 Haoyu Pan1,2
Haoyu Pan1,2 Hui Deng
Hui Deng Cheng Song
Cheng Song- 1College of Biological and Pharmaceutical Engineering, West Anhui University, Luan, China
- 2School of Life Science, Anhui Agricultural University, Hefei, China
With the rapid advancement of high-throughput sequencing technology, it is now possible to identify individual gene families from genomes on a large scale in order to study their functions. WRKY transcription factors are a key class of regulators that regulate plant growth and abiotic stresses. Here, a total of 74 WRKY genes were identified from Dendrobium officinale Kimura et Migo genome. Based on the genome-wide analysis, an in-depth analysis of gene structure and conserved motif was performed. The phylogenetic analysis indicated that DoWRKYs could be classified into three main groups: I, II, and III, with group II divided into five subgroups: II-a, II-b, II-c, II-d, and II-e. The sequence alignment indicated that these WRKY transcriptional factors contained a highly conserved WRKYGQK heptapeptide. The localization analysis of chromosomes showed that WRKY genes were irregularly distributed across several chromosomes of D. officinale. These genes comprised diverse patterns in both number and species, and there were certain distinguishing motifs among subfamilies. Moreover, the phylogenetic tree and chromosomal location results indicated that DoWRKYs may have undergone a widespread genome duplication event. Based on an evaluation of expression profiles, we proposed that DoWRKY5, 54, 57, 21, etc. may be involved in the transcriptional regulation of the JA signaling pathway. These results provide a scientific reference for the study of DoWRKY family genes.
Introduction
External environment seriously affect plant growth and food security (Waqas et al., 2019). Among them, abiotic stresses, such as UV-B, drought, chilling, heavy metals, etc., inevitably lead to the decline of medicinal material quality and the accumulation of harmful substances (Jiang et al., 2020). The discovery of large-scale gene families that are involved in abiotic stress opens up new ways to deal with and control harmful conditions (Song et al., 2022a). Bioinformatics is an emerging field developed by integrating biology with computer science and mathematics (Kan et al., 2021). It tackles the barrier of processing huge amounts of biological data by applying computer science and technology and statistical methodologies thoroughly. With the advancement of high-throughput sequencing technology, genome assembly and sequencing, in conjunction with bioinformatics algorithms, have catapulted life sciences into the “omics” era (Jing and Liu, 2018; Song et al., 2021). The quantity of data contained in these omics metadata is enormous, and the relationships between the datasets are intricate. To comprehend the relationship between the structure and function of regulatory elements, it is often necessary to study several genes simultaneously (Patro et al., 2017). This mainly includes structural genomes, comparative genomes, and functional genomes. The investigation of the differentially expressed genes generated from the experiment involves large-scale data analysis of online services and local area networks, providing researchers with reference and optional data analysis tools (Kang et al., 2020). Computational biology is frequently employed to decode medicinal plant genomes, pan-genomes and molecular markers, as well as to explore the origin and evolution of related species, structural variation of genomes, neofunctionalization of gene families, and systematic biology, among other applications (Nadler et al., 1997). In terms of genome assembly, the assembly strategy and an algorithm based on overlapping graphs are applied to construct the process. PacBio CCS sequencing with Hi-C technology, Oxford Nanopore, and other third-generation sequencing technologies has been extensively employed to sequence non-model species in recent years (Sun et al., 2020; Ye et al., 2021). In terms of gene prediction, de novo sequencing based on hidden Markov models enhances prediction accuracy (Wang F et al., 2021). Regarding functional gene mining, bioinformatics can closely integrate the metabolome, genome, transcriptome and proteome, and examine the natural variation of metabolic regulatory genes using principle component analysis, hierarchical clustering analysis, and correlation analysis (Srivastava et al., 2018). Some researchers also use metabolome-genome wide association analysis (mGWAS) to investigate the underlying genetic basis of metabolite biosynthesis pathways (Zhang et al., 2020).
D. officinale is a perennial herb in the orchid family that belongs to the Dendrobium genus (Wang Y et al., 2021). It is mainly distributed in the Ta-pieh Mountain of Anhui, Zhengjiang, Guangzhou, Guangxi, Yunnan, and other provinces (Song et al., 2022b). D. officinale has considerable effects on immunity and hematopoiesis, as well as anti-oxidation, anti-tumor, anti-fatigue, and hypoglycemic properties (Song et al., 2022d). WRKY transcription factor family is one of the most extensive transcription factor families found in higher plants (Li et al., 2020). It is a crucial class of transcription factors that regulates plant growth, development, and stress tolerance (Guo et al., 2019; Jue et al., 2018). The representative aspect of WRKY transcription factors is the conserved WRKY domain, which consists of the highly conserved WRKYGQK heptapeptide and C2-H2 (C-X4-C-X22-23-H-X1-H) or C2-HC (C-X4-C-X23-H-X1-C) type zinc finger motifs (Cui et al., 2018; Jiao et al., 2018). WRKY transcription factors have a strong affinity for the common W-box element, which has a structural base consisting of a heptapeptide sequence and a zinc finger motif (Nan and Gao, 2019). Accordingly, WRKY proteins are classified into three major groups (I-III) based on the number of WRKY domains and the structure of their zinc finger motifs. The group II is further divided into five subclasses: II-a, II-b, II-c, II-d, and II-e. Since the first WRKY gene SPF1, was cloned from sweet potato, a vast number of WRKY members have been discovered in plants (He et al., 2017; Hu et al., 2021).
Genome-wide identification of gene families clarified the structure and function of each gene, and provided a theoretical foundation for future functional verification (Tang et al., 2021). Simultaneously, using the homologous gene alignment of many species, related gene sequences with high affinity for their motifs may be determined, as they are regarded as the probable functions of homologous genes. In 2015, Chen and his collaborators started developing BioCJava (the origin of TBtools). It is a biological large-scale data analysis software written in Java with over 140 functional modules and a good human-computer interface. The JIGplot engine provides a user-friendly interface for displaying graphs (Chen et al., 2020). Here, bioinformatics methods were used to identify the DoWRKY transcription factors. The amino acid alignment, gene structure analysis, phylogenetic tree construction, and chromosomal location were performed by TBtools (v.1.097), MEGA (v.6.06), and some other bioinformatics analysis softwares. The conserved motifs, cis-acting regions, and amino acid binding site found in the data can help enrich our understanding of WRKY genes and roles, as well as provide some theoretical references for further investigation.
