
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 10 January 2023
Sec. Cancer Genetics and Oncogenomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1097234
 Cheng Xin1†
Cheng Xin1† Yi Lai2†
Yi Lai2† Liqiang Ji†Ye Wang1Shihao Li1
Liqiang Ji†Ye Wang1Shihao Li1 Liqiang Hao1Wei Zhang1Ronggui Meng1Jun Xu3*Yonggang Hong1*Zheng Lou1*
Liqiang Hao1Wei Zhang1Ronggui Meng1Jun Xu3*Yonggang Hong1*Zheng Lou1*Background: Individualized recurrence risk prediction in patients with stage II/III colorectal cancer (CRC) is crucial for making postoperative treatment decisions. However, there is still a lack of effective approaches for identifying patients with stage II and III CRC at a high risk of recurrence. In this study, we aimed to establish a credible gene model for improving the risk assessment of patients with stage II/III CRC.
Methods: Recurrence-free survival (RFS)-related genes were screened using Univariate Cox regression analysis in GSE17538, GSE39582, and GSE161158 cohorts. Common prognostic genes were identified by Venn diagram and subsequently subjected to least absolute shrinkage and selection operator (LASSO) regression analysis and multivariate Cox regression analysis for signature construction. Kaplan-Meier (K-M), calibration, and receiver operating characteristic (ROC) curves were used to assess the predictive accuracy and superiority of our risk model. Single-sample gene set enrichment analysis (ssGSEA) was employed to investigate the relationship between the infiltrative abundances of immune cells and risk scores. Genes significantly associated with the risk scores were identified to explore the biological implications of the 9-gene signature.
Results: Survival analysis identified 347 RFS-related genes. Using these genes, a 9-gene signature was constructed, which was composed of MRPL41, FGD3, RBM38, SPINK1, DKK1, GAL3ST4, INHBB, CTB-113P19.1, and FAM214B. K-M curves verified the survival differences between the low- and high-risk groups classified by the 9-gene signature. The area under the curve (AUC) values of this signature were close to or no less than the previously reported prognostic signatures and clinical factors, suggesting that this model could provide improved RFS prediction. The ssGSEA algorithm estimated that eight immune cells, including regulatory T cells, were aberrantly infiltrated in the high-risk group. Furthermore, the signature was associated with multiple oncogenic pathways, including cell adhesion and angiogenesis.
Conclusion: A novel RFS prediction model for patients with stage II/III CRC was constructed using multicohort validation. The proposed signature may help clinicians better manage patients with stage II/III CRC.
Colorectal cancer (CRC) is a common and fatal gastrointestinal malignant tumor, with an estimated 147,950 new cases in 2020 (Siegel et al., 2020). Although great advances have been achieved in the perioperative management of CRC patients, a high postoperative recurrence rate remains a challenge that hampers remarkable improvement in patient outcomes (Xie et al., 2021). Most patients with CRC are diagnosed at stage II/III and present with resectable tumors (Brenner et al., 2014). After radical resection and subsequent adjuvant chemotherapy, 30%–60% of patients relapse within 5 years, with a dismal prognosis (Manfredi et al., 2006). Moreover, the individual benefits of adjuvant therapy are still questionable, resulting in potential over-treatment (Gray et al., 2007; Schippinger et al., 2007; Varghese, 2015). Therefore, accurate recurrence risk stratification and personalized adjuvant chemotherapy are of great significance for the long-term survival of patients with CRC.
To date, the most commonly used clinicopathologic factor for identifying high-risk stage II/III patients is using the tumor-node-metastasis (TNM) system. However, drug responses and clinical outcomes can vary widely in patients with CRC at the same TNM stage because of high genetic and epigenetic heterogeneity (Weiser et al., 2011; Ji et al., 2018; Lahoz et al., 2022). This clinical challenge indicates that the conventional TNM staging system is inadequate for risk evaluation, highlighting the urgent need to exploit novel and reliable molecular classifiers. With recent advances in high-throughput techniques, risk assessment and prognostic prediction have been dramatically improved by the use of gene expression profiling and bioinformatics technology (Koncina et al., 2020; Ahluwalia et al., 2021; Ghafouri-Fard et al., 2021). For stage II/III CRC, several prognostic signatures with predictive capacity for recurrence risk have been established by analyzing key cancer-associated pathways, such as autophagy and the tumor microenvironment (Mo et al., 2019; Zhang et al., 2021; Ren et al., 2022; Zhang et al., 2022). All of these models exhibit favorable predictive performance, but their credibility remains to be improved because they are mostly derived from a single GSE39582 dataset and lack multi-cohort and cross-platform validation.
In the current study, we explored genes with the potential to predict CRC recurrence and established a prognostic 9-gene signature with cross-cohort compatibility. The proposed model showed elevated accuracy and efficiency compared with previous models and clinical parameters for risk assessment. Moreover, this signature is closely related to multiple oncogenic pathways such as cell adhesion and angiogenesis. These findings might be meaningful in guiding postoperative prognostic stratification and in understanding the recurrence mechanisms of patients with stage II/III CRC.
Five independent CRC cohorts with TNM stage information were collected for survival analysis in the current study. Gene expression profiles and clinical data were obtained from the Gene Expression Omnibus (GEO) database. Among these cohorts, the GSE39582 cohort (N = 464) was used for training, while the GSE17536 (N = 110), GSE17538 (N = 142), GSE37892 (N = 129), and GSE161158 (N = 151) cohorts were used for validation. To ensure accuracy, samples with RFS <30 days were excluded from the subsequent analyses. In addition, 54 pairs of tumor and adjacent normal specimens were gained from patients who were diagnosed with stage II/III CRC and underwent surgical treatments at the Department of Colorectal Surgery at Shanghai Changhai Hospital. None of the patients received any local or systemic treatment before the surgery. Written informed consent for the use of clinical samples in medical research was obtained from all patients. All clinical procedures were approved by the Ethics Committee of Shanghai Changhai Hospital.
Three cohorts with the largest number of CRC samples (GSE17538, GSE39582, and GSE161158) were selected to screen reliable genes indicative of RFS. Univariate Cox regression analysis was conducted using the ‘survival’ package in the R environment (version 3.5.2) to screen for prognostic genes (p < .05) in three independent cohorts. The Venn diagram (Bardou et al., 2014) was subsequently used to screen for common genes with the ability to predict RFS in these three cohorts. The identified genes were then sent for Gene Ontology (GO) analysis for functional annotation on the Database for Annotation, Visualization, and Integrated Discovery (DAVID) online website (Sherman et al., 2022).
Using the commonly prognostic genes screened above, least absolute shrinkage and selection operator (LASSO) regression analysis based on the “glmnet” R package combined with multivariate Cox regression analysis based on the “survival” R package were performed to generate an optimal prognostic signature. The risk score was calculated as follows: Risk score = (coefficient 1 × expression level of gene 1) + (coefficient 2 × expression level of gene 2) + ... + (coefficient n × expression level of gene n). Each patient was assigned a risk score, and patients were classified into low- and high-risk groups according to the medium value of the risk scores. Kaplan–Meier (K-M) survival curves plotted by the “survminer” R package, calibration curves plotted by the ‘rms’ R package, and time-dependent receiver operating characteristic (ROC) curves plotted by the “timeROC” package were utilized to assess the prognostic ability of this signature. The area under the curve (AUC) values calculated using the K-M ROC R package were used to compare the predictive performance of our signature with clinical factors and two previously reported prognostic signatures that predicted postoperative recurrence in stage II/III CRC patients (Cheng et al., 2018; Zhang et al., 2022). Additionally, univariate Cox regression analyses were conducted to identify independent survival indicators in each cohort.
