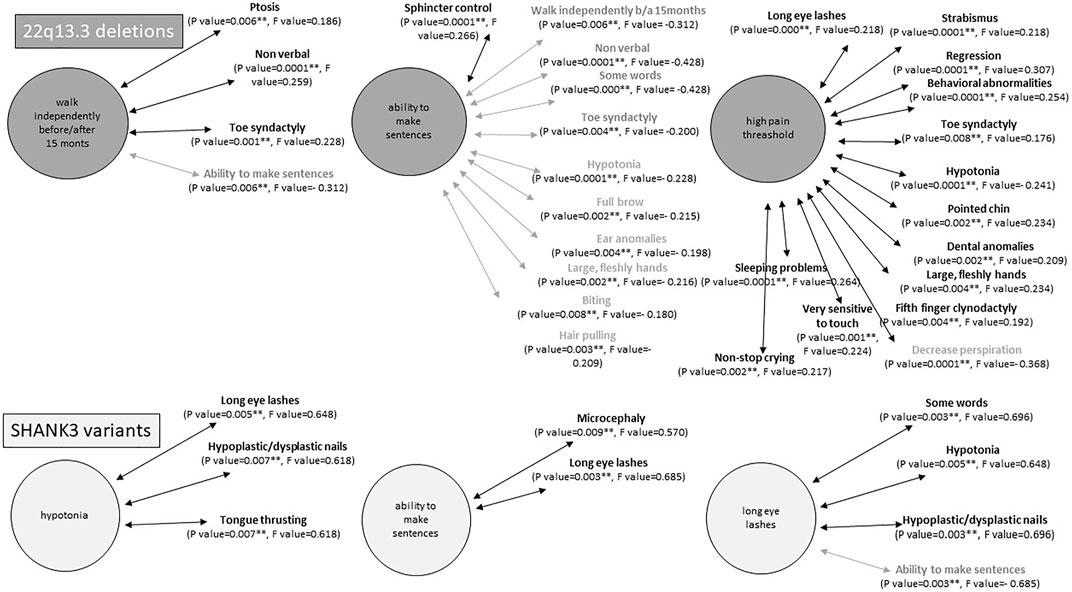
FIGURE 2. Examples of statistically significant correlations (p < 0.001) between intercategorical variables in individuals with 22q13.3 deletions (top) or SHANK3 variants (bottom). Statistical analyses were performed using Kendal tau_b correlation coefficient. In bold, positive correlations and in gray negative correlations.
Brain MRI studies were performed in 51% (95/187) of individuals in the microdeletion group and 62% (13/21) in the SHANK3 sequence variant group with abnormal findings found in 38% (36/95) and 15% (2/13), respectively (Table 1a). Abnormal findings included hypoplasia/atrophy of the cerebellar vermis, abnormalities of the corpus callosum (ranging from thinness to agenesis or dysgenesis), abnormalities of the white matter, arachnoid cysts, and hydrocephalus. We also found other abnormalities, such as ventriculomegaly, enlarged cisterna-magna and vermis, prominent metopic suture, cerebral dysplasia with lateral ventricular dilatation, and frontal cerebral hypertrophy.
Speech abilities (evaluated only in patients ≥3 years old; n = 199/210, 94.8%) showed severe abnormalities in most of the patients evaluated (148/199, 74.4%). Thirty-five percent of patients (70/199) had no speech at all, around 39% (78/199) had an elementary vocabulary of 10 words or less, and around 26% (51/199) were reported to have a significant vocabulary and the ability to use limited phrases for a short and comprehensible conversation (Table 1a). Table 1 segregates the numbers by deletions and SHANK3 variants. Remarkably, most of the verbally fluent individuals in the microdeletion group have small deletions.
The main reason for referral to a genetic consultation in patients with microdeletions was developmental delay, whereas in individuals with sequence variants, ASD and language delay were the most frequent reasons for referral (Figure 3 and Table 1). Similarly, ASD and delayed or absent speech were the main cause of genetic consultation among patients with smaller deletions (≤0.25 Mb). We compared these groups by Chi-square test and z-test (post hoc, corrected by Bonferroni). We choose 0.25 Mb as the size of the deletions with the minimal telomeric lost segment, including the SHANK3 gene. The chi-square test revealed differences between groups constituted by large deletions (>0.25 Mb, 153 cases), small deletions (<0.25 Mb, 36 cases), and SHANK3 variants (21 cases) for the first- and second-main reasons for referral to genetic consultation (p = .0001, F = 43.491 and p = .0001, F = 37.491, respectively). These differences were mainly observed between deletions >0.25 Mb and both smaller deletions and variants in SHANK3 (Figure 3). In addition, hypotonia and dysmorphic features were the main reasons for referral in individuals with medium-size deletions (2.5–5.0 Mb). In patients with deletions ≥5 Mb, the main reason for genetic consultation was severe ID and developmental delay with other severe comorbidities (data not shown).
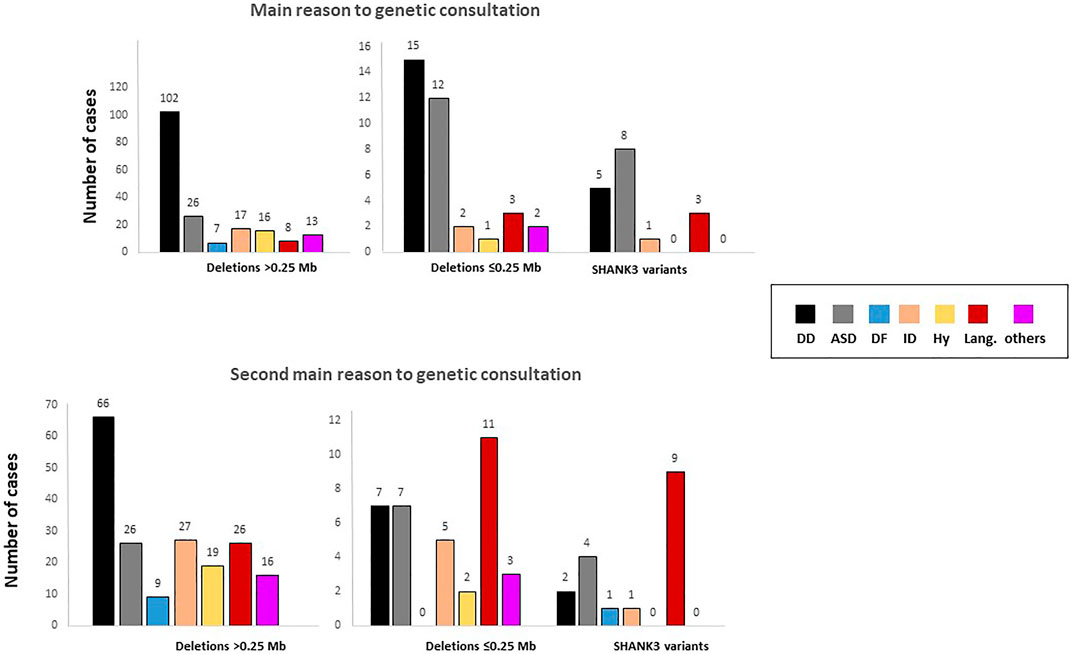
FIGURE 3. Reasons for referral for genetic evaluation stratified according to the type of genetic defect. Analyses were performed by one-way ANOVA. DD, developmental delay; ASD, autism spectrum disorder; DF, dysmorphic features; ID, intellectual disability; Hy, hypotonia; Lang., language.
We applied different CMA platforms and MLPA approaches to confirm and establish the size of the deletions. Figure 4 illustrates the need to use MLPA for a complete characterization of patients with deletions. This is explained by the lack of probes at the end of the 22q13.33 band in commercial microarrays versus customized microarrays (Supplementary Figure S2). A compilation of additional examples is shown in Supplementary Figure S3.
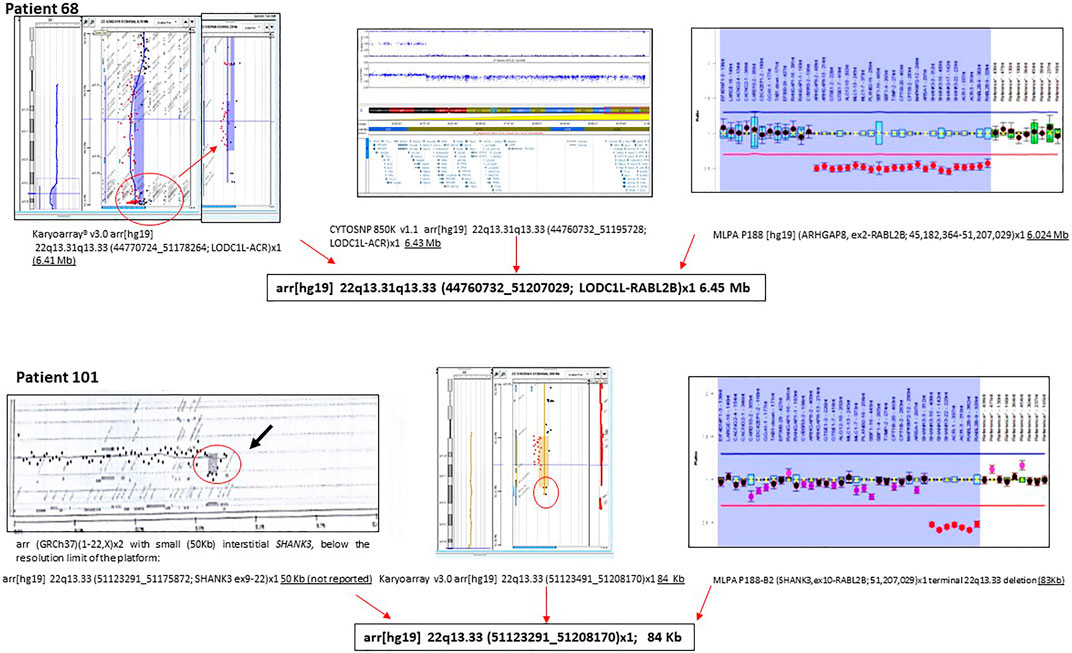
FIGURE 4. Examples of molecular characterization of two individuals with PMS. Different molecular approaches were used, including CGH-array, SNP array, and MLPA.