Materials and methods
Identification of the WRKY genes in D. officinale
The latest D. officinale genome and annotation files were obtained from the NCBI genome database (BioProject accession: PRJNA662181). The AtWRKY sequence files and seed files (PF03106) from the Arabidopsis Information Resource (https://www.arabidopsis.org) and Pfam (http://pfam.xfam.org/) web services allow us to download the hmm file of the hidden Markov model containing the WRKY typical conserved domains (Supplementary Table S1). MEGA, and TBtools software are used for further sequence analysis (Tamura et al., 2013; Chen et al., 2020). The hmmbuild command in the hmmer package was used to create the WRKY.hmm file from the seed file. Following the acquisition of the WRKY.hmm file, the hmmsearch command was used to retrieve the WRKY.out file. To verify the comparison results, the TBtools software was used to extract the protein sequence of the WRKY proteins from the.out file to obtain the WRKY.fas file, resulting in a screened sequence file only containing the target protein. These proteins were submitted to the batch sequence search of pfam for verification. Subsequently, all candidate DoWRKYs were verified using Pfam and SMART to confirm that they contained the core domains. Based on the sequence alignments generated by the ClustalX software (http://www.clustal.org/clustal2/), all potentially redundant WRKY sequences were discarded. A preprint has previously been published (Pan et al., 2022).
The phylogenetic analysis and amino acid alignment of WRKY transcription factors
MEGA 6 (v.6.06) was used to create a protein sequence alignment project (Tamura et al., 2013). The screened DoWRKYs and the AtWRKYs were concurrently imported to construct an alignment. After the alignment, the sequence differences of the WRKY transcription factors of the two species are retrieved, and the genetic relationship between the different WRKY transcription factors of the two species can be detected. The phylogenetic tree was constructed based on the WRKY conserved domain of D. officinale and A. thaliana using a neighbour-joining method (execution parameters: Poission correction, pairwise deletion, and bootstrap of 1,000 repetitions). The related DoWRKY proteins can be classified by referring to the classification of defined AtWRKY proteins. The IQ-TREE software (v.1.6.12) was used to further determine the phylogeny relationship between DoWRKYs and AtWRKYs (Nguyen et al., 2014). Our best-fit model to determine sequence alignment through method optimization is VT + R7. Using this model, we construct the phylogenetic tree based on the maximum likelihood method. iqtree.exe -s./bidui.fas-m VT + R7-bb 1000-alrt 1000-nt AUTO was the running parameter. To analyze the amino acid sequence of DoWRKYs and determine the conserved domains, the GeneDoc (v. 2.7) (https://genedoc.software.informer.com/2.7/) was used. The phylogenetic trees were used to assess the homology of DoWRKYs with other WRKY proteins.
The conserved motif analysis of WRKY transcription factors
The screened DoWRKY protein sequence.fa file was submitted to the MEME online web service (http://meme.nbcr.net/meme/tools/meme) for searching the conserved motif. The following settings were set: The number of motifs is set to 20. The width of the advanced option motifs was in the range of 6–200. All of the other parameters were set to default. The xml file can be obtained after submitting it to the MEME-suite service (https://meme-suite.org/meme/tools/meme), or by utilizing the MEME suite Wrpper from the TBtools package (Chen et al., 2020). To obtain a map of conserved motif analysis, the biosequence structures illustrator was used to display a motif pattern with the default settings.
The gene structure analysis and chromosomal localization of WRKY transcription factors
Using the biosequence structures illustrator tool, the gene structure of DoWRKYs was obtained based on the gene annotation file. The settings are made in order to obtain the generic structural map. The color and height of the gene structure of CDS and UTR regions can be adjusted. Using the genes on chromosomes function module of TBtools, the gene location was visualized based on the gene annotation file. All of the WRKY genes on the pseudochromosomes can be found by putting in the file after the gene names have been changed.
The expression profile of DoWRKY genes under different MeJA treatment
The expression pattern of WRKY gene family was further analyzed using the transcriptome data of D. officinale. First of all, the clean data were mapped to the latest genome of D. officinale (https://www.ncbi.nlm.nih.gov/genome/31795?genome_assembly_id = 1672529). Then, the HISAT2 software was applied to align the obtained clean data with the reference genome, and the comparison efficiency was calculated to evaluate the assembly quality of the selected reference genome (Pertea et al., 2016). The StringTie software was used to assemble the aligned reads, and the obtained unigenes were quantitatively analyzed (Pertea et al., 2015). The gene expression levels of DoWRKY genes were represented using the fragments per kilobase of transcript per million mapped reads (FPKM) method (Sims et al., 2014). TBtools software was used to build a heatmap illustrating the expression profile of groups with drastically altered genes (Chen et al., 2020).
Results
Identification and category of DoWRKY genes
To create the WRKY.hmm file from the seed file, the hmmbuild function of the hmmer software was employed. Using the hmmsearch function, the WRKY.out file was retrieved based on the WRKY.hmm file. TBtools software was used to extract the protein sequence of the WRKY protein id in the initial alignment of the.out file, obtain the WRKY transcription factor sequence file, submit the DoWRKY protein sequence file to the Pfam website for verification, and then compare the NCBI database to remove those that do not meet the requirements (Table 1). We renamed DoWRKY1-DoWRKY74 after the analysis of chromosomal location. The basic properties of these genes include subfamily classification, type of zinc finger structure, number of amino acids, molecular weight, and isoelectric point. The full name of the protein spans from 72 to 1,583 aa (DoWRKY60 and DoWRKY33). Some truncated proteins may be alternative splices of their paralogs, such as DoWRKY60, DoWRKY9, and DoWRKY31. DoWRKY33 has longer amino acids, suggesting that it may contain LTR-retrotransposons. It was indicated that DoWRKY transcription factors could be classified into three types: group I, group II, and group III, with group II further subdivided into five subclasses: II-a, II-b, II-c, II-d, and II-e (Figure 1). Both groups I and II have two highly conserved WRKY amino acid sequences. Group II has a WRKY amino acid sequence. Both groups I and II have a C2-H2 type of zinc finger structure, and group III has a WRKY amino acid sequence. There are 13 class I WRKY proteins, eight class II-a WRKY proteins, three class II-b WRKY proteins, 16 class II-c WRKY proteins, and six class d WRKY proteins, 11 class II-e WRKY proteins, 17 class III WRKY proteins.