The single-sample gene set enrichment analysis (ssGSEA) method was conducted by the “GSVA” package to estimate the immune infiltration values of 23 immune cells in CRC samples. The relationships between immune infiltrative levels and risk scores were determined using Pearson correlation analysis.
To clarify the close association between the risk signature and patient prognosis, Pearson correlation analysis was performed to identify genes correlated with (p < .05) risk scores in the GSE39582 cohort. Based on the correlation coefficients, the top 1000 genes with positive or negative correlations were subjected to Gene Ontology-Biological Process (GO-BP) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment using the DAVID online website. The enriched items and corresponding p-values were visualized by the “ggplot2” package.
IHC assays were performed as previously described (Chen and Zhang, 2018). Sections were incubated with specific primary antibodies against MRPL41 (1:200 dilution, ab121821, Abcam, Cambridge, United Kingdom) and RBM38 (1:50 dilution, ab200403, Abcam, Cambridge, United Kingdom) at 4°C overnight. After incubation with the appropriate secondary antibodies at room temperature for 1 h, sections were stained with diaminobenzidine and hematoxylin. IHC scores were quantified as previously described (Chen and Zhang, 2018).
Statistical analyses were performed using GraphPad Prism 6 software. Parametric data were analyzed using the Student’s t-test. Statistical log-rank p < .05 was considered significant.
A total of 207 common genes with an HR > 1 (Figure 1A) and 140 common genes with an HR < 1 (Figure 1B) were selected for model construction. GO-BP analysis demonstrated that these 347 genes were closely related to functions of cell adhesion, proliferation, and angiogenesis (Figure 1C). LASSO regression analysis of these prognostic genes identified 24 candidate genes for subsequent analysis (Figures 1D, E). To avoid overfitting, multivariate Cox regression analysis was performed on these 24 genes to generate a 9-gene signature (Figure 1F). The risk score formula was developed as follows: Risk score = −0.82604 × expression level of MRPL41–0.68172 × expression level of FGD3—0.35399 × expression level of RBM38–0.06949 × expression level of SPINK1 + 0.09875 × expression level of DKK1 + 0.35278 × expression level of GAL3ST4 + 0.38681 × expression level of INHBB +0.50069 × expression level of CTB-113P19.1 + 1.01899 × expression level of FAM214B.
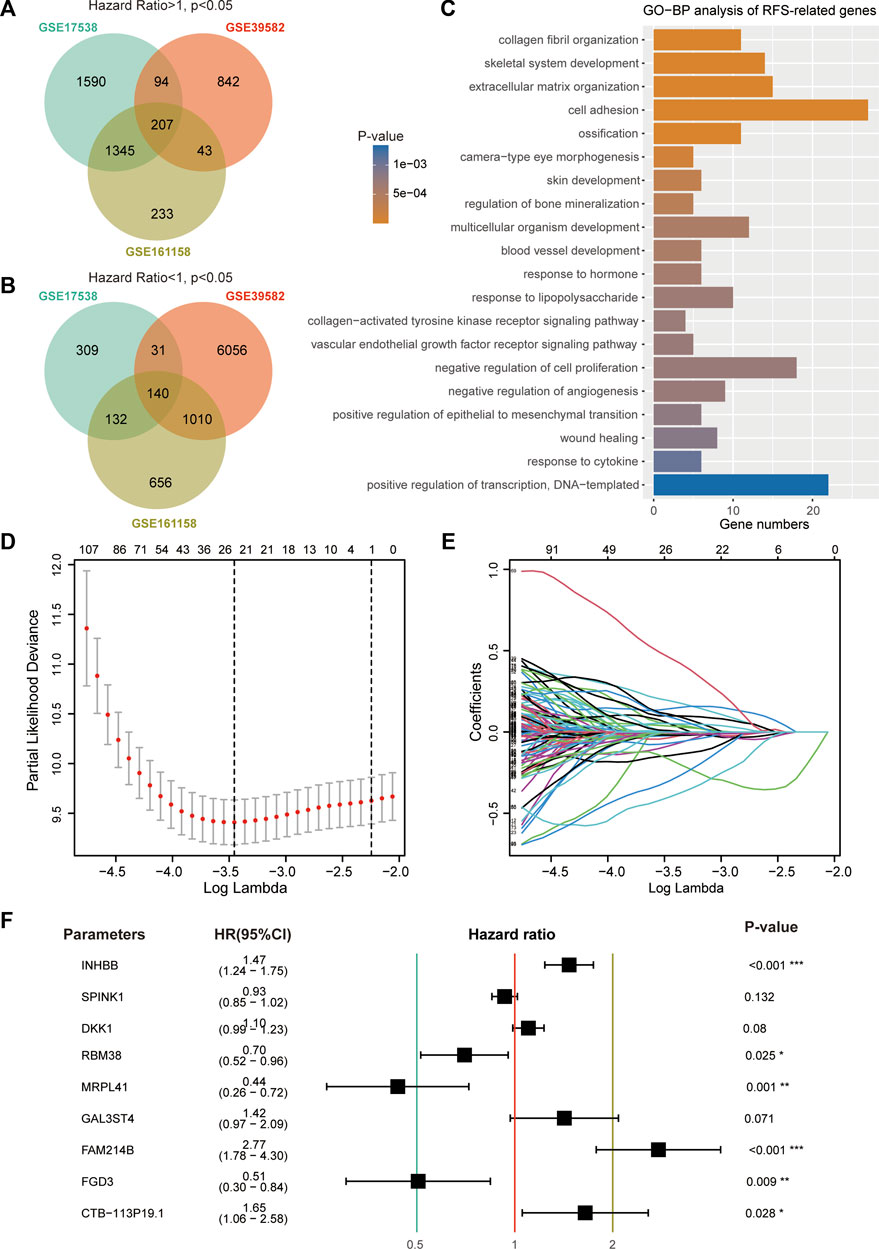
FIGURE 1. Construction of prognostic signature using RFS-related genes. (A) Venn diagram screened 207 prognostic genes with a hazard ratio >1 in three CRC cohorts. (B) Venn diagram screened 140 prognostic genes with a hazard ratio <1. (C) Top 20 enriched GO-BP terms of 347 RFS-related genes. (D) Cross-validation for tuning parameter (lambda) screening in the LASSO regression model. (E) LASSO coefficients of 24 RFS-related genes. (F) Hazard ratio, 95% CI, and p-value of the nine genes.
In the GSE39582 cohort, the K-M survival curve showed a significantly decreased RFS time in patients in the high-risk group (Figure 2A, left panel). Patients in the high-risk group had markedly higher recurrence rates than those in the low-risk group (Figure 2A, middle panel). The calibration curve analysis showed that the survival probabilities predicted by our model were in good agreement with the actual survival probabilities (Figure 2A, right panel). In addition to the training cohort, the proposed signature also precisely estimated the different survival times and events of low- and high-risk patients in the GSE17538 (Figure 2B), GSE161158 (Figure 2C), GSE17536 (Figure 2D), and GSE37892 (Figure 2E) cohorts.