One-hundred eighty-nine out of 210 individuals carried deletions at 22q13.3 (90%), all of them including SHANK3 (Table 3). Table 3 also summarizes how the different genomic rearrangements were distributed in the cohort. The number of individuals with ring chromosome 22 (r(22), 20 cases), post-zygotic mosaicism (17 cases), or additional genomic rearrangements (40 cases, including variants of uncertain significance (VUS) and clinically relevant variants in other chromosomes as well as 12 cases with other rearrangements at chromosome 22), is remarkable. Supplementary Table S2 shows the genomic coordinates of the 22q13 deletions and other CNVs identified in the cohort. The mean 22q13 deletion size was 3.52 ± 2.83 Mb (median: 3.29 Mb), ranging from 12 Kb within the last exon of SHANK3 (individual 51) to 10.30 Mb (individual 170) from the telomere. To our knowledge, the latter is the largest deletion reported so far and was likely not lethal because it is in mosaic form. Cytogenetic data of most of these individuals are shown in Supplementary Table S2.
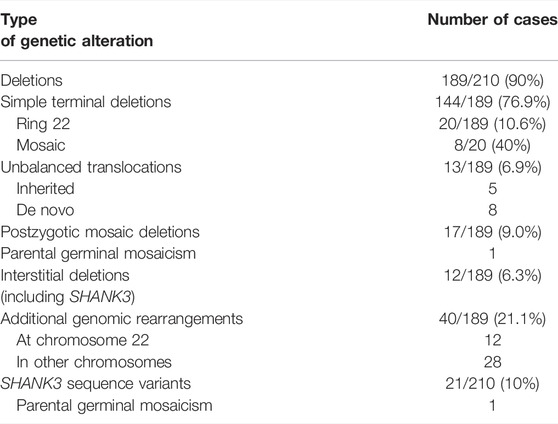
TABLE 3. Summary of genetic findings from the cohort.
The use of combined SNP arrays and MLPA allowed finding different degrees of post-zygotic mosaicism in microdeletion cases. We found 17 patients with mosaicism ranging from 10% to 82% (Figure 5). In addition, the finding of two siblings with the same deletion (a 48 Kb-interstitial microdeletion with breakpoints within genes SHANK3 and RABL2B, Supplementary Figure S4) suggests parental germinal mosaicism, which was later confirmed as paternal after haplotype analysis using SNP arrays (CytoScan 850K, Illumina).
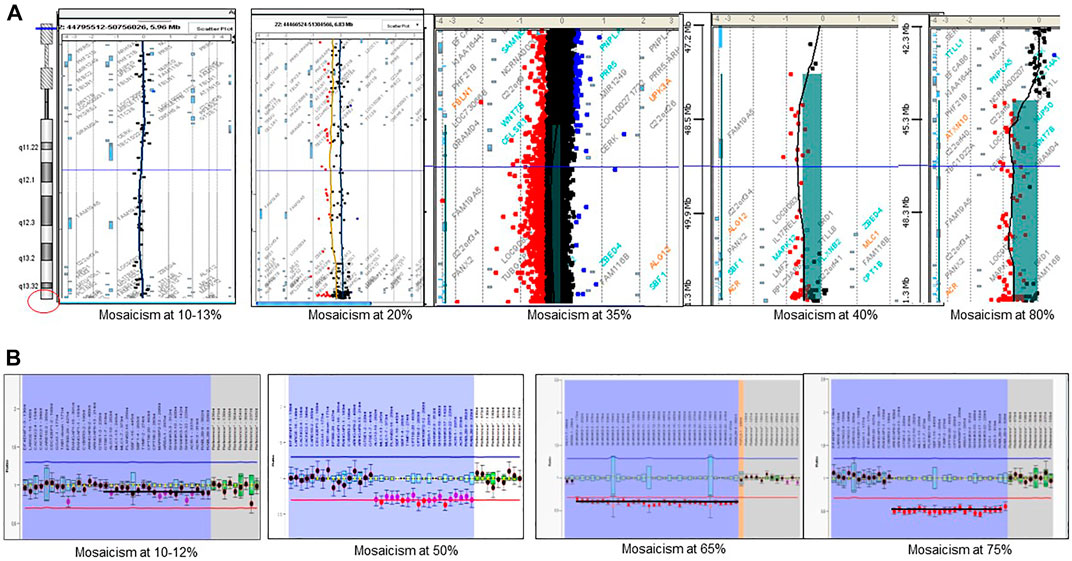
FIGURE 5. Detection of post-zygotic mosaicism in PMS by using microarrays and MLPA. (A) examples of mosaicism detected by CGH-array; (B) examples of mosaicism detected by MLPA.
Breakpoint analyses showed a recurrent 5′breakpoint hot spot, apparently the same described by Bonaglia M. C. et al. (2001). We observed a similar breakpoint in 22 individuals with smaller deletions (coordinates 51123505 to telomere, GCRh37, Supplementary Figure S5). This region is rich in SINEs and LINEs, such as Alu sequences, which could be involved in causing these rearrangements by various mechanisms (Bonaglia et al., 2011; Cooper et al., 2011; Oberman et al., 2015). Our data also point out two additional 3′ recurrent breakpoints (Supplementary Figure S5), which are also extremely rich in Alu sequences. The first recurrent breakpoint was located between coordinates 51146663 and 51175872 (GCRh37; patients 94, 99, and 117) and the second one was located between intron 19 and the end of the last exon of SHANK3 (NM_001372044.1; patients 31, 57, 75, and 77). Both hypothetical breakpoints were close to the one predicted in a patient reported by Bonaglia M. C. et al. (2001). Additional cases are needed to confirm these new hot spot breakpoints.
We tested six highly polymorphic short tandem repeats (STR) to identify the parental origin of the deleted chromosome in 86 trios. In 35 cases (40.7%), the results were noninformative. Among 51 trios with informative findings, we found that deletions originated from the paternally inherited chromosome in 76.5% (39/51) and the maternally inherited chromosome in 23.5% of cases (12/51).
In this cohort, we also evaluated 21 patients (10%) carrying SHANK3 variants (Table 4). All of them were de novo; 19 variants were within the penultimate exon (NM_001372044.2), one affected the canonical splicing site at exon 24, and one was located in exon 20. There were 17 frameshift, one nonsense, one splice site, and two missense variants. Some of the variants (Table 4) have been previously described in public databases (ClinVar, LOVD, Varsome) and several publications and are recurrent in our patients (Leblond et al., 2014; Bramswig et al., 2015; Holder & Quach 2016; Thevenon et al., 2016; Yuen et al., 2017; De Rubeis et al., 2018; Zhou et al., 2019; Kaplanis et al., 2020; Feliciano et al., 2019; Lelieveld et al., 2016; Retterer et al., 2016; O´Roak et al., 2014; Farwell et al., 2015; Durand et al., 2007), suggesting several hot spots for de novo variants.
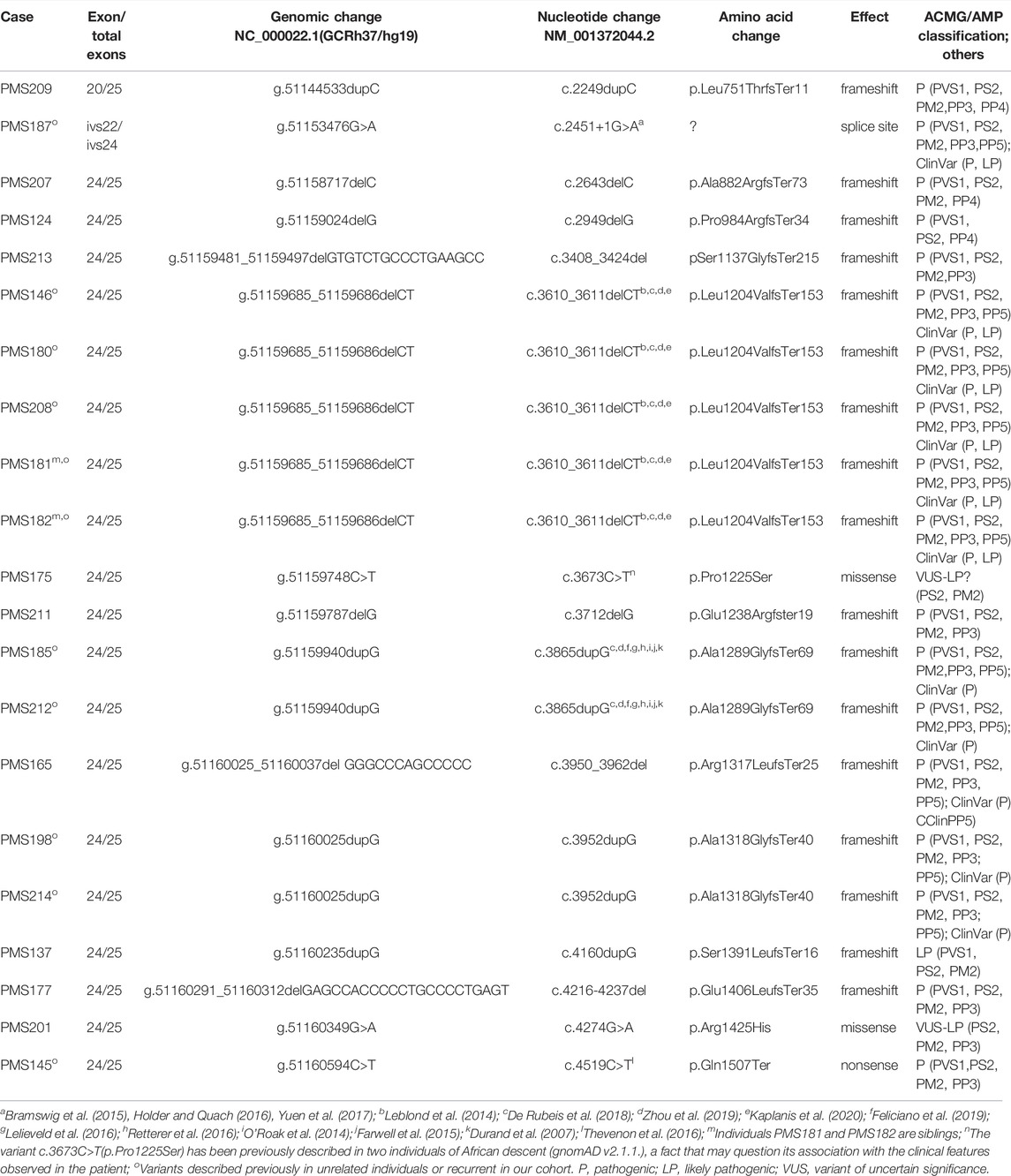
TABLE 4. SHANK3 sequence variants identified in this study.