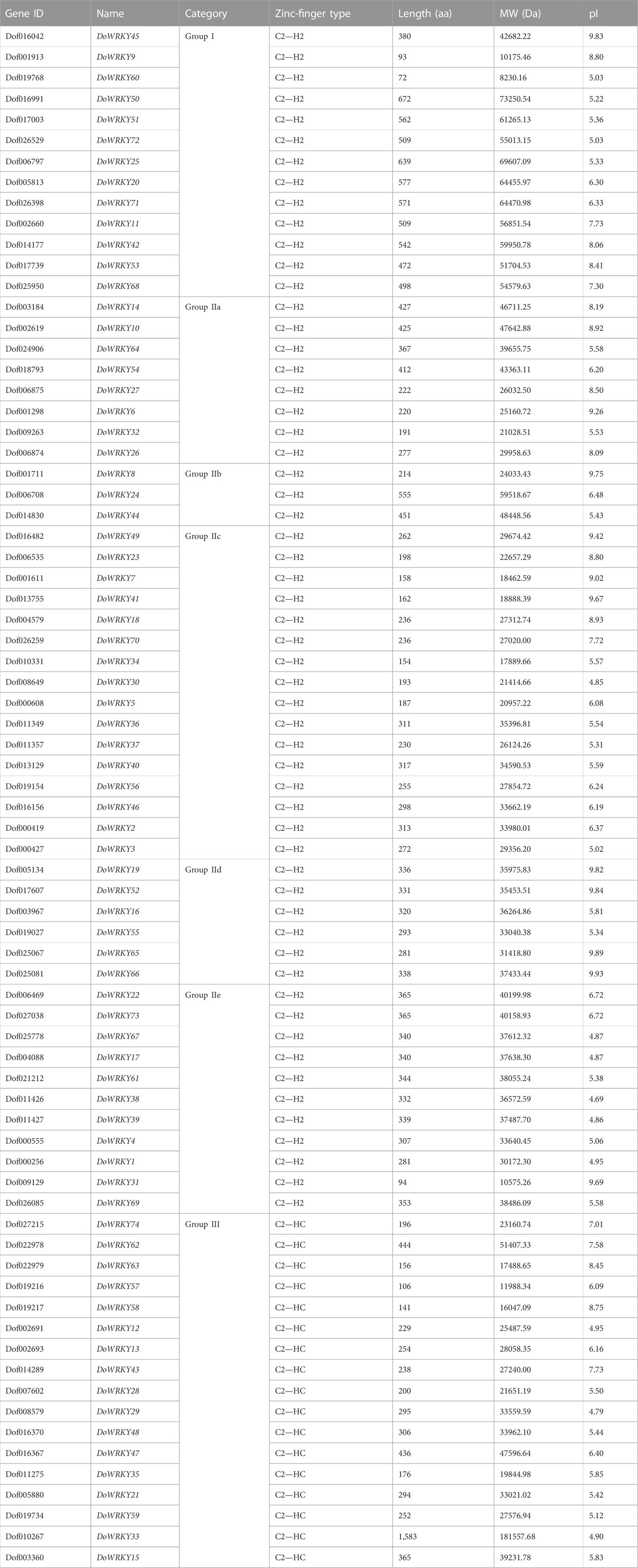
TABLE 1. Classification and structural properties of DoWRKYs.

FIGURE 1. Multiple sequence alignment of the WRKY domain among DoWRKY proteins. Red indicates conserved WRKY domains. Purple indicates zinc finger motifs and dashes indicate gaps.
The phylogenetic analysis of DoWRKY genes
We imported the WRKY transcription factors into MEGA 6.0. The Dendrobium.fa file and the WRKY transcription factor aligned the muscular function of A. thaliana to compare and analyze the protein sequences of the two WRKY transcription factors, and we acquired the WRKY transcription factors of the two species after alignment. The NJ method was used to generate a phylogenetic tree map from sequence differences. An analogy was made to the related DoWRKY transcription factors based on the grouping of distinct WRKY transcription factors in the evolutionary tree, alluding to the obvious classification of AtWRKY transcription factors. In Dendrobium, WRKY transcription factors can be found in different parts of the plant, and the comparison and analysis of amino acids were fully described (Figure 2). The results indicated that all DoWRKYs could be divided into three groups and clustered separately from AtWRKYs. However, due to the distant relationship between Arabidopsis and Dendrobium, it can be seen that orthologs on the same branch, such as DoWRKY45 and AtWRKY10, DoWRKY49 and AtWRKY13, DoWRKY23 and AtWRKY12, DoWRKY35 and AtWRKY30, etc., have no similarity greater than 80%. To more accurately display the degree of homology between DoWRKY and other AtWRKY10 members, we used IQ-TREE software to construct an ML tree (Figure 3). The results show that these DoWRKYs can still be dispersed into three groups and five subgroups (group II). The clustering of the phylogenetic tree constructed by the ML method was basically consistent with the evolutionary tree constructed by the NJ method. Interestingly, we found that DoWRKY from group IIc could be further divided into two branches; we temporarily call them group IIc-1 (left) and group IIc-2 (right). In group IIc-1 subclade, there are 10 DoWRKYs clustered in one branch, while only seven DoWRKYs were in group IIc-2 subclade.
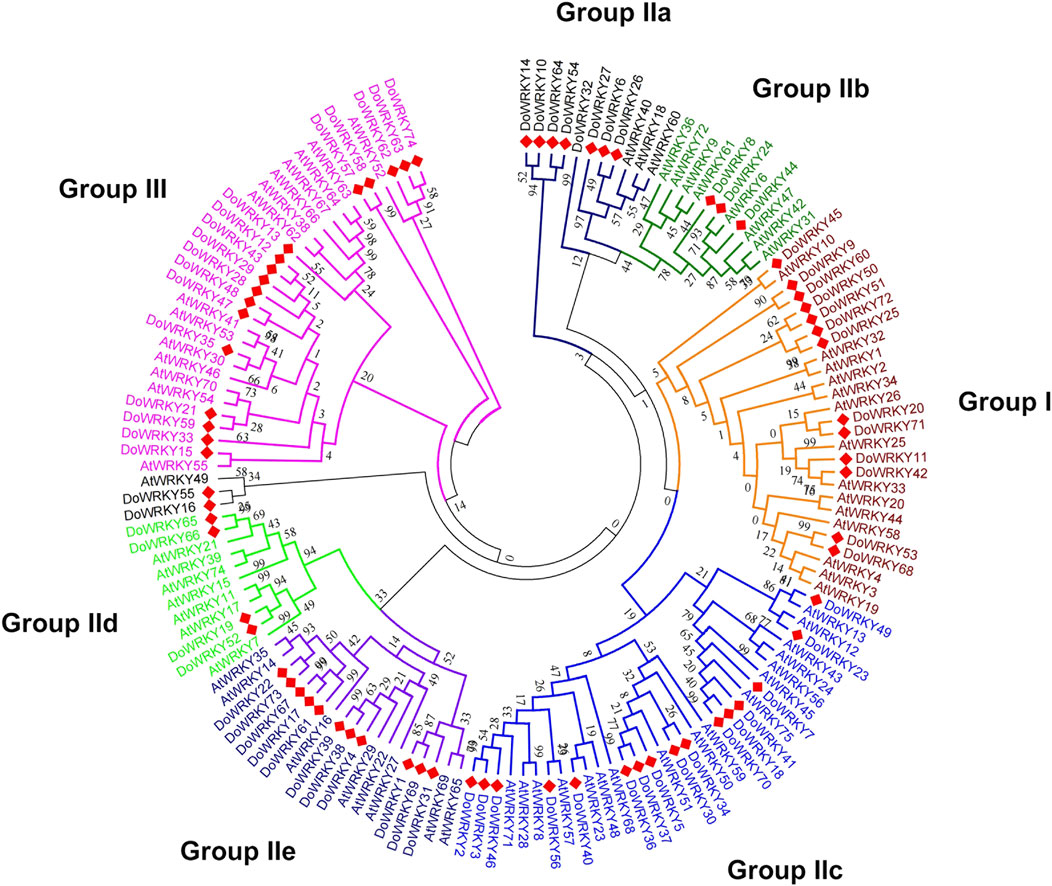
FIGURE 2. The phylogenetic tree of DoWRKYs and AtWRKYs by neighbor-joining method.