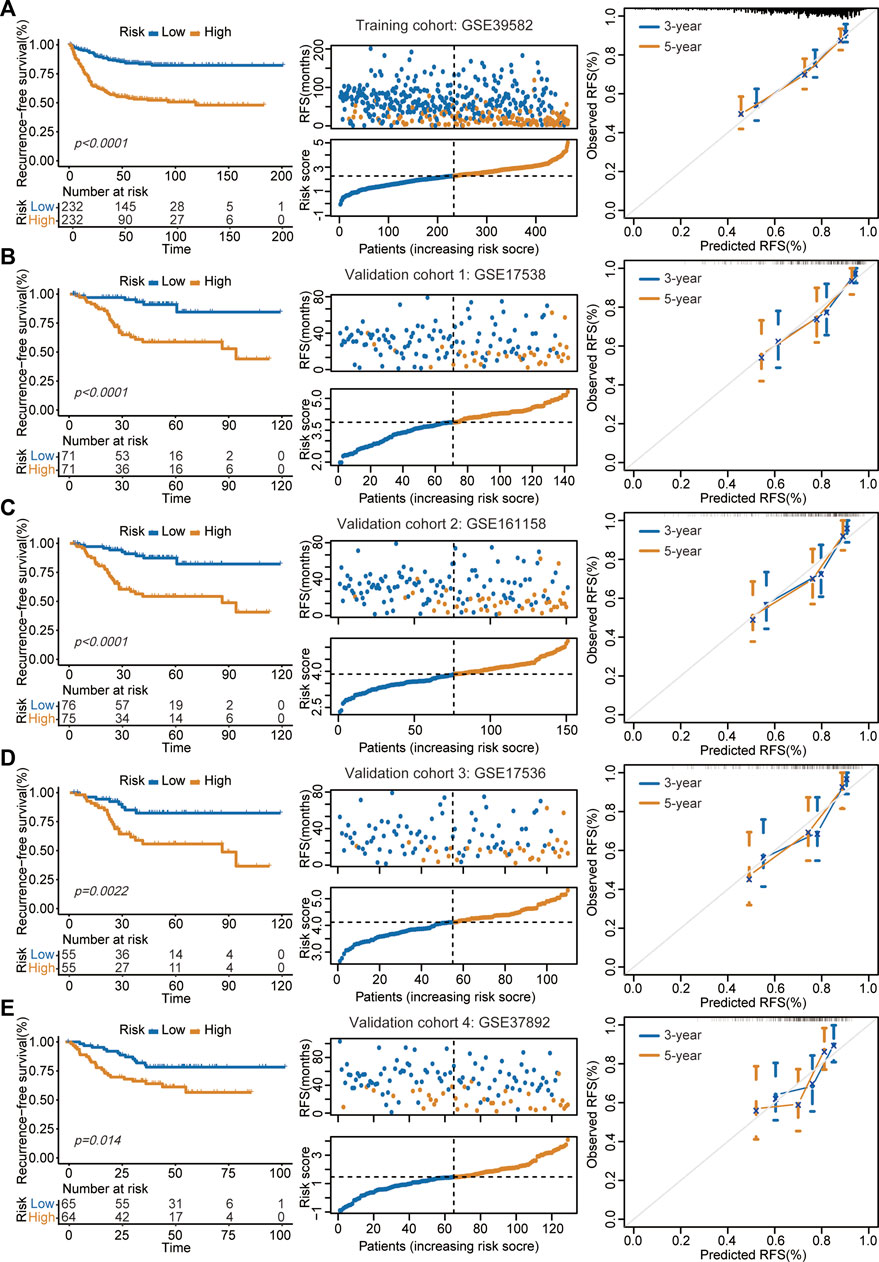
FIGURE 2. Assessment of prognostic performance in training and validation cohorts. (A–E) The proposed signature precisely captured the survival differences between two risk groups in GSE39582 (A), GSE17538 (B), GSE161158 (C), GSE17536 (D), and GSE37892 (E) cohorts, respectively. Left panel: K-M curves estimated the RFS differences between two risk groups. Middle panel: From top to bottom was the distribution of survival status and risk scores. The orange dots represented recurrence while the blue dots represented non-recurrence. Right panel: Calibration curves for the 9-gene signature.
Multiple prognostic signatures for stage II/III CRC recurrence risk stratification were established, and we wondered whether our signature outperformed the published signatures in risk prediction. An AUC value analysis was adopted, and a high AUC value indicated high predictive accuracy. In addition to the second highest predictive accuracy in the GSE17536 cohort (AUC = 0.744, Figure 3A), the proposed signature showed the strongest prediction precision for 2 year RFS in the GSE17538 cohort (AUC = 0.777, Figure 3B), GSE37892 cohort (AUC = 0.719, Figure 3C), GSE39582 cohort (AUC = 0.747, Figure 3D), and GSE161158 cohort (AUC = 0.789, Figure 3E).
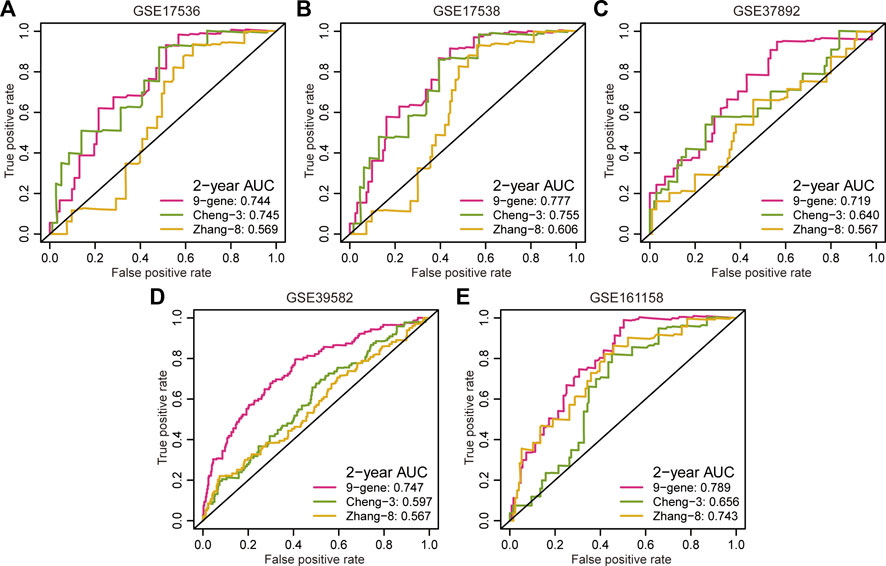
FIGURE 3. Comparison of predictive accuracy between our signature and previous signatures. (A–E) ROC curves investigated the predictive accuracy of gene signatures for 2 year RFS prediction in GSE17536 (A), GSE17538 (B), GSE37892 (C), GSE39582 (D), and GSE161158 (E) cohorts, respectively.
The results of the univariate Cox regression analysis demonstrated that the 9-gene signature was associated with unfavorable survival in five independent cohorts (Figure 4A). To investigate the superiority of our signature in predicting RFS, time-dependent ROC analyses were performed to assess the accuracy of each predictor. The 9-gene signature exhibited higher dynamic AUC values than the clinical predictors over time in all the GSE17536 (Figure 4B), GSE17538 (Figure 4C), GSE39582 (Figure 4E), and GSE161158 cohorts (Figure 4F), except for the GSE37892 cohort (Figure 4D). These findings suggest that our signature outperformed clinical indicators for recurrence prediction.