The interpretation of these two missense variants within SHANK3 remains difficult (Table 4). We classified them as VUS-likely pathogenic by following ACMG/AMP criteria based on de novo condition, the individuals’ clinical features, their absence in European non-Finnish population in gnomAD, the domain of the protein affected, in silico pathogenicity scores, and its medium-high level of conservation position in the evolution. However, the missense SHANK3 variant c.3673C>T(p.Pro1225Ser) was observed in two independent individuals of African descent (total allele frequency 7 × 10−6; gnomAD v2.1.1), a finding that may question its association with the clinical features observed in the patient.
Finally, the presence of the same SHANK3 variant in male monochorionic dizygotic twins suggested potential gonadal mosaicism in one of the parents (data not shown). Haplotype analysis using SNP array suggested a paternal origin of the variant. We also have the suspicion for another case with parental mosaicism in a family with two affected twins.
The significant clinical and genetic heterogeneity observed in patients with PMS suggests the type of genetic defect modulates the clinical features. Thus, we propose a numerical score of the GFAP, constructing a continuous variable based on a prioritization array of different “core” clinical weighted-HPO items (see Methods). These variables were based on comorbidity items, developmental delay, speech delay, dysmorphic features, and behavior items. Figure 6A shows the median values for GFAP for the whole cohort and different types of genetic defects. Figures 6B–D shows median values for other continuous variables (age at diagnosis and evaluation and size of deletions) in the different groups.
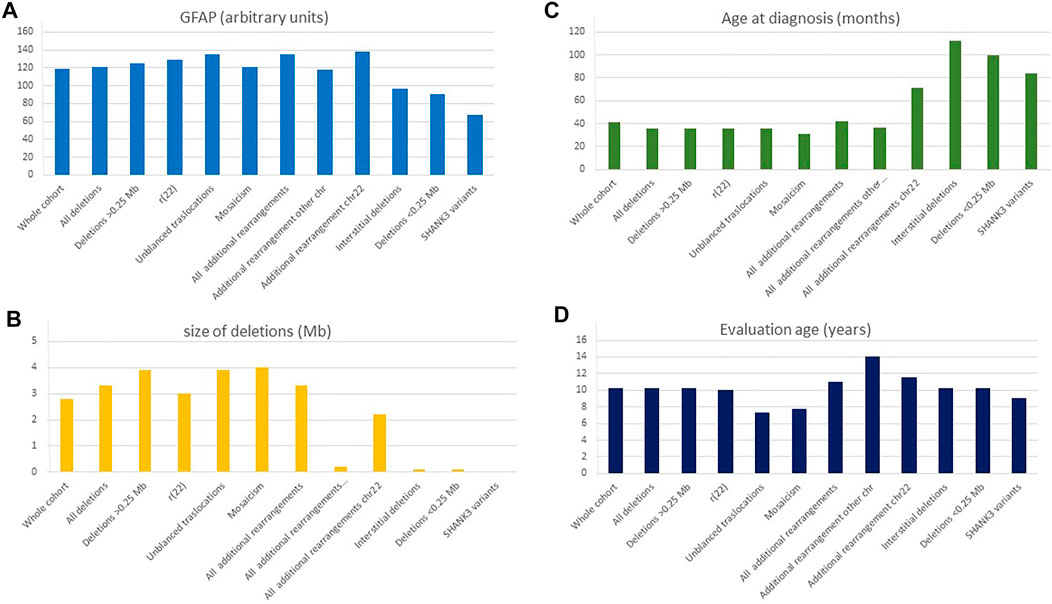
FIGURE 6. Distribution of continuous variables according to the type of genetic defect. (A) GFAP, global function assessment of the patient (arbitrary values); (B) Size of the deletions (Mb); (C) Age at diagnosis (months); (D) Age at evaluation (years). *ASD diagnosis according to the psychiatrists of the referring institutions.
We compared 10 subgroups of individuals with different types of genetic defects: 1) large deletions (>0.25 Mb; mean size ± SD, 4.29 ± 2.50), 2) smaller deletions (≤0.25 Mb, 0.10 ± 0.05), 3) interstitial deletions (1.94 ± 3.55 Mb), 4) SHANK3 sequence variants, 5) ring 22 (3.53 ± 2.44 Mb), 6) unbalanced translocations (3.69 ± 1.61 Mb), 7) mosaic deletions (3.5 ± 3.48 Mb), 8) additional rearrangement at chromosome 22 (3.32 ± 2.02 Mb), 9) additional rearrangement in other chromosomes (2.62 ± 2.26 Mb), and 10) all cases with additional rearrangements (2.99 ± 2.26 Mb) (Table 5). Bonferroni or T3-Dunnett post hoc tests reveal that the significant differences in the variable “size of deletion” were mainly due to differences between large (>0.25 Mb) and small (≤0.25 Mb) or interstitial deletions, and between small deletions and ring 22 or unbalanced translocations (Table 5).
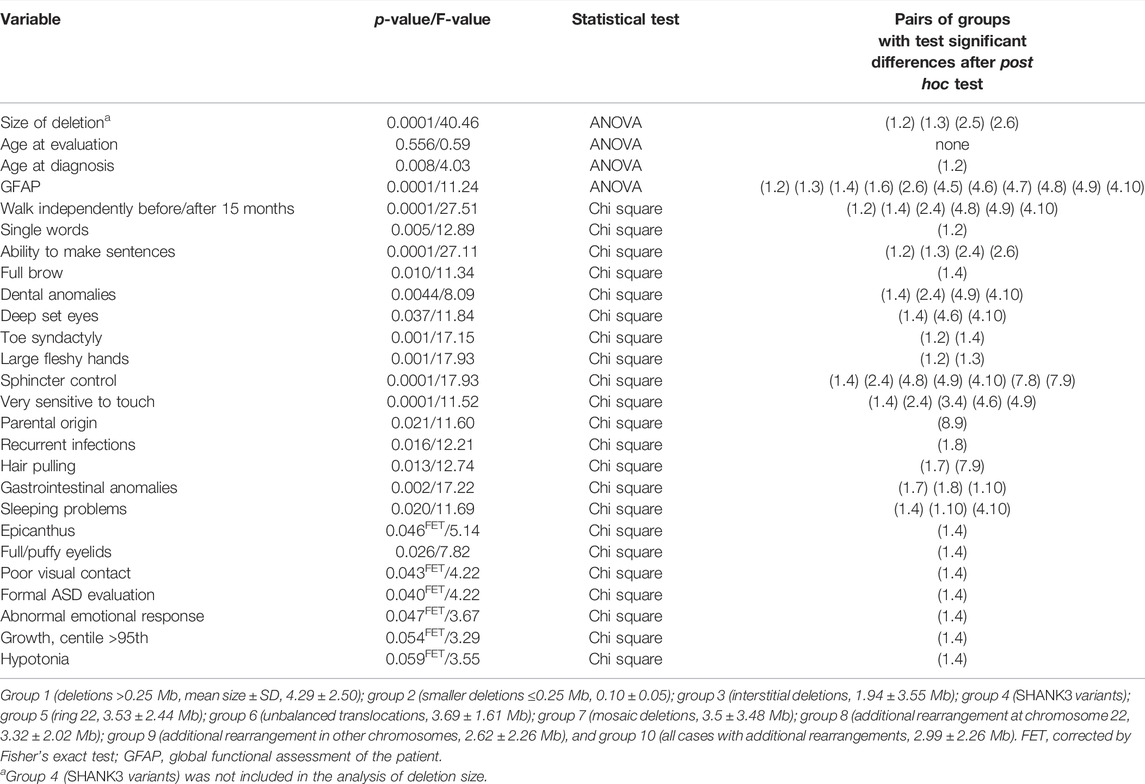
TABLE 5. Comparison between groups with different types of genetic alterations.
Using the GFAP, we observed significant differences mainly between patients with large deletions compared with patients with small deletions, interstitial deletions, and sequence variants (Table 5). Remarkably, no significant differences were detected between small deletions and individuals with sequence variants in SHANK3 (Table 5).
Pearson statistical analysis was performed to explore correlations between these continuous variables. We observed significant direct correlations between size of the deletion and GFAP (Pearson value = 0.33, p = .0001) as well as inverse correlations between age at diagnosis and size of the deletions (Pearson value = −0.240, p = .001) and GFAP (Pearson value = −0.133, p = .03). Altogether, our data suggest that the age at diagnosis seems to be inversely related to the degree of difficulty at diagnosis. Indeed, patients with small deletions (below 0.25 Mb; mean 0.10 ± 0.05 Mb) were diagnosed later (mean 7.61 ± 4.47 years) than those with large-size deletions (˃0.25 Mb, 4.35 ± 2.62 Mb, mean age at diagnosis: 5.52 ± 7.87 years). This fact was also observed in patients with interstitial deletions (mean age at diagnosis 9.75 ± 8.07 years and 1.91 ± 3.51 Mb for deletion size) and SHANK3 gene variants (mean age at diagnosis 7.86 ± 4.49 years).
Individuals with r(22), mosaic deletions, and unbalanced translocations affecting the 22q13 band were diagnosed significantly earlier than the average (mean ages 5.59, 4.41, and 3.57 years, respectively) even though the mean deletion size in those cases was 3–4 Mb (3.19, 3.24, and 3.91 Mb, respectively) similar to the average of the cohort (median 3.08 Mb).