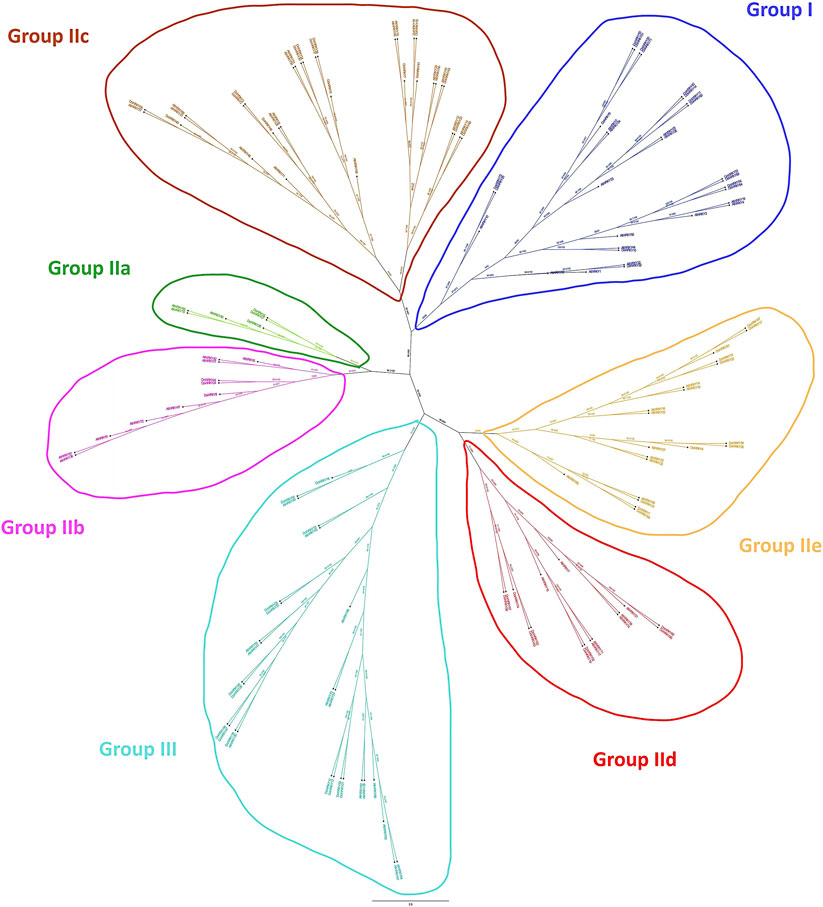
FIGURE 3. The phylogenetic tree of DoWRKYs and AtWRKYs by maximum likelihood method.
The conserved motif analysis of DoWRKY genes
To investigate the conserved motifs of DoWRKY genes, the screened DoWRKY protein sequence.fa file was submitted to the MEME online analysis website. Rom the xml files, the TBtools software was used to visualize motif patterns, set and retain 20 motifs, and obtain the map of conserved motif analysis. The analysis results show that the number and types of motif structures of different DoWRKY transcription factors are quite different, but the number and types of motif structures of the same type of DoWRKY genes are very close. According to the phylogenetic tree, the conserved motifs of DoWRKY genes were as follows: motif 1 and motif three are characteristic motifs containing WRKYGQK, which are in line with phylogenetic classification. The number of motifs contained in each WRKY transcription factor varies from 1 to 11, among which class I DoWRKY generally has the most motifs. Some DoWRKY genes, like motifs 9, 17, 19, etc., have subfamily-specific motif sequences. The distribution of specific motif was shown in Figure 4.
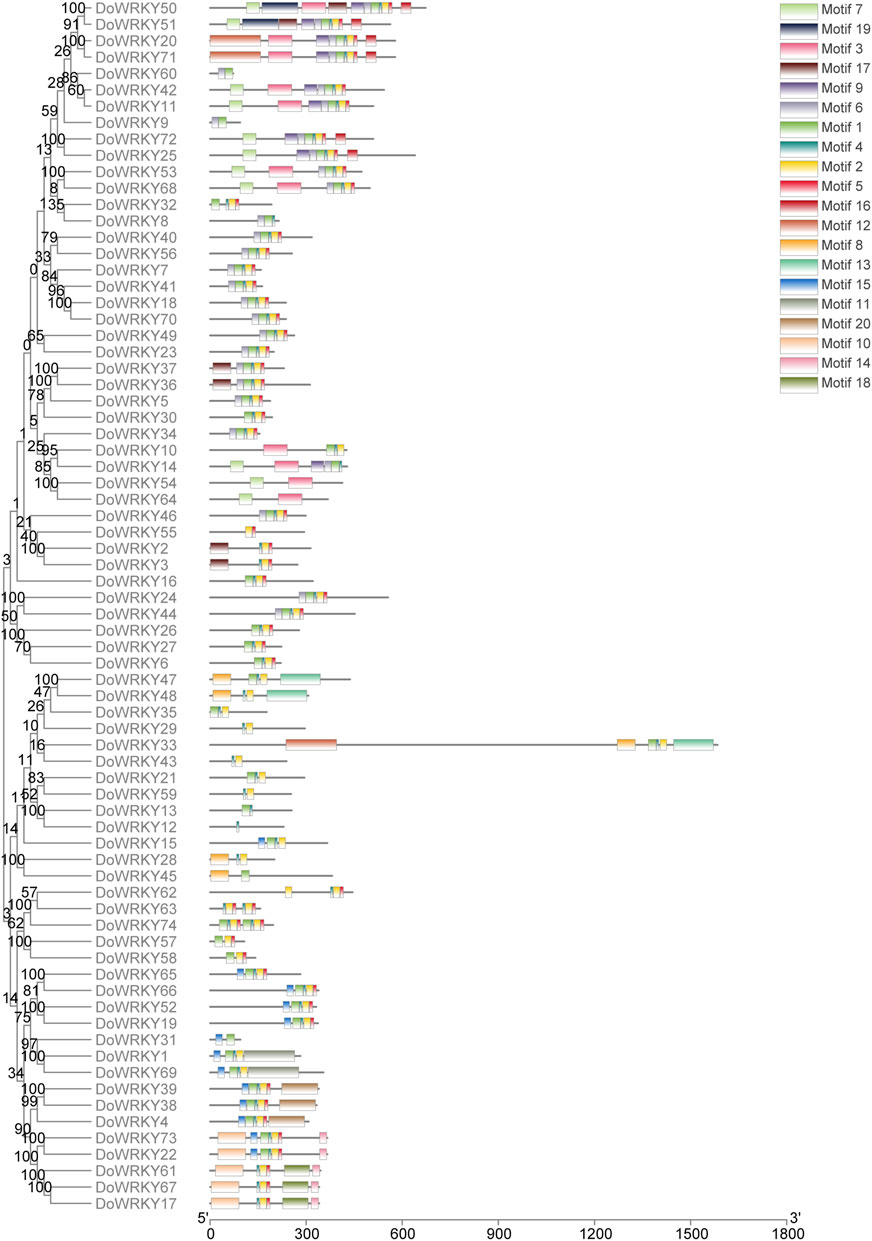
FIGURE 4. Analysis of the conserved motifs of DoWRKY genes.