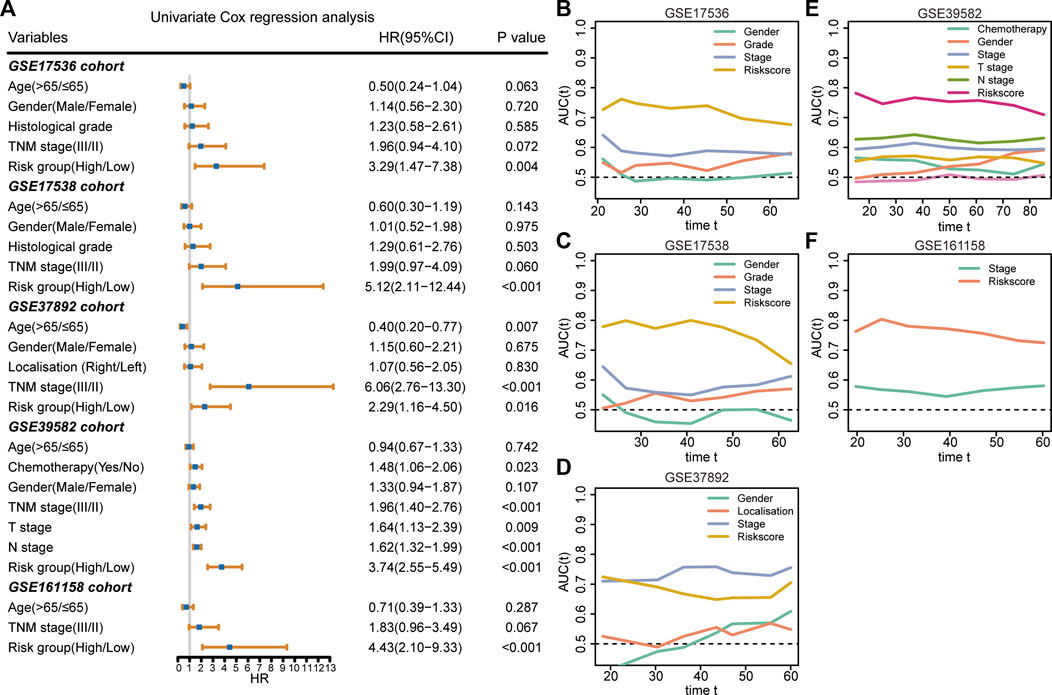
FIGURE 4. Superiority evaluation of the 9-gene signature compared with clinical factors (A) Univariate Cox regression analyses identified independent prognostic factors in each cohort. (B–F) Time-dependent AUC values of prognostic factors in GSE17536 (B), GSE17538 (C), GSE37892 (D), GSE39582 (E), and GSE161158 (F) cohorts, respectively.
The immune infiltrative differences between the low- and high-risk groups were determined by ssGSEA analysis in each cohort (Figures 5A–E). The intersecting differences with the same alternative trends in these five cohorts were regarded as signature-related immunological changes. The results showed that eight immune cells, including γδ T cells, immature dendritic cells, macrophages, mast cells, natural killer T cells, regulatory T cells, T follicular helper cells, and Type 1 T helper cells, were upregulated in the high-risk group. Correlation analyses illustrated that in the GSE17536 and GSE17538 cohorts, the risk score was negatively associated with the infiltrative level of activated CD8+ T cells, whereas it was positively associated with the infiltrative level of regulatory T cells in all five cohorts (Figure 5F). We then investigated the association between the risk score and several immune checkpoints, such as CD274, CTLA4 and IDO1. As shown in supplementary Figures 1A–E, a substantial increase in the expression of HAVCR2 was found in the high-risk group in each CRC cohort. In addition, a significant decrease in the expression of GZMB was observed in the high-risk group in multiple cohorts.
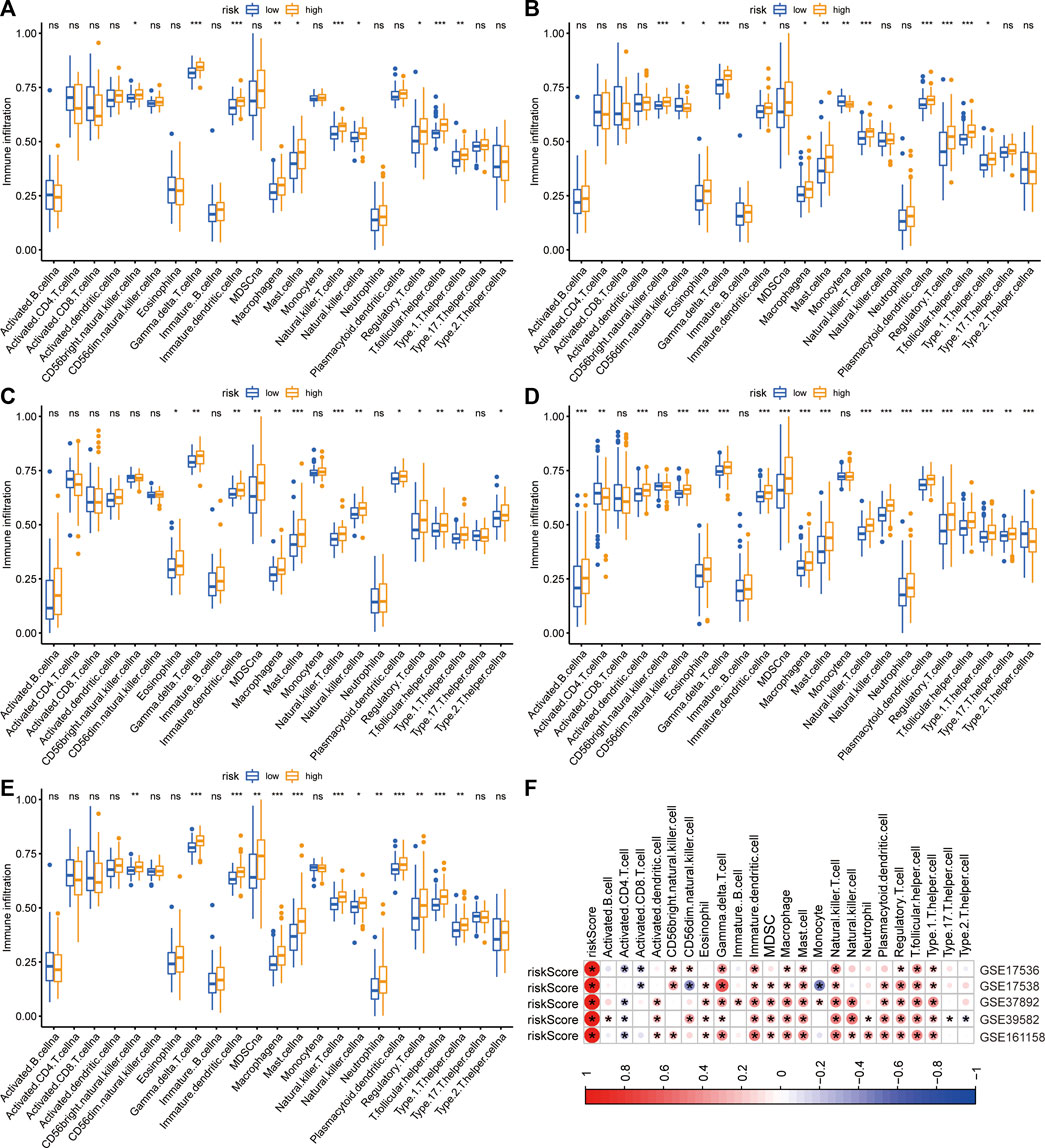
FIGURE 5. Differences in immune infiltration between two risk groups. (A–E) The immune infiltrative differences between two risk groups in GSE17536 (A), GSE17538 (B), GSE37892 (C), GSE39582 (D), and GSE161158 (E) cohorts, respectively. (F) The heatmap of correlations between immune infiltration and risk signature in each cohort. The blue color indicated negatively related and the red indicated positively related. *p < .05.
To elucidate why high-risk scores lead to poor prognosis, biological process and KEGG enrichment analyses were performed on the top 1000 genes that were positively or negatively correlated with risk scores, respectively. In the biological process analyses, genes with positive correlations were found to be related to extracellular matrix organization, cell adhesion, and angiogenesis (Figure 6A), while negatively correlated genes were primarily associated with cell division, DNA repair, and cell cycle (Figure 6C). In the pathway enrichment analyses, genes with positive correlation were principally involved in ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathways (Figure 6B), whereas genes with negative correlation were mainly enriched in the cell cycle and DNA replication (Figure 6D).