Although individuals with small deletions and SHANK3 variants showed similar findings in most of the categorical variables (Table 5), a remarkable difference was observed in “the ability to make sentences” between the two groups, with 30/65 (46.2%, Supplementary Table S3) of individuals with deletions below 0.25 Mb able to make sentences compared with 5/18 (27.7%, Table 1a) among those with SHANK3 variants. Interestingly, we also found significant differences in the variable “parental origin” between groups with additional rearrangements (at chromosome 22 vs. other chromosomes). As expected, significant differences were found between all deletions and individuals with SHANK3 variants, mostly affecting dysmorphic features (Table 5).
No statistically significant differences were detected between gender and continuous variables (size of the deletion, age of diagnosis, age of evaluation or GFAP, Student’s t-test, data not shown). However, significant differences were observed between gender and several categorical variables (seizures, decreased perspiration, microcephaly, fifth finger clinodactyly, and lymphedema; chi-square test, p = 0.023, 0.056, 0.008, 0.029 and 0.001, respectively; data not shown), with higher frequencies in females.
Finally, we observed significant differences between parental origin and GFAP (p = 0.048, Student’s t-test) and two categorical variables, high pain threshold and lymphedema (chi-square test, p = 0.039 and 0.027, respectively, n = 51). In all cases, maternal origin (n = 12) was associated with higher GFAP values and with a worse prognosis (Table 5).
We applied Ward’s hierarchical cluster analysis using deletion size as the unique variable to test how individuals with microdeletions group according to their deletion size. Individuals were grouped into four clusters (the number was established by BIC and AIC algorithms) as follows: cluster 1: 0.52 ± 0.51 Mb (64 individuals), cluster 2: 3.39 ± 0.77 Mb (66 individuals), cluster 3: 6.10 ± 0.69 Mb (29 individuals), and cluster 4: 8.27 ± 0.74 Mb (28 individuals). Extended variable frequencies in each cluster are shown in Supplementary Table S3. One-way ANOVA followed by a post hoc test (Bonferroni or T3-Dunnett) revealed statistically significant differences between age at diagnosis, GFAP, and size of deletions in different clusters (p = 0.009, 0.0001, and 0.0001, respectively, Table 6). Supplementary Figure S6 shows that some clinical findings, such as “ability to make sentences” or “walk independently before/after 15 months,” were preferentially associated with cluster 1. In fact, in cluster 1 (deletions 0.52 ± 0.51 Mb), 53.8% of these individuals were able to make sentences (35/65), followed by 15.6% (10/64) in cluster 2 and only 3.7% (1/27) in clusters 3 and 4. The chi-square test followed by z post hoc test with Bonferroni correction showed significant differences among clusters for several categorical variables (Table 6).
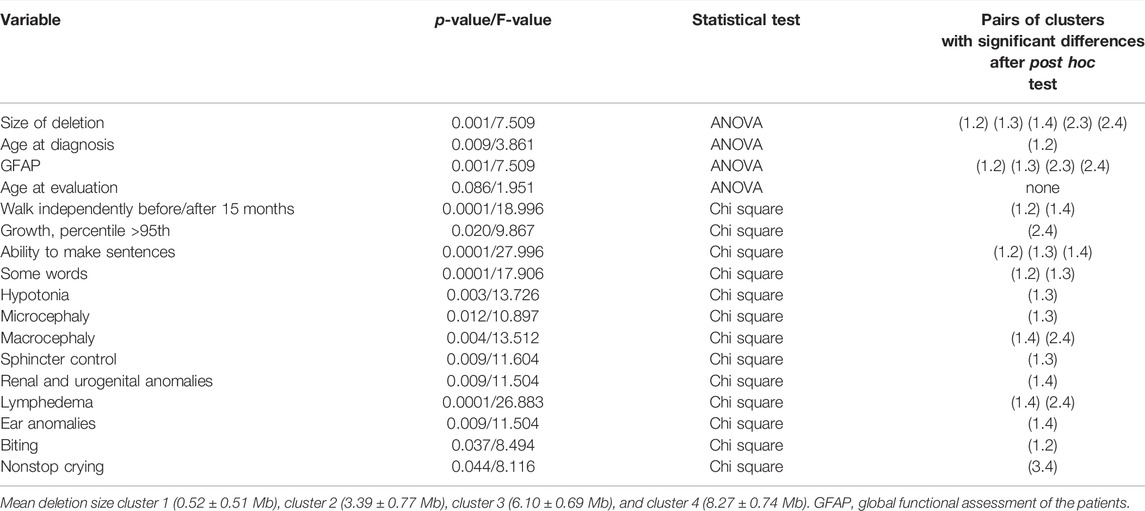
TABLE 6. Comparison between Ward’s clusters obtained using deletion size.
When Ward’s clusters were dissected by frequencies of these variables (in percentages), we observed higher frequencies of several core features, considered as a better prognosis, in cluster 1 than in other clusters (Supplementary Table S3). On the other hand, higher percentages of other core items, reflecting comorbidity (normally associated with a worse prognosis; renal and urogenital abnormalities, hearing problems, lymphedema, no words, or growth above the 95th percentile, Supplementary Table S3) mapped preferentially in cluster 4, which is associated with large deletions. Finally, other items seemed to correlate directly (toe syndactyly, ear anomalies, GFAP, MRI anomalies, abnormal emotional response, or renal and urogenital anomalies) or inversely (age at diagnosis) to the size of the deletions (Supplementary Table S3).
Linear regression was used to obtain a coefficient of correlation to deletion size at 22q13 for each feature (Table 7). The coefficient of correlation ranged between 0 and 0.7. “F value” was examined to determine if the coefficient of correlation was significant. For most features, no correlation to deletion size was found. However, several clinical features were found to have a statistically significant correlation with the size of the deletion (Table 7), including the ability to make sentences, lymphedema, macrocephaly, renal and urogenital anomalies, and brain MRI anomalies. At a significance level of 0.05, one would expect 1 in 20 significant correlations by chance, whereas 14/61 (23%) correlations for the size of deletion were obtained. With a similar approach, we identified 6/61 (9.8%) correlations with age at diagnosis and 8/61 (13.1%) with age at evaluation.
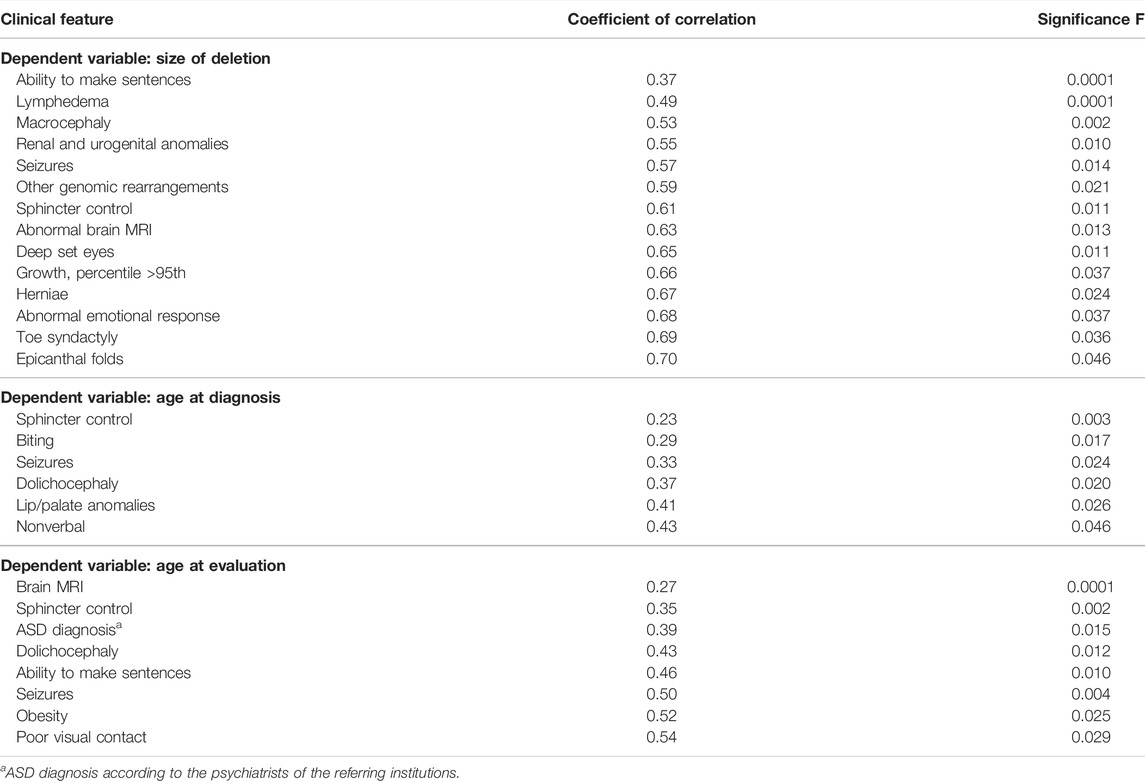
TABLE 7. Comparison of clinical features and the size of the 22q13 deletion, age at diagnosis and age at evaluation using linear regression to obtain a coefficient of correlation.