The gene structure analysis of DoWRKY genes
The diversity of gene structures is favorable for the emergence of large gene families. The genomic structure of each DoWRKY gene was mapped. To better understand the gene structure, a phylogenetic tree was made based on the full-length DoWRKY proteins. Following the extraction of the gene ID corresponding to the screened DoWRKY transcription factors, the TBtools software was employed to visualize the gene structure based on the gene annotation file. Different types of DoWRKY genes vary greatly in length, numbers of exons and introns, and starting sites. All DoWRKY genes have 1-6 exons. WRKY-like transcription factor genes have the largest number of exons. The number of introns in DoWRKYs is between 0 and 3, and most of them have 1-2 introns. The same type of gene structure in DoWRKYs was very similar. Most of them have the same number of exons. CDS start sites and UTR were also very close. The distribution of exons and introns is very different between different subclasses, such as class I and class III. There are differences in gene length between classes, and large differences in the distribution of exons and introns (Figure 5).

FIGURE 5. The gene structure analysis of DoWRKY genes.
The chromosomal location of DoWRKY genes
From the Dendrobium genome annotation information gff3 file, TBtools software was used to perform the chromosomal location analysis based on the gene annotation file. After gene renaming, those genes can be introduced into the file to obtain the location of the WRKY transcription factor gene on the genome. It is found that 74 DoWRKY genes are irregular and uneven. The distribution of DoWRKY genes on each chromosome segment, some of which are not located on the chromosome, may be caused by the fact that these DoWRKY genes are not annotated on the chromosome. The chromosomal location of the gene was shown in Figure 6. As DoWRKY genes were unevenly distributed throughout all chromosomes, and the number of genes on each chromosome was unrelated to its length. Eight genes, including DoWRKY67, DoWRKY69, DoWRKY70, etc., were dispersed throughout eight large segments while the majority of genes were distributed across 19 scaffolds. Chromosome 1 contained the largest number of DoWRKY genes. Several patterns of gene duplication have been observed, such as whole-genome duplication (or segmental duplication) and tandem duplication. The genes from tandem duplication, including DoWRKY2 and DoWRKY3, DoWRKY12 and DoWRKY13, DoWRKY26 and DoWRKY27, DoWRKY38 and DoWRKY39, etc., are scattered throughout the genome. Those genes with higher homology but not located on the same chromosome may be attributed to segmental duplication.
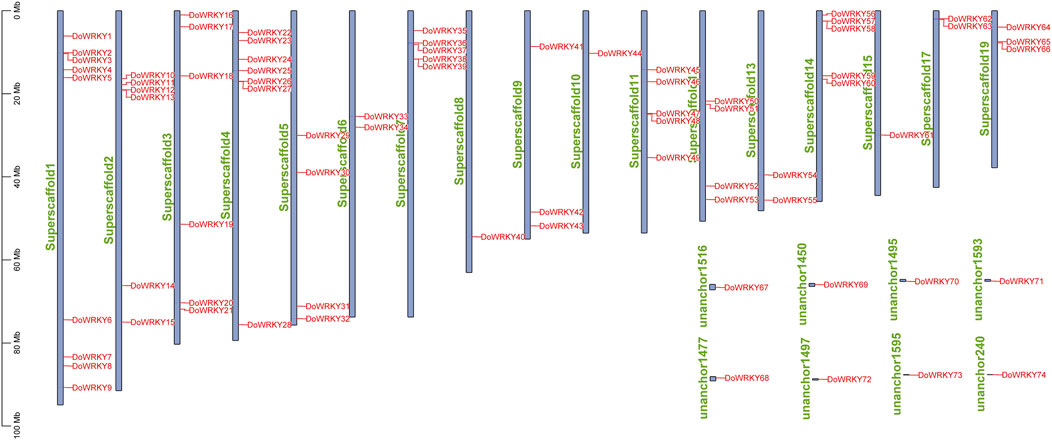
FIGURE 6. The chromosome location analysis of DoWRKY genes.
The expression profile of DoWRKY genes under MeJA treatment
To further clarify the roles of the WRKY genes in abiotic stress, we conducted a comparative transcriptomic analysis of MeJA-treated D. officinale at different stages (Jiao et al., 2022). The results showed that WRKY family members were differentially expressed at different treatment times and showed two expression patterns (Figure 7). The first is that most WRKY genes are not expressed before and after treatment, and a small number of WRKY genes are down-regulated; the second is that this group of WRKY genes is significantly up-regulated, especially DoWRKY5. Whether in the control or MeJA treatment groups, a portion of the WRKY genes, such as DoWRKY54, 57, 21, and 58 reached the highest expression level at 4 h and gradually decreased at 24 h but did not return to the initial level (Supplementary Table S2). Therefore, the latter may be involved in the regulation of JA-responsive genes. The relevant transcriptional regulation mechanism needs further validation.

FIGURE 7. The expression profiles of DoWRKYs after MeJA treatment. The expression level of genes were normalized by log2 (FPKM value).