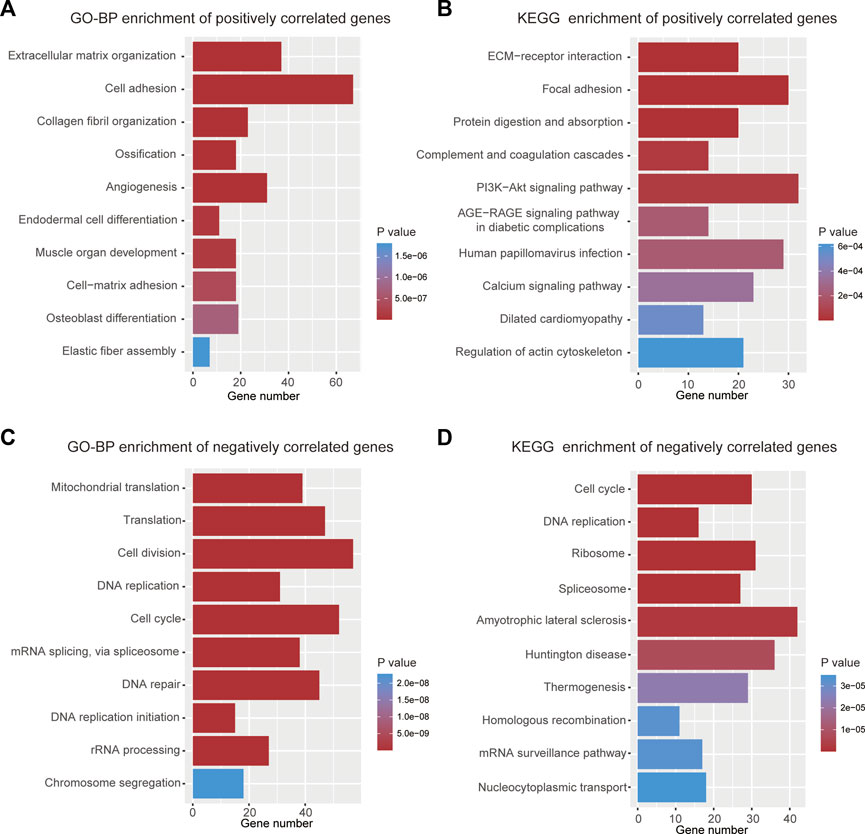
FIGURE 6. Functional analyses of the 9-gene signature. (A) Top 10 terms of GO-BP analysis for genes with positive correlation. (B) Top 10 terms of KEGG analysis for genes with positive correlation. (C) Top 10 terms of GO-BP analysis for genes with negative correlation. (D) Top 10 terms of KEGG analysis for genes with negative correlation.
We subsequently explored the differences in the expression of nine genes between normal and CRC tissues by analyzing TCGA data using the TIMER website (Li et al., 2016). Information about CTB-113P19.1. is not available on the website. As protective prognostic genes, MRPL41 and RBM38 were significantly downregulated, while FGD3 was upregulated and SPINK1 was not differentially expressed in CRC tissues. Among the risk prognostic genes, DKK1 and INHBB were markedly upregulated, while GAL3ST4 and FAM214B were markedly downregulated in CRC tissues (Figure 7).
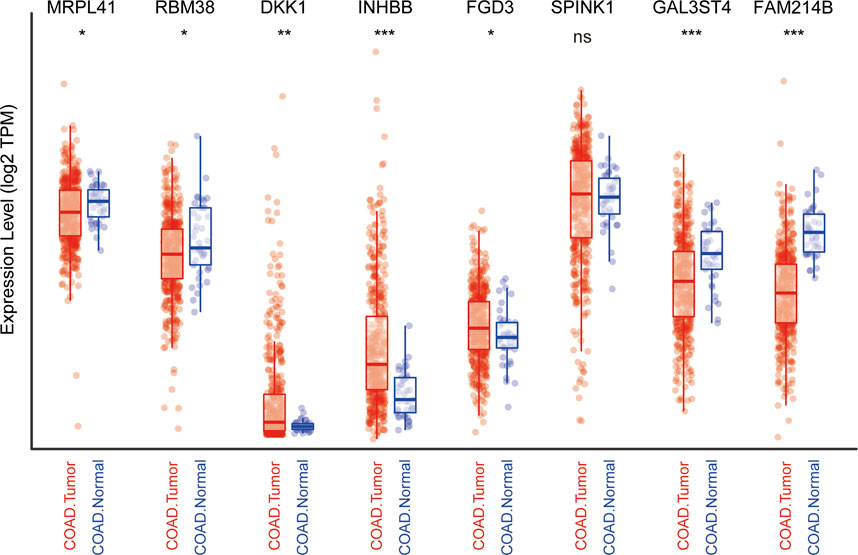
FIGURE 7. Expression profile of the nine genes in TCGA cohort. *p < .05, **p < .01, ***p < .001, ns: not significant.
From the above analyses of public RNA-sequencing data, we discovered that mRNA expression levels of MRPL41 and RBM38 were significantly decreased, whereas mRNA expression levels of DKK1 and INHBB were markedly elevated in CRC tissues. Protein expression levels of DKK1 and INHBB were also increased in CRC patients according to the literature (Sui et al., 2021; Zhou et al., 2022), but protein expression patterns of MRPL41 and RBM38 have rarely been reported. By analyzing the results of IHC staining assays from the Human Protein Atlas database (Uhlén et al., 2015), we found that protein expression levels of MRPL41 and RBM38 were dramatically decreased in CRC tissues (Figures 8A, B). The IHC assay further validated the downregulation of MRPL41 and RBM38 in CRC samples from our center (Figures 8C–F). These findings indicated that MRPL41 and RBM38 may play suppressive roles in CRC progression.
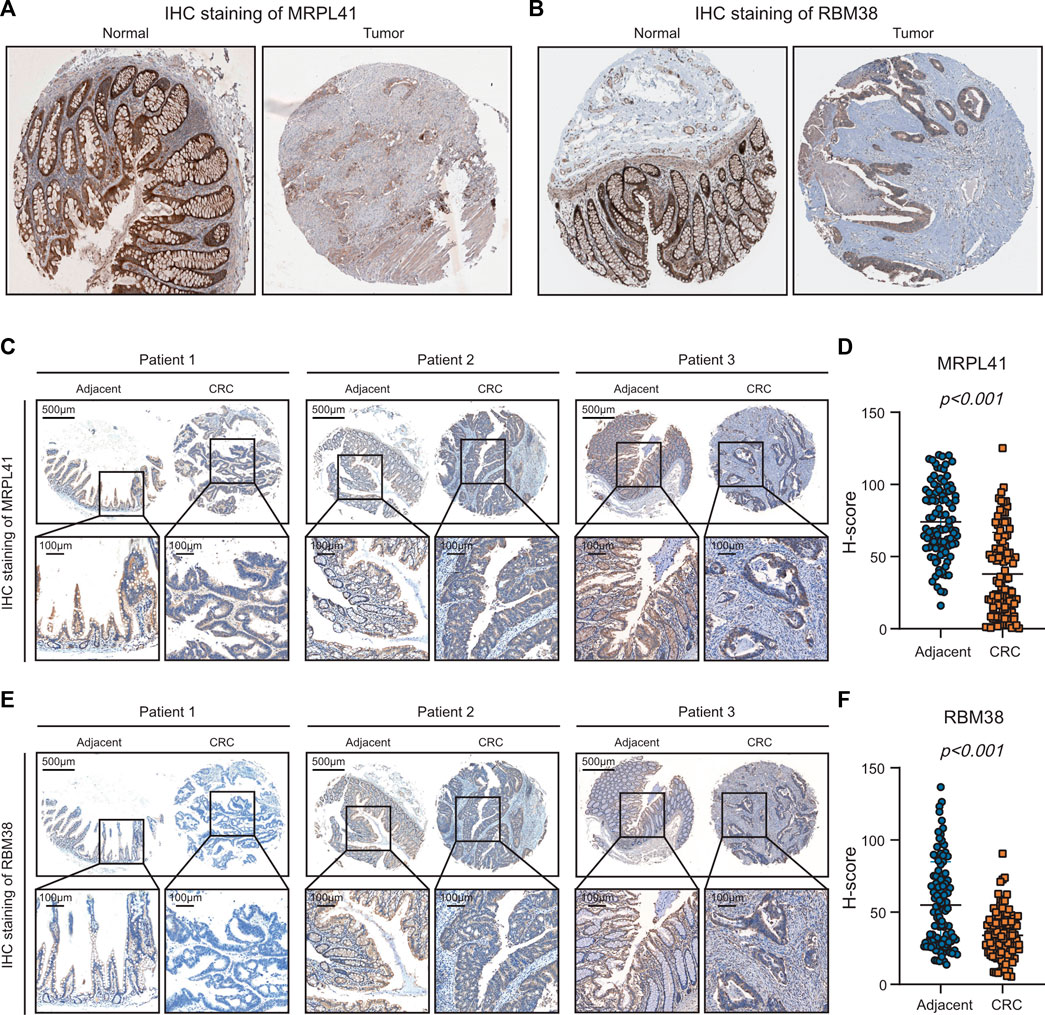
FIGURE 8. Experimental validations of MRPL41 and RBM38 expression patterns. (A) Representative IHC staining images of MRPL41 retrieved from the HPA database. (B) Representative IHC staining images of RBM38 downloaded from the HPA database. (C) Representative IHC staining images of MRPL41. (D) The quantitative H-scores of MRPL41 staining. (E) Representative IHC staining images of RBM38. (F) The quantitative H-scores of RBM38 staining.