We describe one of the largest series of patients with PMS characterized by CMA and other genetic approaches, including karyotype, MLPA, and FISH. We also explored the high genetic, and phenotypic variability observed in PMS individuals. Although the true prevalence of this rare disease is still unknown, it is among the most common subtelomeric microdeletion syndromes (Delahaye et al., 2009). Previous findings show that PMS is diagnosed in around 0.5% of individuals with ASD and ID (Cooper et al., 2011; Betancur and Buxbaum, 2013; Leblond et al., 2014; Chen et al., 2017; Samogy-Costa et al., 2019). Previous data suggest that the prevalence of this syndrome remains underestimated worldwide due to several reasons:
a) The lack of a distinctive phenotype without significant dysmorphic features (Figure 1). In most cases, individuals carrying SHANK3 variants and small deletions do not have a distinctive facial appearance.
b) High genetic and clinical variability. We observed marked intracohort variability. Analysis of GFAP revealed significant differences depending on the type of genetic defect and the type of rearrangements found in individuals. We found additional rearrangements in 21.2% of the cases. Some of them involved other OMIM-related syndromes (Supplementary Table S2), including hereditary neuropathy with liability to pressure palsies (OMIM#162500), affecting PMP22; Chromosome 15q11.2 deletion syndrome BP1-BP2 (OMIM#615656), affecting NIPA1-NIPA2; 15q13.3 deletion syndrome (OMIM#2612001), affecting CHRNA7, and 16p11.2 microdeletion syndrome (OMIM#611913), which may contribute partially to the variability of some individuals. Previous studies also report the presence of additional rearrangements with putative clinical relevance in individuals with PMS (Tabet et al., 2017; Samogy-Costa et al., 2019). Interestingly, our data show that individuals with additional rearrangements and, in particular, those with small 22q13 deletions had higher values of GFAP (associated with worse prognosis) than cases with simple small deletions. In our series, some of the patients carried the same additional CNVs reported by Tabet and others (2017), in most cases inherited from a reportedly healthy parent. We do not know the consequences of these findings or if it is just a coincidence. Most of these and other similar CNVs (15q11.2 deletions and duplications, 15q13.3 deletions and duplications, 16p13.11 deletions, 16p12.1 deletions, 16p11.2 proximal and distal deletions, 17q12 deletions and duplications, and 22q11.21 duplications) are linked to susceptibility loci for a variety of pediatric diseases (Girirajan and Eichler, 2010; Cooper et al., 2011). For some of these CNVs, the enrichment in affected individuals (mainly ID, ASD, or DD cases) in comparison with healthy controls seems to give them a putative pathogenic classification (Rosenfeld et al., 2013).
c) The difficulty in detecting chromosome 22 microdeletions in routine cytogenetic analysis even at the 550–850 band level of resolution. Our data show that small terminal deletions, interstitial deletions, and SHANK3 variants were diagnosed later than those carrying other type of rearrangements, such as ring chromosomes, mosaic deletions, or unbalanced translocations. Thus, most cases were diagnosed in tertiary hospitals that applied CMA testing as a first-tier test through its laboratory routines for individuals with ID, ASD, and congenital malformations, following international guidelines (Miller et al., 2010). Misdiagnosis or underdiagnosis of mosaicism could be observed when using CMA as a unique tool. Mosaicism lower than 15% cannot be easily detected by CMA (Figure 3) owing to the variability of the assay and the fact that most of the commercial CMA platforms do not have a significant number of probes at the end of the telomere of chromosome 22 (Supplementary Figure S5). FISH or MLPA combined with CMA must be applied in suspected patients. We found an unexpectedly high number of post-zygotic mosaicism (17/189; 9.0%) in patients with microdeletions when compared with a previous report, which established a mosaic frequency of around 2.5%–5.8% for deletions at 22q13.3 (Samogy-Costa et al., 2019). It is not easy to predict the expected clinical features in patients with mosaicism though patients with <10% of mosaicism in blood can present a complete manifestation of the disease (Phelan et al., 2018). We also found two independent families with suspected gonadal mosaicism. This aspect is important because it complicates genetic counseling. Germinal mosaicism in PMS is not frequent, but it has been described in a few families (Tabolacci et al., 2005; Durand et al., 2007; Gauthier et al., 2009; Nemirovsky et al., 2015; Zwanenburg et al., 2016).
In PMS individuals with terminal deletions diagnosed with CMA, it is essential to rule out the presence of r(22). Confirmation of r(22) has significant implications for clinical management because individuals with r(22) have an increased risk of tumors in the nervous system due to biallelic loss of the NF2 (neurofibromatosis type 2) gene (Lyons-Warren et al., 2017; Ziats et al., 2020). We observed three out of 20 patients with r(22) with neurofibromatosis type 2; these three individuals were included in the series reported by Zyats and others (2020). The prevalence of tumors associated with r(22) is unknown. Thus, we recommend follow-up of PMS patients carrying r(22) and highlight the importance of karyotyping individuals with terminal deletions of the long arm of chromosome 22.
d) Difficulties in testing SHANK3 variants. Implementing exome or panels to analyze SHANK3 variants was rare and expensive during the period of recruitment of this cohort in our country. However, in recent years (2019–2020), we recruited 18 patients with SHANK3 variants.
We propose an algorithm for laboratory management of individuals with PMS (Figure 7). We recommend CMA as a first-tier test for patients with ID and ASD to determine the exact deletion size, define the deletion breakpoints, and detect additional genomic rearrangements, such as terminal duplications in other chromosomes. Most patients also need other molecular approaches, such as MLPA or FISH, for accurate laboratory characterization (Supplementary Figure S5). Terminal deletions need karyotyping to rule out a r(22), and FISH is mandatory in parents when suspicion of unbalanced or balanced translocation is suspected. Low-grade mosaicism may be detected by applying FISH in the proband. When other techniques, such as FISH or MLPA, established the diagnosis of PMS as the first test (Figure 7), CMA is still mandatory to complete the diagnosis of individuals (to determine the affected genes, deletion size, other rearrangements, etc.). Finally, when all cytogenetic and molecular approaches are negative in individuals with ID or ASD with other clinical features of PMS, we recommend an exome-analysis (trio or singleton) with extensive analysis of SHANK3 sequence variants (Figure 7).
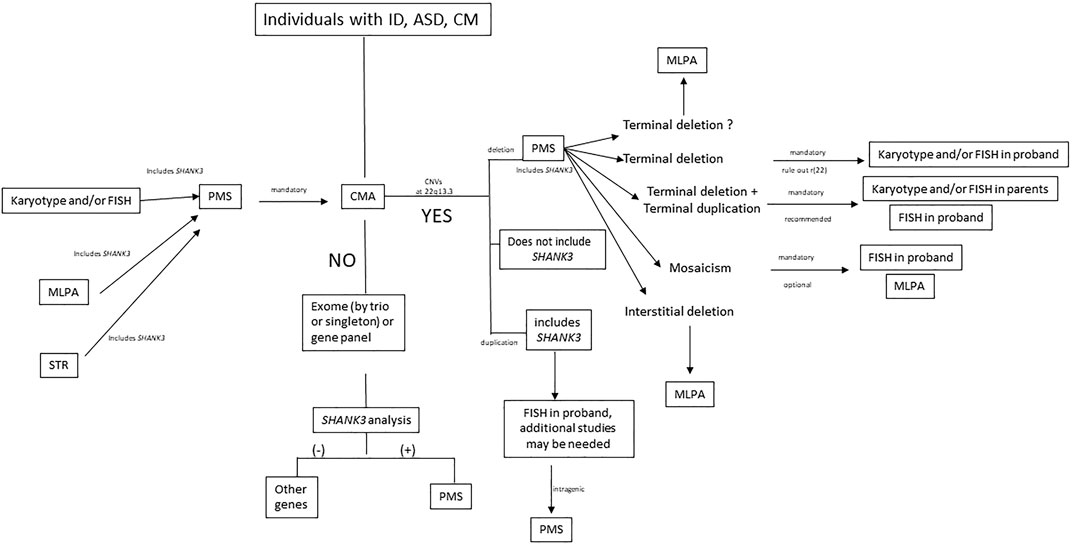
FIGURE 7. Laboratory algorithm for management of samples suspected of PMS. ID, intellectual disability; ASD, autism spectrum disorder; CM, congenital malformations; PMS, Phelan-McDermid syndrome; CMA, chromosome microarray analysis; MLPA, multiplex ligation-dependent probe amplification; STR, short tandem repeat; CNV, copy number variation; r(22), ring chromosome 22.
It is also remarkable that, although formal ASD studies were only performed in 20% (43/210) of the cohort, 29/43 (67%) of them have an ASD diagnosis according to the psychiatrists of the referring institutions. Thus, for PMS individuals, formal ASD evaluation is mandatory. Sixty individuals of this cohort are included in a recent study of the behavioral profile in PMS performed by our colleagues (Burdeus-Olavarrieta et al., 2021).
It is suggested that the haploinsufficiency of SHANK3 is the most significant contributor to PMS. We believe that SHANK3 is a major contributor to the neurocognitive features of the syndrome, but not the only one. Other genes may contribute to the PMS phenotype by modulating SHANK3 action. Several authors review a possible effect of different genes in the PMS phenotype (Tabet et al., 2017; Mitz et al., 2018; Ziats et al., 2019; Li et al., 2020; Ricciardello et al., 2021), but how those genes contribute is still unknown.
Only a few studies investigate putative relations between the size of the deletions and clinical features of PMS, and the causality remains unclear (Cusmano-Ozog et al., 2007; Dhar et al., 2010; Sarasua et al., 2011; Soorya et al., 2013; Sarasua et al., 2014a; Sarasua et al., 2014b; Tabet et al., 2017; Samogy-Costa et al., 2019). The clinical features of patients with pathogenic variants in SHANK3 overlap with those of individuals with deletions, giving this gene an important role in the spectrum of clinical features of PMS.
We found that speech skills, one of the main features of the syndrome, might be directly associated with the size and/or mapping of the deletion. Indeed, most individuals who can make sentences (aged older than 3 years) had smaller deletions, supporting previously described observations (Sarasua et al., 2014a; Samogy-Costa et al., 2019; Brignell et al., 2021). In addition, among individuals with SHANK3 variants, 27% (5/18) of patients in this study were able to maintain short conversations, compared with 18% (3/17) and 38% (3/8) of individuals verbally fluent reported by De Rubeis et al. (2018) and Xu et al. (2020), respectively.
Our data also support significant differences between individuals with SHANK3 variants and small deletions in the ability to make sentences. Thus, other genes or some interaction nearby could modulate language abilities. In fact, a recent study also showed that SHANK3 seemed necessary but not exclusive for expressive language in PMS individuals (Brignell et al., 2021).
Additional differences between individuals with SHANK3 variants and those with small deletions were also observed for cognitive features, such as sleeping anomalies or sphincter control, with higher frequencies in individuals with SHANK3 variants than in the smaller deletion group. As expected, differences in several facial dysmorphic features were observed between individuals with deletions and SHANK3 variants.