Discussion
As a crucial family of transcription factors in the regulation of plant growth, development, and stress resistance, WRKY transcription factors are involved in a variety of physiological processes in plants. The genome-wide investigation of the WRKY gene family has been undertaken thoroughly in hundreds of species, and the genomes of many species have begun to be sequenced gradually (Li et al., 2019). Owing to the achievement of cutting-edge high-throughput sequencing technology, some Dendrobium species have been widely sequenced in the last 5 years. In this study, the WRKY genes in the Dendrobium genome were identified, and 74 of them met the criteria. This was less than the 91 WRKY transcription factors found in the Arabidopsis genome. The majority of them shared similar domains with AtWRKYs, and they all have the WRKYGQK and zinc finger structures with ZF. The heptapeptide sequences of DoWRKY include WTKTGQK, WKKYGQK, WRKYGRD, WRKYGKK, and other types. The group II WRKY transcription factors in Dendrobium have a maximum of 44 members, whereas groups I and III merely have 13 and 17 members, respectively. The phylogenetic analysis indicated that groups I and III of Dendrobium are more closely related than groups II-a and II-b. The same branch is closely connected, and II-d and II-e are likewise related. The conserved domains showed that DoWRKY genes contain multiple conserved domains. There are additional subgroups of typical conserved domains in addition to the core WRKUGQK conserved domains. The conserved domains of DoWRKY are mostly similar. The group I DoWRKYs have the most conserved domains and may have significant regulatory functions (He et al., 2017; Tang et al., 2021). In Arabidopsis, the proteins AtWRKY25 and AtWRKY23 may interact with MAP kinase four substrate 1 (MKS1), which is essential for the regulation of SA-dependent resistance. The SA-related defense gene PR1 was identified to be expressed at a higher level in an Atwrky33 mutant (Andreasson et al., 2005). AtWRKY50 and AtWRKY51 proteins controled both SA- and low oleic acid-dependent inhibition of JA signaling, resulting in greater resistance to Alternaria brassicicola but increased susceptibility to Botrytis cinerea (Gao et al., 2011). AtWRKY57 regulates the plant immune response process and competes with AtWRKY33 by binding to the promoters of JAZ1 and JAZ5 (Jiang and Yu 2016). DoWRKY11 and DoWRKY14 clustered with AtWRKY25 and 33 in one clade, while DoWRKY40 clustered with AtWRKY23 in another subclade. DoWRKY30, DoWRKY34, and AtWRKY50/51 merged a single branch. These results imply that DoWRKY11, 14, 40 and DoWRKY30, 34 have different regulatory effects on the target genes involved in SA signaling. In addition to being involved in phytohormone signaling, WRKY genes are involved in leaf senescence. AtWRKY22, AtWRKY54, AtWRKY70, AtWRKY57, AtWRKY45, AtWRKY75, AtWRKY6, AtWRKY46, AtWRKY25, AtWRKY53, and AtWRKY55 participate in the progression of leaf senescence. AtWRKY54, 70, has high homology with DoWRKY12, 13, and DoWRKY21, 59, respectively. DoWRKY15 and AtWRKY55 were clustered into one branch, which suggested that they might be involved in the leaves senescence process of D. officinale. Many studies had also shown that some members of the WRKY family helped mediate the biosynthesis of secondary metabolites in plants (Schluttenhofer and Yuan, 2015). D. officinale is also an important Chinese herbal medicine. It will be of great significance if some key DoWRKY genes can be isolated. AtWRKY18 and AtWRKY40 are implicated in camalexin and indole-glucosinolate biosynthesis, and the accumulation of these compounds was required for resistance toward Golovinomyces orontii (Schön et al., 2013). The ectopic expression of AtWRKY18, AtWRK40, and AtMYC2 activated the MEP pathway genes of Salvia sclarea, leading to an increase in abietane diterpene levels (Alfieri et al., 2018). In this study, DoWRKY26 shared a high level of homology with AtWRKY18, 40, and 60; hence, it was suggested that WRKY26 might be involved in the transcriptional regulation of secondary metabolite biosynthetic genes (Figure 8).
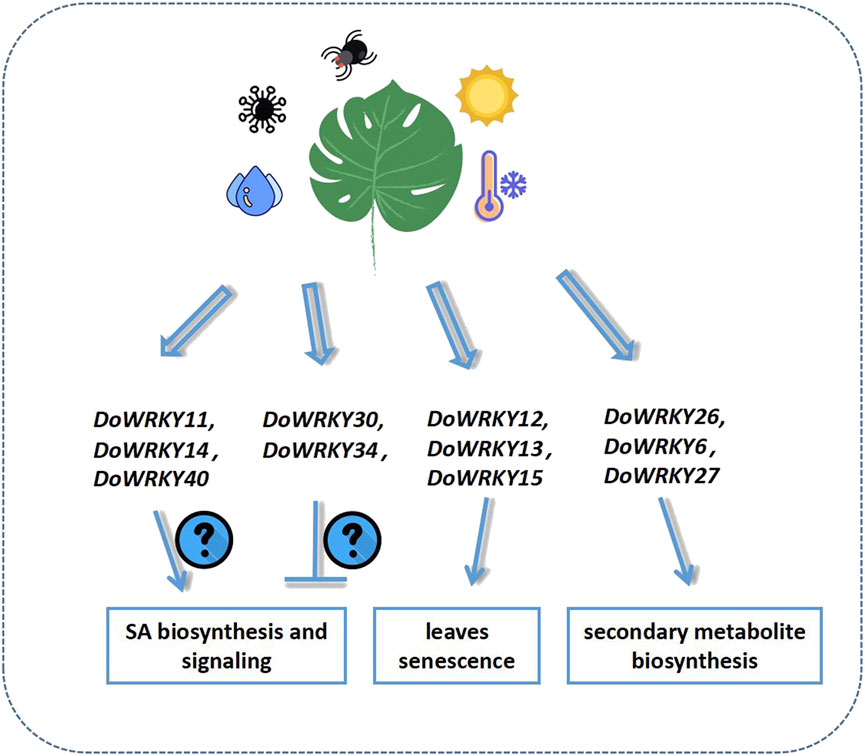
FIGURE 8. The putative roles of some DoWRKYs in D. officinale.
Gene duplications are critical to the rapid expansion of the genome and the evolution of gene families (Song et al., 2022c). Polyploidy drives species adaption, genetic diversity, and genome evolution. In the field of horticulture, chromosomal polyploidy produces a number of excellent orchid varieties (Vilcherrez-Atoche et al., 2022). Much evidence indicated that duplication and expansion of WRKY genes occurred in the interspecies (Yang et al., 2018). Numerous evidence suggested that duplication and expansion of WRKY genes occurred among species (Yang et al., 2018). The sequencing of the whole genome of A. shenzhenica boosts our knowledge of the history and evolution of those subfamilies (Zhang et al., 2017). It is notable that whole genome duplication (WGD) has occurred more than once in plant genomes (Clark and Donoghue, 2018). Large-scale tandem duplication or segmental duplication start driving the generation of new genes and speciation evolution (Clark and Donoghue, 2018). Typically, orchids have experienced WGD repetitively, including a historical WGD event and a recent WGD event shared by all orchids. Mycoheterotrophic and parasitic orchids exist with the great majority of ornamental orchids. The loss and survival of symbiotic genes connected to the evolution of particular symbionts spans from the ancestral arbuscular mycorrhiza to the recent ericoid and orchid mycorrhizae (Barrett et al., 2019; Gao et al., 2020). Here, nine tandem duplication gene pairs of DoWRKY were confirmed in peusodochromosomes. Those distributed across multiple scaffolds with a high degree of similarity may result in segmental duplications. This observation is consistent with that of several model plants. The gene structure of DoWRKYs within the same family is comparable, as are the number of CDS and initiation sites, as well as the number and location of introns, despite the major differences within families, such as group I. The number of CDSs and intron sites differs significantly between group I and III, implying that class I WRKY transcription factors have more functions. DoWRKYs are unevenly located on different chromosomes or segments, and some have not yet been located on the chromosome, such as DoWRKY54/59.