The high rates of postoperative recurrence and mortality after recurrence emphasize the importance of improving individual recurrence risk prediction for patients with stage II/III CRC (Ju et al., 2019). To date, multiple efforts have been made to develop reliable predictors of postoperative recurrence in CRC (Wang et al., 2015; Yang et al., 2020; Zhang et al., 2020). However, CRC features high degrees of genomic and transcriptional heterogeneity, and the suitability of current biomarkers for individual patients remains debatable (Kyrochristos and Roukos, 2019). As cancer treatment enters the area of precision medicine, efficacious recurrence assessment that considers genetic and genomic features is of great importance to guide clinicians in individualized follow-up and therapeutic strategies.
Accumulated evidence has proved that prognostic gene signatures have vast capabilities to aid clinical decision-making (Yu et al., 2019; James et al., 2022; Oliveira et al., 2022). We constructed and verified a prognostic 9-gene signature to predict the response of stage II/III CRC. In multiple cohorts, the risk model exhibited significant predictive performance. Univariate Cox regression analyses, together with the Venn diagram, initially screened 347 credible genes with the capacity to predict RFS. A 9-gene signature was developed using these genes. Survival analysis demonstrated the impressive predictive ability of this risk model. ROC curves together with time-dependent AUC values confirmed that our model was superior to previous models and clinical predictors in terms of predicting recurrence.
We then investigated the immunological correlation and biological function of the 9-gene signature. The results showed that the 9-gene signature was negatively associated with the infiltrative values of CD8+ T cell while positively correlated with those of regulatory T cell. Furthermore, high risk score was indicative of decreased GZMB expression and elevated HAVCR2 expression. These findings suggested that the 9-gene signature might be able to predict the response of stage II/III CRC patients to immunotherapy. To clarify the mechanism of gene signature’s effect on cellular immunology, we performed function analyses of genes correlated with this signature. Multiple signaling pathways related to the gene signature played fundamental roles in immune cell function. For example, overexpression of calcium-permeable channels at various locations of T cells is necessary for T cell activation, maturation and secretion of cytokines (Trebak and Kinet, 2019; Vaeth et al., 2020). Moreover, the PI3K-AKT pathway have long been considered to play an important role in the regulation of immune cell metabolism, growth, or survival (O’Donnell et al., 2018; Weichhart and Säemann, 2008; Hand et al., 2010). Above findings might partly explain how this signature influences the infiltration of immune cells and expression of immune checkpoint genes.
Among these nine genes, RBM38, SPINK1, DKK1, and INHBB are implicated in CRC tumorigenesis. RBM38 is downregulated in CRC cell lines and inhibits colorectal cancer cell growth and stemness by competitively binding to PTEN (Guan et al., 2021). However, the function of SPINK1 in CRC development remains unclear. Several studies have reported that SPINK1 is overexpressed in CRC and contributes to cell proliferation, migration, and invasion (Gouyer et al., 2008; Ida et al., 2015; Tiwari et al., 2015; Chen et al., 2022). However, some studies have found that SPINK1 is downregulated in CRC and indicates a favorable prognosis (Koskensalo et al., 2012; Koskensalo et al., 2013; Chen et al., 2016). Moreover, SPINK1 can reduce cetuximab resistance in CRC cells by effectively preventing PRSS1 from cleaving cetuximab (Tan et al., 2020). DKK1 is a well-known oncogene that fosters CRC cell growth, metastasis, chemotherapy resistance, and immune evasion (Qi et al., 2021; Sui et al., 2021; Zhao et al., 2021). INHBB is an unfavorable prognostic biomarker for CRC (Yuan et al., 2020). However, the specific biological functions of MRPL41, FGD3, GAL3ST4, CTB-113P19.1, and FAM214B in CRC remain largely unknown.
Accurate prediction of recurrence risk can help determine the applicability of adjuvant therapy, reduce overtreatment-related adverse effects, and avoid unnecessary medical expenses (Chan et al., 2021). The suitability of gene expression profiles for identifying patients with a high risk of recurrence has been proven in human cancers. The U.S. Food and Drug Administration (FDA) has approved a successfully developed genetic test called MammaPrint to evaluate the recurrence risk in stage I/II breast cancer patients (van ’t Veer et al., 2002). Our study is the first to establish a recurrence predictive signature for stage II/III CRC patients via credible RFS-related genes. Validation in five independent public cohorts, including American and French populations, enhances clinical compatibility. We hope to translate this gene signature into a commercial kit for easy clinical application.
Based on the retrospective data, the current study has several limitations. First, all cohorts used for survival analyses had a relatively small sample size, and the prognostic performance of the 9-gene signature needs larger cohorts and prospective studies to confirm the results. Second, patient treatment data were not available for several cohorts. The individual therapeutic benefits for patients in different risk groups are unclear. Third, the biological function and clinical relevance of the five genes in this signature remain largely unknown, and further in vivo and in vitro experiments are required.
In conclusion, we proposed a recurrence prediction model for patients with stage II/III CRC and comprehensively assessed the predictive capacity of this model. Therefore, it is desirable to guide personalized treatment and prolong patient survival.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Material preparation, public data collection and bioinformatic analyses were performed by CX, YL, and LJ. YW, SL, and LH designed and performed IHC staining assays. CL, YL, WZ, and RM performed statistical analyses and wrote the manuscript. JX, YH, and ZL provided clinical samples and the relevant clinical information. YH and ZL revised the manuscript and supervised the study. All authors read and approved the final manuscript.
This work was supported by grants from Discipline Climbing Peak Project Fund of the First Affiliated Hospital of Naval Military Medical University (2020YXZ043); General cultivation project of Second Military Medical University (2021JCMS15).