The cluster analysis showed a positive correlation between deletion size and GFAP, brain MRI abnormalities, ear anomalies, and toe syndactyly as well as a negative correlation between deletion size and age at diagnosis and abnormal emotional response. It is also clear that several clinical features mapped preferentially in specific regions of the clusters. Indeed, two clear genomic regions can be associated with the size of the cranium. Whereas medium- and large-size deletions seem to be associated with macrocephaly, microcephaly seems to be present only in patients with small deletions. We established an interval between 0.40 and 3.4 Mb linked to microcephaly and between 4.50 and 8 Mb from the telomere related to macrocephaly. This fact suggests the contribution of at least two independent genes for alterations in the cranium size. Interestingly, there are no more than 10 high dosage-sensitive genes (ClinGen, http://www.clinicalgenome.org) in the latter interval. Among them is GRAMD4, which has been established experimentally to have protein-protein interaction with PIAS1 (Supplementary Figure S7). PIAS1 is a member of the ubiquitin protein family, like PIAS4. The PIAS4 gene has been involved in macro/microcephaly in distal 19p13.3 microdeletion/microduplication syndrome (Nevado et al., 2015; Tenorio et al., 2020).
The existence of interstitial deletions not including SHANK3 (Wilson et al., 2008; Disciglio et al., 2014; Ha et al., 2017; this study), which partly overlap some clinical features of PMS (Supplementary Table S4), may also indirectly support a role for additional genes in the clinical spectrum of PMS. At this point, we cannot rule out a positional/regulating effect on SHANK3 in all these cases, nor global alteration of topological chromatin organization (TAD; topological association domains) as is been suggested by others (Kurtas et al., 2018; Srikanth et al., 2021) rather than simply by the deletion of dosage-sensitive genes. This hypothesis needs to be explored in future studies.
A previous large cohort study reported a small but significant increase with age of several clinical findings in PMS, including sensory dysfunction, reduced response to pain, epilepsy, and lymphedema (Sarasua et al., 2014b). Similarly, the risk of psychiatric disorders in PMS increases with age (Denayer et al., 2012; Verhoeven et al., 2012; Kolevzon et al., 2019). Regarding the correlation of clinical features with age, our data cannot support any solid conclusion about the contribution of age to the clinical features of PMS. We found in our cohort six and eight items of 61 that rejected this null hypothesis (∼10% and 13%) for age at diagnosis and age at evaluation, respectively. This is twice the number expected by chance.
Here, we report a large series of Spanish and South American patients with PMS, focusing on phenotype-genotype correlations. The analysis of individuals with sequence variants and their comparison with patients with small deletions support the notion that SHANK3 is essential in most core phenotypic findings of PMS but is not the unique one. Additional genes may modulate the whole phenotype in PMS individuals with microdeletions.
The existence of different types of rearrangements and genomic variations may explain the high variability observed in PMS individuals. Finally, an accurate laboratory approach for PMS individuals using a diagnostic algorithm is proposed to offer appropriate management, follow-up, and genetic counselling to these families.
INGEMM, Madrid, Spain (Rocío Mena, Roser Lleguer, Victoria Fernández-Montaño, Rubén Martín, Blanca Fernández, Fé García-Santiago, Victoria Gómez del Pozo, Carolina Peña; Spanish PMS Association (Norma Alhambra, Carlos García, Juan Ramón Rodríguez); Servicio de Neuropediatría, Hospital Universitario La Paz, Madrid, Spain (Antonio Martínez-Bermejo); Hospital Central de Asturias, Oviedo, Spain (Ignacio Málaga); Hospital San Joan de Deu, Barcelona, Spain (Antonio Federico Martínez-Monseny, Judith Armstrong, Jennifer Anticona, Cristina Hernando-Davalillo, Adrián Alcalá San Martí, Loreto Martorell, Delia Yubero, Tania Nunes, Mar O´Callaghan, Carlos Ortez, Xenia Alonso, Federico Ramos, Jesús Casas López); Hospital Virgen de la Arrixaca, Murcia, Spain (Vanesa López-González, M. Juliana Ballesta); Q- Genomics Laboratory Barcelona, Spain (Lluís Armengol); Hospital Virgen del Rocío, Sevilla, Spain (Antonio González-Meneses; Salud Borrego); Hospital Universitario la Fé, Valencia, Spain (Mónica Roselló); NIM-Genetics, Madrid, Spain (Javier Suela); Hospital Son Espases, Palma de Mallorca, Spain (Ángeles Pérez-Granero); Hospital Clinic, Barcelona, Spain (Laia Rodríguez-Revenga).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DECIPHER Genomics, accession no: 432868 - 433079, IMMGPMS1 - IMMGPMS211.
The studies involving human participants were reviewed and approved by Institutional Review Board of Hospital la Paz, Madrid. Spain PI: 2735 HULP. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JN: Conceived the presented idea, completed data analysis and wrote the manuscript; JN and PL: Designed the study; JN Coordinated data acquisition and collected the data; EV, MP-B, JT, MM and PL: corrected the manuscript; PB assisted with data management; CB statistical analysis; EV, MP-B, JN, PB, SM and MM: managed miocroarrays at INGEMM; EM and IV: performed FISH and karyotyping studies at INGEMM; SG-M, EG-N, JR, MM, MC, FS-S, GO, CO, HP, EG, JC, AM, CS, SC, EG, LP-J, AB, PG-M, FC-S, NG-P, RB-L, MS, JO-E, BG, VS, PT and the Spanish PMS working group: contributed patients from their institutions.
REDES/FIBHULP08. FIBHULP PI: 2735. FIBHULP Auchan Reserch Project. FIBHULP. Raregenomics (B2017/BMD-3721).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank all individuals and families for their participation in this study, as well as Norma Alhambra, Carlos García, and Juan Ramón Rodríguez (Spanish PMS Association) for helping us to develop this project. We thank Rocío Mena, who coordinates the PMS working group. In memoriam of Sol Martin for her valuable support work in the laboratory. This work was supported by a grant from REDES/FIBHULP08 of the Fundación para la Investigación Biomédica Hospital Universitario La Paz (FIBHULP); PI: 2735 from FIBHULP, and AUCHAN Research Project on PMS.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.652454/full#supplementary-material
Anderlid, B.-M., Schoumans, J., Annerén, G., Sahlén, S., Kyllerman, M., Vujic, M., et al. (2002a). Subtelomeric Rearrangements Detected in Patients with Idiopathic Mental Retardation. Am. J. Med. Genet. 107 (4), 275–284. doi:10.1002/ajmg.10029
Anderlid, B.-M., Schoumans, J., Annerén, G., Tapia-Paez, I., Dumanski, J., Blennow, E., et al. (2002b). FISH-mapping of a 100-kb Terminal 22q13 Deletion. Hum. Genet. 110 (5), 439–443. doi:10.1007/s00439-002-0713-7
Betancur, C., and Buxbaum, J. D. (2013). SHANK3 Haploinsufficiency: a "common" but Underdiagnosed Highly Penetrant Monogenic Cause of Autism Spectrum Disorders. Mol. Autism 4 (1), 17. doi:10.1186/2040-2392-4-17
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 Variants in Patients with Different Subtypes of Autism Spectrum Disorders. Eur. J. Hum. Genet. 21, 310–316. doi:10.1038/ejhg.2012.175
Boeckers, T. M. (2006). The Postsynaptic Density. Cell Tissue Res 326 (2), 409–422. doi:10.1007/s00441-006-0274-5
Bonaglia, M. C., Giorda, R., Beri, S., De Agostini, C., Novara, F., Fichera, M., et al. (2011). Molecular Mechanisms Generating and Stabilizing Terminal 22q13 Deletions in 44 Subjects with Phelan/McDermid Syndrome. Plos Genet. 7, e1002173. doi:10.1371/journal.pgen.1002173
Bonaglia, M. C., Giorda, R., Borgatti, R., Felisari, G., Gagliardi, C., Selicorni, A., et al. (2001b). Disruption of the ProSAP2 Gene in a t(12;22)(q24.1;q13.3) Is Associated with the 22q13.3 Deletion Syndrome. Am. J. Hum. Genet. 69 (2), 261–268. doi:10.1086/321293
Bonaglia, M. C., Giorda, R., Mani, E., Aceti, G., Anderld, B. M., Baroncini, A., et al. (2001a). Identification of a Recurrent Breakpoint within the SHANK3 Gene in the 22q13.3 Deletion Syndrome. Am. J. Hum. Genet. 69 (2), 261–268.