Conclusion
The WRKY transcription factor family, as one of the largest TF families, plays a significant and necessary role in plants. Over the years, it has been demonstrated that WRKYs not only contribute to plant growth and development but also exhibit intricate regulatory mechanisms. Here, the identification of DoWRKY reveals the membership of the DoWRKY gene family and elucidates the function and structure of each gene, which could provide a theoretical basis for future functional verification studies. Using the homologous gene alignment of several species, it is possible to simultaneously identify similar homologous genes with great affinity for their motifs as well as infer the potential functions of related subgroup genes. We combined TBtools, MEGA, and other software to identify 74 DoWRKY genes, along with the analysis of amino acid alignment, gene structure, conserved motif, phylogeny, and chromosomal location. Large-scale gene duplication may contribute to the functional preference of the WRKY genes. This includes the emergence of tandem replicated genes. By analyzing conserved motifs and domains, we can divide WRKY genes into three groups. Analysis of phylogenetic tree indicated that some DoWRKY genes shared homology with AtWRKY, which allowed us to speculate on their potential functions. In addition, we preliminarily deduced that DoWRKY5, 54, 57, and 21 may be involved in the JA signaling by analyzing the expression patterns. As a result, we can learn more about WRKY transcription factors and how they work, and we can also use the results of this analysis to learn more about WRKY genes and their functions in other species.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
The authors confirm contribution to the paper as follows: Study conception and design: CS and HP; data collection: CS, HP, and YC; analysis and interpretation of results: HD, YC, JZ, JH, and NS; draft manuscript preparation: CS and HP; funding acquisition: CS and HD. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work is supported by Anhui Provincial Key Research and Development Program (201904f06020007), High-level Talents Research Initiation Funding Project of West Anhui University (WGKQ2022025), Innovation and Entrepreneurship Training Program for College Students (S202110376171, 202110376088X, and S202210376029X), and the key project of the Anhui Provincial Department of Education (KJ2019A0626).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1104968/full#supplementary-material
References
Alfieri, M., Vaccaro, M. C., Cappetta, E., Ambrosone, A., Tommasi, N. De, Leone, A., et al. (2018). Coactivation of MEP - biosynthetic genes and accumulation of abietane diterpenes in Salvia sclarea by heterologous expression of WRKY and MYC2 transcription factors. Sci. Rep. 8, 1–13. doi:10.1038/s41598-018-29389-4
Andreasson, E., Jenkins, T., Brodersen, P., Thorgrimsen, S., Petersen, N. H. T., Zhu, S., et al. (2005). The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J. 24, 2579–2589. doi:10.1038/sj.emboj.7600737
Barrett, C. F., Sinn, B. T., Kennedy, A. H., and Pupko, T. (2019). Unprecedented parallel photosynthetic losses in a heterotrophic orchid genus. Mol. Biol. Evol. 36, 1884–1901. doi:10.1093/molbev/msz111
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi:10.1016/j.molp.2020.06.009
Clark, J. W., and Donoghue, P. C. J. (2018). Whole-genome duplication and plant macroevolution. Trends Plant Sci. 23, 933–945. doi:10.1016/j.tplants.2018.07.006
Cui, Q., Yan, X., Gao, X., Zhang, D., He, H. B., and Jia, G. X. (2018). Analysis of WRKY transcription factors and characterization of two Botrytis cinerea-responsive LrWRKY genes from Lilium regale. Plant Physiology Biochem. 127, 525–536. doi:10.1016/j.plaphy.2018.04.027
Gao, Q., Venugopal, S., Navarre, D., and Kachroo, A. (2011). Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 155, 464–476. doi:10.1104/pp.110.166876
Gao, Y., Zhao, Z., Li, J., Liu, N., Jacquemyn, H., Guo, S., et al. (2020). Do fungal associates of co-occurring orchids promote seed germination of the widespread orchid species Gymnadenia conopsea? Mycorrhiza 30, 221–228. doi:10.1007/s00572-020-00943-1
Guo, H., Zhang, Y., Wang, Z., Lin, L., Cui, M., Long, Y., et al. (2019). Genome-wide identification of WRKY transcription factors in the asteranae. Plants 8 (10), 1–19. doi:10.3390/plants8100393
He, C., Teixeira da Silva, J. A., Tan, J., Zhang, J., Pan, X., Li, M., et al. (2017). A genome-wide identification of the WRKY family genes and a survey of potential WRKY target genes in dendrobium officinale. Sci. Rep. 7 (1), 9200–9214. doi:10.1038/s41598-017-07872-8
Hu, W., Ren, Q., Chen, Y., Xu, G., and Qian, Y. (2021). Genome-wide identification and analysis of WRKY gene family in maize provide insights into regulatory network in response to abiotic stresses. BMC Plant Biol. 21 (1), 1–21. doi:10.1186/s12870-021-03206-z
Jiang, W., Wu, Z., Wang, T., Mantri, N., Huang, H., Li, H., et al. (2020). Physiological and transcriptomic analyses of cadmium stress response in Dendrobium officinale seedling. Plant Physiology Biochem. 148, 152–165. doi:10.1016/j.plaphy.2020.01.010
Jiang, Y., and Yu, D. (2016). The WRKY57 transcription factor affects the expression of jasmonate ZIM-domain genes transcriptionally to compromise Botrytis cinerea resistance. Plant Physiol. 171, 2771–2782. doi:10.1104/pp.16.00747
Jiao, Z., Sun, J., Wang, C., Dong, Y., Xiao, S., Gao, X., et al. (2018). Genome-wide characterization, evolutionary analysis of WRKY genes in Cucurbitaceae species and assessment of its roles in resisting to powdery mildew disease. PLoS ONE 13 (12), 1–19. doi:10.1371/journal.pone.0199851
Jiao, C., Wei, M., Fan, H., Song, C., Wang, Z., Cai, Y., et al. (2022). Transcriptomic analysis of genes related to alkaloid biosynthesis and the regulation mechanism under precursor and methyl jasmonate treatment in Dendrobium officinale. Front. Plant Sci. 13, 1–17. doi:10.3389/fpls.2022.941231
Jing, Z., and Liu, Z. (2018). Genome-wide identification of WRKY transcription factors in kiwifruit (Actinidia spp.) and analysis of WRKY expression in responses to biotic and abiotic stresses. Genes Genomics 40 (4), 429–446. doi:10.1007/s13258-017-0645-1
Jue, D., Sang, X., Liu, L., Shu, B., Wang, Y., Liu, C., et al. (2018). Identification of WRKY gene family from dimocarpus longan and its expression analysis during flower induction and abiotic stress responses. Int. J. Mol. Sci. 19 (8), 1–20. doi:10.3390/ijms19082169
Kan, J., Gao, G., He, Q., Gao, Q., Jiang, C., Ahmar, S., et al. (2021). Genome-wide characterization of wrky transcription factors revealed gene duplication and diversification in populations of wild to domesticated barley. Int. J. Mol. Sci. 22 (10), 5354. doi:10.3390/ijms22105354
Kang, G., Yan, D., Chen, X., Li, Y., Yang, L., and Zeng, R. (2020). Molecular characterization and functional analysis of a novel WRKY transcription factor HbWRKY83 possibly involved in rubber production of Hevea brasiliensis. Plant Physiology Biochem. 155, 483–493. doi:10.1016/j.plaphy.2020.08.013
Li, Y., Zhang, L., Zhu, P., Cao, Q., Sun, J., Li, Z., et al. (2019). Genome-wide identification, characterisation and functional evaluation of WRKY genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.) G. Don. Under abiotic stresses. BMC Genet. 20 (1), 90–15. doi:10.1186/s12863-019-0789-x
Li, W., Pang, S., Lu, Z., and Jin, B. (2020). Function and mechanism of WRKY transcription factors in abiotic stress responses of plants. Plants 9 (11), 1515–15. doi:10.3390/plants9111515
Nadler, S. G., Tritschler, D., Haffar, O. K., Blake, J., Bruce, A. G., and Cleaveland, J. S. (1997). Differential expression and sequence-specific interaction of karyopherin α with nuclear localization sequences. J. Biol. Chem. 272 (7), 4310–4315. doi:10.1074/jbc.272.7.4310
Nan, H., and Gao, L. Z. (2019). Genome-wide analysis of WRKY genes and their response to hormone and mechanic stresses in carrot. Front. Genet. 10, 1–19. doi:10.3389/fgene.2019.00363
Nguyen, L., Schmidt, H. A., Haeseler, A. Von, and Minh, B. Q. (2014). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Pan, H., Feng, Y., Wei, L., Sun, X., Tao, S., and Song, C. (2022). Sequence analysis of WRKY transcription factor family in Dendrobium officinale. New York, NY: bioRxiv.