We thank The National Natural Science Foundation of China for the grant funding. We acknowledge the contributions from GEO, DAVID, TIMER and HPA databases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1097234/full#supplementary-material
AUC, Area under the curve; CRC, Colorectal cancer; EMT, Epithelial-mesenchymal transition; GEO, Gene Expression Omnibus; GO-BP, Gene ontology-biological process; GSVA, Gene set variant analysis; IHC, Immunohistochemical; KEGG, Kyoto Encyclopedia of Genes and Genomes; K-M, Kaplan-Meier; LASSO, Least absolute shrinkage and selection operator; ROC, Receiver operating characteristic; RFS, Recurrence-free survival; ssGSEA, Single-sample gene set enrichment analysis; TCGA, The cancer genome atlas; TNM, Tumor, node, metastasis.
Ahluwalia, P., Kolhe, R., and Gahlay, G. K. (2021). The clinical relevance of gene expression based prognostic signatures in colorectal cancer. Biochimica biophysica acta Rev. cancer 1875 (2), 188513. doi:10.1016/j.bbcan.2021.188513
Bardou, P., Mariette, J., Escudié, F., and Djemiel, C. (2014). jvenn: an interactive Venn diagram viewer. BMC Bioinforma. 15 (1), 293. doi:10.1186/1471-2105-15-293
Brenner, H., Kloor, M., and Pox, C. P. (2014). Colorectal cancer. Lancet (London, Engl. 383 (9927), 1490–1502. doi:10.1016/S0140-6736(13)61649-9
Chan, H. C., Chattopadhyay, A., Chuang, E. Y., and Lu, T. P. (2021). Development of a gene-based prediction model for recurrence of colorectal cancer using an ensemble learning algorithm. Front. Oncol. 11, 631056. doi:10.3389/fonc.2021.631056
Chen, J. J., and Zhang, W. (2018). High expression of WWP1 predicts poor prognosis and associates with tumor progression in human colorectal cancer. Am. J. Cancer Res. 8 (2), 256–265.
Chen, Y. T., Tsao, S. C., Tsai, H. P., Wang, J. Y., and Chai, C. Y. (2016). Serine protease inhibitor Kazal type 1 (SPINK1) as a prognostic marker in stage IV colon cancer patients receiving cetuximab based targeted therapy. J. Clin. pathology 69, 974–978. doi:10.1136/jclinpath-2016-203638
Chen, Y. T., Tseng, T. T., Tsai, H. P., Kuo, S. H., Huang, M. Y., Wang, J. Y., et al. (2022). Serine protease inhibitor kazal type 1 (SPINK1) promotes proliferation, migration, invasion and radiation resistance in rectal cancer patients receiving concurrent chemoradiotherapy: A potential target for precision medicine. Hum. Cell 35, 1912–1927. doi:10.1007/s13577-022-00776-4
Cheng, X., Hu, M., Chen, C., and Hou, D. (2018). Computational analysis of mRNA expression profiles identifies a novel triple-biomarker model as prognostic predictor of stage II and III colorectal adenocarcinoma patients. Cancer Manag. Res. 10, 2945–2952. doi:10.2147/CMAR.S170502
Ghafouri-Fard, S., Hussen, B. M., Gharebaghi, A., Eghtedarian, R., and Taheri, M. (2021). LncRNA signature in colorectal cancer. Pathology, Res. Pract. 222, 153432. doi:10.1016/j.prp.2021.153432
Gouyer, V., Fontaine, D., Dumont, P., de Wever, O., Fontayne-Devaud, H., Leteurtre, E., et al. (2008). Autocrine induction of invasion and metastasis by tumor-associated trypsin inhibitor in human colon cancer cells. Oncogene 27 (29), 4024–4033. doi:10.1038/onc.2008.42
Gray, R., Barnwell, J., McConkey, C., Hills, R. K., Williams, N. S., Kerr, D. J., et al. (2007). Adjuvant chemotherapy versus observation in patients with colorectal cancer: A randomised study. Lancet (London, Engl. 370 (9604), 2020–2029. doi:10.1016/S0140-6736(07)61866-2
Guan, B., Li, G., Wan, B., Guo, X., Huang, D., Ma, J., et al. (2021). RNA-binding protein RBM38 inhibits colorectal cancer progression by partly and competitively binding to PTEN 3'UTR with miR-92a-3p. Environ. Toxicol. 36 (12), 2436–2447. doi:10.1002/tox.23356
Hand, T. W., Cui, W., Jung, Y. W., Sefik, E., Joshi, N. S., Chandele, A., et al. (2010). Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. U. S. A. 107 (38), 16601–16606. doi:10.1073/pnas.1003457107
Ida, S., Ozaki, N., Araki, K., Hirashima, K., Zaitsu, Y., Taki, K., et al. (2015). SPINK1 status in colorectal cancer, impact on proliferation, and role in colitis-associated cancer. Mol. cancer Res. MCR 13 (7), 1130–1138. doi:10.1158/1541-7786.MCR-14-0581
James, N. E., Woodman, M., and Ribeiro, J. R. (2022). Prognostic immunologic signatures in epithelial ovarian cancer. Oncogene 41 (10), 1389–1396. doi:10.1038/s41388-022-02181-5
Ji, D., Qiao, M., Yao, Y., Li, M., Chen, H., Dong, Q., et al. (2018). Serum-based microRNA signature predicts relapse and therapeutic outcome of adjuvant chemotherapy in colorectal cancer patients. EBioMedicine 35, 189–197. doi:10.1016/j.ebiom.2018.08.042
Ju, H. Q., Zhao, Q., Wang, F., Lan, P., Wang, Z., Zuo, Z. X., et al. (2019). A circRNA signature predicts postoperative recurrence in stage II/III colon cancer. EMBO Mol. Med. 11 (10), e10168. doi:10.15252/emmm.201810168
Koncina, E., Haan, S., Rauh, S., and Letellier, E. (2020). Prognostic and predictive molecular biomarkers for colorectal cancer: Updates and challenges. Cancers 12 (2), 319. doi:10.3390/cancers12020319
Koskensalo, S., Hagström, J., Louhimo, J., Stenman, U. H., and Haglund, C. (2012). Tumour-associated trypsin inhibitor TATI is a prognostic marker in colorectal cancer. Oncology 82 (4), 234–241. doi:10.1159/000336080
Koskensalo, S., Louhimo, J., Hagström, J., Lundin, M., Stenman, U. H., and Haglund, C. (2013). Concomitant tumor expression of EGFR and TATI/SPINK1 associates with better prognosis in colorectal cancer. PloS one 8 (10), e76906. doi:10.1371/journal.pone.0076906
Kyrochristos, I. D., and Roukos, D. H. (2019). Comprehensive intra-individual genomic and transcriptional heterogeneity: Evidence-based colorectal cancer precision medicine. Cancer Treat. Rev. 80, 101894. doi:10.1016/j.ctrv.2019.101894
Lahoz, S., Archilla, I., Asensio, E., Hernández-Illán, E., Ferrer, Q., López-Prades, S., et al. (2022). Copy-number intratumor heterogeneity increases the risk of relapse in chemotherapy-naive stage II colon cancer. J. pathology 257 (1), 68–81. doi:10.1002/path.5870
Li, B., Severson, E., Pignon, J. C., Zhao, H., Li, T., Novak, J., et al. (2016). Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17 (1), 174. doi:10.1186/s13059-016-1028-7
Manfredi, S., Bouvier, A. M., Lepage, C., Hatem, C., Dancourt, V., and Faivre, J. (2006). Incidence and patterns of recurrence after resection for cure of colonic cancer in a well defined population. Br. J. Surg. 93 (9), 1115–1122. doi:10.1002/bjs.5349
Mo, S., Dai, W., Xiang, W., Li, Y., Feng, Y., Zhang, L., et al. (2019). Prognostic and predictive value of an autophagy-related signature for early relapse in stages I-III colon cancer. Carcinogenesis 40 (7), 861–870. doi:10.1093/carcin/bgz031