Bowling, K. M., Thompson, M. L., Amaral, M. D., Finnila, C. R., Hiatt, S. M., Engel, K. L., et al. (2017). Genomic Diagnosis for Children with Intellectual Disability And/or Developmental Delay. Genome Med. 9, 43. doi:10.1186/s13073-017-0433-1
Bramswig, N. C., Lüdecke, H.-J., Alanay, Y., Albrecht, B., Barthelmie, A., Boduroglu, K., et al. (2015). Exome Sequencing Unravels Unexpected Differential Diagnoses in Individuals with the Tentative Diagnosis of Coffin-Siris and Nicolaides-Baraitser Syndromes. Hum. Genet. 134 (6), 553–568. doi:10.1007/s00439-015-1535-8
Brignell, A., Gu, C., Holm, A., Carrigg, B., Sheppard, D. A., Amor, D. J., et al. (2021). Speech and Language Phenotype in Phelan-McDermid (22q13.3) Syndrome. Eur. J. Hum. Genet. 29 (4), 564–574. doi:10.1038/s41431-020-00761-1
Burdeus-Olavarrieta, M., San José-Cáceres, A., García-Alcón, A., González-Peñas, J., Hernández-Jusdado, P., and Parellada-Redondo, M. (2021). Characterisation of the Clinical Phenotype in Phelan-McDermid Syndrome. J. Neurodevelop Disord. 13, 26–40. doi:10.1186/s11689-021-09370-5
C Yuen, R. K., Merico, D., Bookman, M., L HoweThiruvahindrapuram, J. B., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole Genome Sequencing Resource Identifies 18 New Candidate Genes for Autism Spectrum Disorder. Nat. Neurosci. 20, 602–611. doi:10.1038/nn.4524
Chen, C.-H., Chen, H.-I., Liao, H.-M., Chen, Y.-J., Fang, J.-S., Lee, K.-F., et al. (2017). Clinical and Molecular Characterization of Three Genomic Rearrangements at Chromosome 22q13.3 Associated with Autism Spectrum Disorder. Psychiatr. Genet. 27 (1), 23–33. doi:10.1097/ypg.0000000000000151
Cooper, G. M., Coe, B. P., Girirajan, S., Rosenfeld, J. A., Vu, T. H., Baker, C., et al. (2011). A Copy Number Variation Morbidity Map of Developmental Delay. Nat. Genet. 43 (9), 838–846. doi:10.1038/ng.909
Cusmano-Ozog, K., Manning, M. A., and Hoyme, H. E. (2007). 22q13.3 Deletion Syndrome: A Recognizable Malformation Syndrome Associated with Marked Speech and Language Delay. Am. J. Med. Genet. 145C, 393–398. doi:10.1002/ajmg.c.30155
De Rubeis, S., Siper, P. M., Durkin, A., Weissman, J., Muratet, F., Halpern, D., et al. (2018). Delineation of the Genetic and Clinical Spectrum of Phelan-McDermid Syndrome Caused by SHANK3 point Mutations. Mol. Autism 9, 31. doi:10.1186/s13229-018-0205-9
Delahaye, A., Toutain, A., Aboura, A., Dupont, C., Tabet, A. C., Benzacken, B., et al. (2009). Chromosome 22q13.3 Deletion Syndrome with a De Novo Interstitial 22q13.3 Cryptic Deletion Disrupting SHANK3. Eur. J. Med. Genet. 52 (5), 328–332. doi:10.1016/j.ejmg.2009.05.004
Denayer, A., Van Esch, H., de Ravel, T., Frijns, J. P., Van Buggenhout, G., Vogels, A., et al. (2012). Neuropsychopathology in 7 Patients with the 22q13 Deletion Syndrome: Presence of Bipolar Disorder and Progressive Loss of Skills. Mol. Syndromol 3 (1), 14–20. doi:10.1159/000339119
Dhar, S. U., del Gaudio, D., German, J. R., Peters, S. U., Ou, Z., Bader, P. I., et al. (2010). 22q13.3 Deletion Syndrome: Clinical and Molecular Analysis Using Array CGH. Am. J. Med. Genet. 152A, 573–581. doi:10.1002/ajmg.a.33253
Disciglio, V., Rizzo, C. L., Mencarelli, M. A., Mucciolo, M., Marozza, A., Di Marco, C., et al. (2014). Interstitial 22q13 Deletions Not Involving SHANK3 Gene: a New Contiguous Gene Syndrome. Am. J. Med. Genet. 164 (7), 1666–1676. doi:10.1002/ajmg.a.36513
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the Gene Encoding the Synaptic Scaffolding Protein SHANK3 Are Associated with Autism Spectrum Disorders. Nat. Genet. 39 (1), 25–27. doi:10.1038/ng1933
Farwell, K. D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E. C., Tippin Davis, B., et al. (2015). Enhanced Utility of Family-Centered Diagnostic Exome Sequencing with Inheritance Model-Based Analysis: Results from 500 Unselected Families with Undiagnosed Genetic Conditions. Genet. Med. 17 (7), 578–586. doi:10.1038/gim.2014.154
Feliciano, P., Zhou, X., Astrovskaya, I., Turner, T. N., Wang, T., Brueggeman, L., et al. (2019). Exome Sequencing of 457 Autism Families Recruited Online Provides Evidence for Autism Risk Genes. NPJ Genom Med. 4, 19. doi:10.1038/s41525-019-0093-8
Gauthier, J., Spiegelman, D., Piton, A., Lafrenière, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel De Novo SHANK3 Mutation in Autistic Patients. Am. J. Med. Genet. 150B, 421–424. doi:10.1002/ajmg.b.30822
Girirajan, S., and Eichler, E. E. (2010). Phenotypic Variability and Genetic Susceptibility to Genomic Disorders. Hum. Mol. Genet. 19 (R2), R176–R187. doi:10.1093/hmg/ddq366
Ha, J. F., Ahmad, A., and Lesperance, M. M. (2017). Clinical Characterization of Novel Chromosome 22q13. Microdeletions Int. J. Pediatr. Otorhinolaryngol. 95, 121–126.
Heilstedt, H. A., Ballif, B. C., Howard, L. A., Lewis, R. A., Stal, S., Kashork, C. D., et al. (2003). Physical Map of 1p36, Placement of Breakpoints in Monosomy 1p36, and Clinical Characterization of the Syndrome. Am. J. Hum. Genet. 72 (5), 1200–1212. doi:10.1086/375179
Holder, J. L., and Quach, M. M. (2016). The Spectrum of Epilepsy and Electroencephalographic Abnormalities Due to SHANK3 Loss-Of-Function Mutations. Epilepsia 57 (10), 1651–1659. doi:10.1111/epi.13506
Kaplanis, J., Samocha, K. E., Wiel, L., Zhang, Z., Arvai, K. J., Eberhardt, R. Y., et al. (2020). Evidence for 28 Genetic Disorders Discovered by Combining Healthcare and Research Data. Nature 586 (7831), 757–762. doi:10.1038/s41586-020-2832-5
Kolevzon, A., Delaby, E., Berry-Kravis, E., Buxbaum, J. D., and Betancur, C. (2019). Neuropsychiatric Decompensation in Adolescents and Adults with Phelan-McDermid Syndrome: a Systematic Review of the Literature. Mol. Autism 10, 50. doi:10.1186/s13229-019-0291-3
Kurtas, N., Arrigoni, F., Errichiello, E., Zucca, C., Maghini, C., D’Angelo, M. G., et al. (2018). Chromothripsis and Ring Chromosome 22: a Paradigm of Genomic Complexity in the Phelan-McDermid Syndrome (22q13 Deletion Syndrome). J. Med. Genet. 55 (4), 269–277. doi:10.1136/jmedgenet-2017-105125
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. Plos Genet. 10, e1004580. doi:10.1371/journal.pgen.1004580
Lelieveld, S. H., Reijnders, M. R. F., Pfundt, R., Yntema, H. G., Kamsteeg, E.-J., de Vries, P., et al. (2016). Meta-analysis of 2,104 Trios Provides Support for 10 New Genes for Intellectual Disability. Nat. Neurosci. 19, 1194–1196. doi:10.1038/nn.4352
Li, S., Xi, K.-w., Liu, T., Zhang, Y., Zhang, M., Zeng, L.-d., et al. (2020). Fraternal Twins with Phelan-McDermid Syndrome Not Involving the SHANK3 Gene: Case Report and Literature Review. BMC Med. Genomics 13, 146–153. doi:10.1186/s12920-020-00802-0
Lim, E. T., Uddin, M., Uddin, M., De Rubeis, S., Chan, Y., Kamumbu, A. S., et al. (2017). Rates, Distribution and Implications of Postzygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 20 (9), 1217–1224. doi:10.1038/nn.4598
Lyons-Warren, A. M., Cheung, S. W., and Holder, J. L. (2017). Clinical Reasoning: A Common Cause for Phelan-McDermid Syndrome and Neurofibromatosis Type 2. Neurology 89, e205–e209. doi:10.1212/wnl.0000000000004573
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R. R., Carter, N. P., et al. (2010). Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 86, 749–764. doi:10.1016/j.ajhg.2010.04.006
Mitz, A. R., Philyaw, T. J., Boccuto, L., Shcheglovitov, A., Sarasua, S. M., Kaufmann, W. E., et al. (2018). Identification of 22q13 Genes Most Likely to Contribute to Phelan McDermid Syndrome. Eur. J. Hum. Genet. 26, 293–302. doi:10.1038/s41431-017-0042-x
Nemirovsky, S. I., Cordoba, M., Zaiat, J. J., Completa, S. P., Vega, P. A., Gonzalez-Moron, D., et al. (2015). Whole-genome Sequencing Reveals a De Novo SHANK3 Mutation in Familial Autism Spectrum Disorder. PLoS One 10, e0116358. doi:10.1371/journal.pone.0116358
Nevado, J., Rosenfeld, J. A., Mena, R., Palomares-Bralo, M., Vallespín, E., Ángeles Mori, M., et al. (2015). PIAS4 Is Associated with Macro/microcephaly in the Novel Interstitial 19p13.3 Microdeletion/microduplication Syndrome. Eur. J. Hum. Genet. 23 (12), 1615–1626. doi:10.1038/ejhg.2015.51
O'Roak, B. J., Stessman, H. A., Boyle, E. A., Witherspoon, K. T., Martin, B., Lee, C., et al. (2014). Recurrent De Novo Mutations Implicate Novel Genes Underlying Simplex Autism Risk. Nat. Commun. 5, 5595. doi:10.1038/ncomms6595
Oberman, L. M., Boccuto, L., Cascio, L., Sarasua, S., and Kaufmann, W. E. (2015). Autism Spectrum Disorder in Phelan-McDermid Syndrome: Initial Characterization and Genotype-Phenotype Correlations. Orphanet J. Rare Dis. 10, 105. doi:10.1186/s13023-015-0323-9
Phelan, K., Rogers, R. C., and Boccuto, L. (2018). 22q13.3 Deletion Syndrome, Chromosome 22q13.3 Deletion Syndrome, Deletion 22q13 Syndrome. Genereviews.