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14 (4), 417–419. doi:10.1038/nmeth.4197
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi:10.1038/nbt.3122
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi:10.1038/nprot.2016-095
Schluttenhofer, C., and Yuan, L. (2015). Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 167, 295–306. doi:10.1104/pp.114.251769
Schön, M., Töller, A., Diezel, C., Roth, C., Westphal, L., Wiermer, M., et al. (2013). Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant-Microbe Interact. 26, 758–767. doi:10.1094/MPMI-11-12-0265-R
Sims, D., Sudbery, I., Ilott, N. E., Heger, A., and Ponting, C. P. (2014). Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 15, 121–132. doi:10.1038/nrg3642
Song, C., Li, G., Dai, J., and Deng, H. (2021). Genome-wide analysis of PEBP genes in dendrobium huoshanense: Unveiling the antagonistic functions of FT/TFL1 in flowering time. Front. Genet. 12, 1–11. doi:10.3389/fgene.2021.687689
Song, C., Cao, Y., Dai, J., Li, G., Manzoor, M. A., Chen, C., et al. (2022a). The multifaceted roles of MYC2 in plants: Toward transcriptional reprogramming and stress tolerance by jasmonate signaling. Front. Plant Sci. 13, 1–14. doi:10.3389/fpls.2022.868874
Song, C., Ma, J., Li, G., Pan, H., Zhu, Y., Jin, Q., et al. (2022b). Natural composition and biosynthetic pathways of alkaloids in medicinal dendrobium species. Front. Plant Sci. 13, 1–15. doi:10.3389/fpls.2022.850949
Song, C., Wang, Y., Manzoor, M. A., Mao, D., Wei, P., Cao, Y., et al. (2022c). In-depth analysis of genomes and functional genomics of orchid using cutting-edge high-throughput sequencing. Front. Plant Sci. 13, 1–15. doi:10.3389/fpls.2022.1018029
Song, C., Zhang, Y., Chen, R., Zhu, F., Wei, P., Pan, H., et al. (2022d). Label-free quantitative proteomics unravel the impacts of salt stress on dendrobium huoshanense. Front. Plant Sci. 13, 1–12. doi:10.3389/fpls.2022.874579
Srivastava, R., Kumar, S., Kobayashi, Y., Kusunoki, K., Tripathi, P., Kobayashi, Y., et al. (2018). Comparative genome-wide analysis of WRKY transcription factors in two Asian legume crops: Adzuki bean and Mung bean. Sci. Rep. 8 (1), 1–19. doi:10.1038/s41598-018-34920-8
Sun, W., Ma, Z., Chen, H., and Liu, M. (2020). Genome-wide investigation of WRKY transcription factors in Tartary buckwheat (Fagopyrum tataricum) and their potential roles in regulating growth and development. PeerJ 8 (3), e8727. doi:10.7717/peerj.8727
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi:10.1093/molbev/mst197
Tang, Y., Guo, J., Zhang, T., Bai, S., He, K., and Wang, Z. (2021). Genome-wide analysis of WRKY gene family and the dynamic responses of key WRKY genes involved in Ostrinia furnacalis attack in Zea mays. Int. J. Mol. Sci. 22 (23), 13045. doi:10.3390/ijms222313045
Vilcherrez-Atoche, J. A., Iiyama, C. M., and Cardoso, J. C. (2022). Polyploidization in orchids: From cellular changes to breeding applications. Plants 11, 469. doi:10.3390/plants11040469
Wang, F., Li, X., Zuo, X., Li, M., Miao, C., Zhi, J., et al. (2021). Transcriptome-wide identification of WRKY transcription factor and functional characterization of RgWRKY37 involved in acteoside biosynthesis in rehmannia glutinosa. Front. Plant Sci. 12, 739853. doi:10.3389/fpls.2021.739853
Wang, Y., Dai, J., Chen, R., Song, C., Wei, P., Wang, Y., et al. (2021). Long noncoding RNA-based drought regulation in the important medicinal plant Dendrobium huoshanense. Acta Physiol. Plant. 43 (11), 144. doi:10.1007/s11738-021-03314-1
Waqas, M., Azhar, M. T., Rana, I. A., Azeem, F., Ali, M. A., Nawaz, M. A., et al. (2019). Genome-wide identification and expression analyses of WRKY transcription factor family members from chickpea (Cicer arietinum L.) reveal their role in abiotic stress-responses. Genes Genomics 41 (4), 467–481. doi:10.1007/s13258-018-00780-9
Yang, X., Li, H., Yang, Y., Wang, Y., Mo, Y., Zhang, R., et al. (2018). Identification and expression analyses of WRKY genes reveal their involvement in growth and abiotic stress response in watermelon (Citrullus lanatus). PLoS ONE 13 (1), e0191308. doi:10.1371/journal.pone.0191308
Ye, H., Qiao, L., Guo, H., Guo, L., Ren, F., Bai, J., et al. (2021). Genome-wide identification of wheat WRKY gene family reveals that TaWRKY75-A is referred to drought and salt resistances. Front. Plant Sci. 12, 1–17. doi:10.3389/fpls.2021.663118
Zhang, G. Q., Liu, K. W., Li, Z., Lohaus, R., Hsiao, Y. Y., Niu, S. C., et al. (2017). The Apostasia genome and the evolution of orchids. Nature 549, 379–383. doi:10.1038/nature23897
Keywords: computational analysis, bioinformatics, in-depth analysis, expression profile, WRKY transcription factor
Citation: Pan H, Chen Y, Zhao J, Huang J, Shu N, Deng H and Song C (2023) In-depth analysis of large-scale screening of WRKY members based on genome-wide identification. Front. Genet. 13:1104968. doi: 10.3389/fgene.2022.1104968
Received: 28 November 2022; Accepted: 23 December 2022;
Published: 09 January 2023.
Edited by:
Dawei Xue, Hangzhou Normal University, ChinaReviewed by:
Jen-Tsung Chen, National University of Kaohsiung, TaiwanIrfan Ali Sabir, Shanghai Jiao Tong University, China
Copyright © 2023 Pan, Chen, Zhao, Huang, Shu, Deng and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Deng, ZGh1cEBxcS5jb20=; Cheng Song, bGFubmlhbzgxMjMyOTIxOEAxNjMuY29t