O'Donnell, J. S., Massi, D., Teng, M. W. L., and Mandala, M. (2018). PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Seminars cancer Biol. 48, 91–103. doi:10.1016/j.semcancer.2017.04.015
Oliveira, L. J. C., Amorim, L. C., Megid, T. B. C., de Resende, C. A. A., and Mano, M. S. (2022). Gene expression signatures in early breast cancer: Better together with clinicopathological features. Crit. Rev. oncology/hematology 175, 103708. doi:10.1016/j.critrevonc.2022.103708
Qi, L., Chen, J., Zhou, B., Xu, K., Wang, K., Fang, Z., et al. (2021). HomeoboxC6 promotes metastasis by orchestrating the DKK1/Wnt/β-catenin axis in right-sided colon cancer. Cell death Dis. 12 (4), 337. doi:10.1038/s41419-021-03630-x
Ren, H., Bösch, F., Pretzsch, E., Jacob, S., Westphalen, C. B., Holch, J. W., et al. (2022). Identification of an EMT-related gene signature predicting recurrence in stage II/III colorectal cancer - a retrospective study in 1780 patients. Ann. Surg. 276, 897–904. doi:10.1097/SLA.0000000000005644
Schippinger, W., Samonigg, H., Schaberl-Moser, R., Greil, R., Thödtmann, R., Tschmelitsch, J., et al. (2007). A prospective randomised phase III trial of adjuvant chemotherapy with 5-fluorouracil and leucovorin in patients with stage II colon cancer. Br. J. cancer 97 (8), 1021–1027. doi:10.1038/sj.bjc.6604011
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., et al. (2022). David: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50 (W1), W216–W221. doi:10.1093/nar/gkac194
Siegel, R. L., Miller, K. D., Goding Sauer, A., Fedewa, S. A., Butterly, L. F., Anderson, J. C., et al. (2020). Colorectal cancer statistics, 2020. CA a cancer J. Clin. 70 (3), 145–164. doi:10.3322/caac.21601
Sui, Q., Liu, D., Jiang, W., Tang, J., Kong, L., Han, K., et al. (2021). Dickkopf 1 impairs the tumor response to PD-1 blockade by inactivating CD8+ T cells in deficient mismatch repair colorectal cancer. J. Immunother. cancer 9 (3), e001498. doi:10.1136/jitc-2020-001498
Tan, Z., Gao, L., Wang, Y., Yin, H., Xi, Y., Wu, X., et al. (2020). PRSS contributes to cetuximab resistance in colorectal cancer. Sci. Adv. 6 (1), eaax5576. doi:10.1126/sciadv.aax5576
Tiwari, R., Pandey, S. K., Goel, S., Bhatia, V., Shukla, S., Jing, X., et al. (2015). SPINK1 promotes colorectal cancer progression by downregulating Metallothioneins expression. Oncogenesis 4 (8), e162. doi:10.1038/oncsis.2015.23
Trebak, M., and Kinet, J. P. (2019). Calcium signalling in T cells. Nat. Rev. Immunol. 19 (3), 154–169. doi:10.1038/s41577-018-0110-7
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347 (6220), 1260419. doi:10.1126/science.1260419
Vaeth, M., Kahlfuss, S., and Feske, S. (2020). CRAC channels and calcium signaling in T cell-mediated immunity. Trends Immunol. 41 (10), 878–901. doi:10.1016/j.it.2020.06.012
van 't Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415 (6871), 530–536. doi:10.1038/415530a
Varghese, A. (2015). Chemotherapy for stage II colon cancer. Clin. colon rectal Surg. 28 (4), 256–261. doi:10.1055/s-0035-1564430
Wang, L., Shen, X., Wang, Z., Xiao, X., Wei, P., Wang, Q., et al. (2015). A molecular signature for the prediction of recurrence in colorectal cancer. Mol. cancer 14 (1), 22. doi:10.1186/s12943-015-0296-2
Weichhart, T., and Säemann, M. D. (2008). The PI3K/Akt/mTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. rheumatic Dis. 67 (3), 70–74. doi:10.1136/ard.2008.098459
Weiser, M. R., Gönen, M., Chou, J. F., Kattan, M. W., and Schrag, D. (2011). Predicting survival after curative colectomy for cancer: Individualizing colon cancer staging. J. Clin. Oncol. official J. Am. Soc. Clin. Oncol. 29 (36), 4796–4802. doi:10.1200/JCO.2011.36.5080
Xie, H., Mahoney, D. W., Foote, P. H., Burger, K. N., Doering, K. A., Taylor, W. R., et al. (2021). Novel methylated DNA markers in the surveillance of colorectal cancer recurrence. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 27 (1), 141–149. doi:10.1158/1078-0432.CCR-20-2589
Yang, W. J., Wang, H. B., Wang, W. D., Bai, P. Y., Lu, H. X., Sun, C. H., et al. (2020). A network-based predictive gene expression signature for recurrence risks in stage II colorectal cancer. Cancer Med. 9 (1), 179–193. doi:10.1002/cam4.2642
Yu, F., Quan, F., Xu, J., Zhang, Y., Xie, Y., Zhang, J., et al. (2019). Breast cancer prognosis signature: Linking risk stratification to disease subtypes. Briefings Bioinforma. 20 (6), 2130–2140. doi:10.1093/bib/bby073
Yuan, J., Xie, A., Cao, Q., Li, X., and Chen, J. (2020). INHBB is a novel prognostic biomarker associated with cancer-promoting pathways in colorectal cancer. Biomed. Res. Int. 2020, 6909672. doi:10.1155/2020/6909672
Zhang, W., Zhang, X., Zhao, D., Hu, M., Ge, X., and Xia, L. (2022). An individualized EMT-related gene signature to predict recurrence-free survival in stage II/III colorectal cancer patients. Dig. Dis. Sci. 67, 5116–5126. doi:10.1007/s10620-021-07338-y
Zhang, Z., Feng, Q., Jia, C., Zheng, P., Lv, Y., Mao, Y., et al. (2020). Analysis of relapse-associated alternative mRNA splicing and construction of a prognostic signature predicting relapse in I-III colon cancer. Genomics 112 (6), 4032–4040. doi:10.1016/j.ygeno.2020.07.002
Zhang, Z., Wu, Q., Zhu, D., He, G., Feng, Q., and Xu, J. (2021). Tumor microenvironment derived signature predicting relapse-free survival in I-III cancer and preliminary experiment verification. Int. Immunopharmacol. 91, 107243. doi:10.1016/j.intimp.2020.107243
Zhao, Y., Wang, C., and Goel, A. (2021). Andrographis overcomes 5-fluorouracil-associated chemoresistance through inhibition of DKK1 in colorectal cancer. Carcinogenesis 42 (6), 814–825. doi:10.1093/carcin/bgab027
Keywords: stage II/III colorectal cancer, risk score, recurrence-free survival, prognostic signature, immune infiltration
Citation: Xin C, Lai Y, Ji L, Wang Y, Li S, Hao L, Zhang W, Meng R, Xu J, Hong Y and Lou Z (2023) A novel 9-gene signature for the prediction of postoperative recurrence in stage II/III colorectal cancer. Front. Genet. 13:1097234. doi: 10.3389/fgene.2022.1097234
Received: 18 November 2022; Accepted: 19 December 2022;
Published: 10 January 2023.
Edited by:
Bo Feng, Shanghai Jiao Tong University, ChinaReviewed by:
Jiewei Lin, Tongji University, ChinaCopyright © 2023 Xin, Lai, Ji, Wang, Li, Hao, Zhang, Meng, Xu, Hong and Lou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Xu, aHN1amFzb24zNEAxNjMuY29t; Yonggang Hong, MTc2MzcxODMyQHFxLmNvbQ==; Zheng Lou, bG91emhlbmdwcm9AMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.