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genet. Med. 18, 696–704. doi:10.1038/gim.2015.148
Ricciardello, A., Tomaiuolo, P., and Persico, A. M. (2021). Genotype-phenotype Correlation in Phelan‐McDermid Syndrome: A Comprehensive Review of Chromosome 22q13 Deleted Genes. Am. J. Med. Genet. 185, 2211–2233. doi:10.1002/ajmg.a.62222
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Rosenfeld, J. A., Coe, B. P., Eichler, E. E., Cuckle, H., and Shaffer, L. G. (2013). Estimates of Penetrance for Recurrent Pathogenic Copy-Number Variations. Genet. Med. 15 (6), 478–481. doi:10.1038/gim.2012.164
Samogy-Costa, C. I., Varella-Branco, E., Monfardini, F., Ferraz, H., Fock, R. A., Barbosa, R. H. A., et al. (2019). A Brazilian Cohort of Individuals with Phelan-McDermid Syndrome: Genotype-Phenotype Correlation and Identification of an Atypical Case. J. Neurodevelop Disord. 11 (1), 13. doi:10.1186/s11689-019-9273-1
Sarasua, S. M., Boccuto, L., Sharp, J. L., Dwivedi, A., Chen, C.-F., Rollins, J. D., et al. (2014a). Clinical and Genomic Evaluation of 201 Patients with Phelan-McDermid Syndrome. Hum. Genet. 133 (7), 847–859. doi:10.1007/s00439-014-1423-7
Sarasua, S. M., Dwivedi, A., Boccuto, L., Chen, C.-F., Sharp, J. L., Rollins, J. D., et al. (2014b). 22q13.2q13.32 Genomic Regions Associated with Severity of Speech Delay, Developmental Delay, and Physical Features in Phelan-McDermid Syndrome. Genet. Med. 16, 318–328. doi:10.1038/gim.2013.144
Sarasua, S. M., Dwivedi, A., Boccuto, L., Rollins, J. D., Chen, C.-F., Rogers, R. C., et al. (2011). Association between Deletion Size and Important Phenotypes Expands the Genomic Region of Interest in Phelan-McDermid Syndrome (22q13 Deletion Syndrome). J. Med. Genet. 48, 761–766. doi:10.1136/jmedgenet-2011-100225
Sauer, K. A., Bockmann, J., Steionestel, K., Boeckers, T. M., and Grabucker, A. M. (2019). Altered Intestinal Morphology and Microbiota Composition in the Autism Spectrum Disorders Associated SHANK3 Mouse Model. Int. J. Mol. Sci. 20 (09), 2134. doi:10.3390/ijms20092134
Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D., Diepvens, F., and Pals, G. (2002). Relative Quantification of 40 Nucleic Acid Sequences by Multiplex Ligation-dependent Probe Amplification. Nucleic Acids Res. 30 (12), e57. doi:10.1093/nar/gnf056
Shao, L., Shaw, C. A., Lu, X.-Y., Sahoo, T., Bacino, C. A., Lalani, S. R., et al. (2008). Identification of Chromosome Abnormalities in Subtelomeric Regions by Microarray Analysis: a Study of 5,380 Cases. Am. J. Med. Genet. 146A (17), 2242–2251. doi:10.1002/ajmg.a.32399
Soorya, L., Kolevzon, A., Zweifach, J., Lim, T., Dobry, Y., Schwartz, L., et al. (2013). Prospective Investigation of Autism and Genotype-Phenotype Correlations in 22q13 Deletion Syndrome and SHANK3 Deficiency. Mol. Autism 4 (4), 18. doi:10.1186/2040-2392-4-18
Srikanth, S., Jain, L., Zepeda-Mendoza, C., Cascio Kelly Jones, L., Pauly, R., DuPont, B., et al. (2021). Position Effects of 22q13 Rearrangements on Candidate Genes in Phelan- McDermid Syndrome. PLoS One 16 (7), e0253859. doi:10.1371/journal.pone.0253859
Tabet, A.-C., Rolland, T., Ducloy, M., Lévy, J., Buratti, J., Mathieu, A., et al. (2017). A Framework to Identify Contributing Genes in Patients with Phelan-McDermid Syndrome. Npj Genom. Med. 2, 32. doi:10.1038/s41525-017-0035-2
Tabolacci, E., Zollino, M., Lecce, R., Sangiorgi, E., Gurrieri, F., Leuzzi, V., et al. (2005). Two brothers with 22q13 Deletion Syndrome and Features Suggestive of the Clark???Baraitser Syndrome. Clin. Dysmorphol. 14, 127–132. doi:10.1097/00019605-200507000-00004
Tenorio, J., Nevado, J., González‐Meneses, A., Arias, P., Dapía, I., Venegas‐Vega, C. A., et al. (2020). Further Definition of the Proximal 19p13.3 Microdeletion/microduplication Syndrome and Implication of PIAS4 as the Major Contributor. Clin. Genet. 97 (3), 467–476. doi:10.1111/cge.13689
Thevenon, J., Duffourd, Y., Masurel-Paulet, A., Lefebvre, M., Feillet, F., El Chehadeh-Djebbar, S., et al. (2016). Diagnostic Odyssey in Severe Neurodevelopmental Disorders: toward Clinical Whole-Exome Sequencing as a First-Line Diagnostic Test. Clin. Genet. 89 (6), 700–707. doi:10.1111/cge.12732
Vallespín, E., Palomares Bralo, M., MoriÁ., M. Á., Martín, R., García-Miñaúr, S., Fernández, L., et al. (2013). Customized High Resolution CGH-Array for Clinical Diagnosis Reveals Additional Genomic Imbalances in Previous Well-Defined Pathological Samples. Am. J. Med. Genet. 161 (8), 1950–1960. doi:10.1002/ajmg.a.35960
Verhoeven, W. M. A., Egger, J., Willemsen, M. H., de Leijer, G. J., and Kleefstra, T. (2012). Phelan-McDermid Syndrome in Two Adult brothers: Atypical Bipolar Disorder as its Psychopathological Phenotype? Ndt 8, 175–179. doi:10.2147/ndt.s30506
Wilson, H. L., Crolla, J. A., Walker, D., Artifoni, L., Dallapiccola, B., Takano, T., et al. (2008). Interstitial 22q13 Deletions: Genes Other Than SHANK3 Have Major Effects on Cognitive and Language Development. Eur. J. Hum. Genet. 16, 1301–1310. doi:10.1038/ejhg.2008.107
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular Characterisation of the 22q13 Deletion Syndrome Supports the Role of Haploinsufficiency of SHANK3/PROSAP2 in the Major Neurological Symptoms. J. Med. Genet. 40, 575–584. doi:10.1136/jmg.40.8.575
Xu, N., Lv, H., Yang, T., Du, X., Sun, Y., Xiao, B., et al. (2020). A 29 Mainland Chinese Cohort of Patients with Phelan-McDermid Syndrome: Genotype-Phenotype Correlations and the Role of SHANK3 Haploinsufficiency in the Important Phenotypes. Orphanet J. Rare Dis. 15 (1), 335. doi:10.1186/s13023-020-01592-5
Zhang, Y., Kong, W., Gao, Y., Liu, X., Gao, K., Xie, H., et al. (2015). Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/developmental Disabilities. PLoS One 10, e0141782. doi:10.1371/journal.pone.0141782
Zhou, W. Z., Zhang, J., Li, Z., Lin, X., Li, J., Wang, S., et al. (2019). Targeted Resequencing of 358 Candidate Genes for Autism Spectrum Disorder in a Chinese Cohort Reveals Diagnostic Potential and Genotype-Phenotype Correlations. Hum. Mutat. 40 (6), 801–815. doi:10.1002/humu.23724
Ziats, C. A., Grosvenor, L. P., Sarasua, S. M., Thurm, A. E., Swedo, S. E., Mahfouz, A., et al. (2019). Functional Genomics Analysis of Phelan-McDermid Syndrome 22q13 Region during Human Neurodevelopment. PLoS One 14, e0213921. doi:10.1371/journal.pone.0213921
Ziats, C. A., Jain, L., McLarney, B., Vandenboom, E., DuPont, B. R., Rogers, C., et al. (2020). Neurofibromatosis Type 2 in Phelan-McDermid Syndrome: Institutional Experience and Review of the Literature. Eur. J. Med. Genet. 63 (11), 104042. doi:10.1016/j.ejmg.2020.104042
Zwanenburg, R. J., Ruiter, S. A. J., van den Heuvel, E. R., Flapper, B. C. T., and Van Ravenswaaij-Arts, C. M. A. (2016). Developmental Phenotype in Phelan-McDermid (22q13.3 Deletion) Syndrome: a Systematic and Prospective Study in 34 Children. J. Neurodevelop Disord. 8, 16–28. doi:10.1186/s11689-016-9150-0
Keywords: autistic behavior, 22q13 deletion syndrome, Phelan-McDermid syndrome (PMS), SHANK3, subtelomeric deletion syndrome, intellectual disabilities (ID)
Citation: Nevado J, García-Miñaúr S, Palomares-Bralo M, Vallespín E, Guillén-Navarro E, Rosell J, Bel-Fenellós C, Mori MÁ, Milá M, Campo Md, Barrúz P, Santos-Simarro F, Obregón G, Orellana C, Pachajoa H, Tenorio JA, Galán E, Cigudosa JC, Moresco A, Saleme C, Castillo S, Gabau E, Pérez-Jurado L, Barcia A, Martín MS, Mansilla E, Vallcorba I, García-Murillo P, Cammarata-Scalisi F, Gonçalves Pereira N, Blanco-Lago R, Serrano M, Ortigoza-Escobar JD, Gener B, Seidel VA, Tirado P and Spanish PMS Working Group (2022) Variability in Phelan-McDermid Syndrome in a Cohort of 210 Individuals. Front. Genet. 13:652454. doi: 10.3389/fgene.2022.652454
Received: 12 January 2021; Accepted: 16 February 2022;
Published: 12 April 2022.
Edited by:
Katalin Komlosi, Medical Center University of Freiburg, GermanyReviewed by:
Andreas Martin Grabrucker, University of Limerick, IrelandCopyright © 2022 Nevado, García-Miñaúr, Palomares-Bralo, Vallespín, Guillén-Navarro, Rosell, Bel-Fenellós, Mori, Milá, Campo, Barrúz, Santos-Simarro, Obregón, Orellana, Pachajoa, Tenorio, Galán, Cigudosa, Moresco, Saleme, Castillo, Gabau, Pérez-Jurado, Barcia, Martín, Mansilla, Vallcorba, García-Murillo, Cammarata-Scalisi, Gonçalves Pereira, Blanco-Lago, Serrano, Ortigoza-Escobar, Gener, Seidel, Tirado, Lapunzina and Spanish PMS Working Group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián Nevado, am5ldmFkb0BzYWx1ZC5tYWRyaWQub3Jn
†ORCID: Julian Nevado, orcid.org/0000-0001-5611-2659; Pablo Lapunzina, orcid.org/0000-0002-6324-4825; Luis Pérez-Jurado, orcid.org/0000-0002-1988-3005
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.