 Chunyan Tong1,2
Chunyan Tong1,2 Fengling Shi
Fengling Shi- 1College of Grassland, Resources and Environment, Inner Mongolia Agricultural University, Hohhot, China
- 2Key Laboratory of Grassland Resources (IMAU), Ministry of Education, Hohhot, China
Medicago ruthenica, important forage in the legume family, possesses high nutritional value and carries abundant tolerance genes. This study used whole-genome data of M. ruthenica to perform a genome-wide analysis of the nucleotide-binding site-leucine-rich repeat receptor (NLR) gene family, which is the largest family of plant disease resistance genes (R genes). A total of 338 NLR genes were identified in the M. ruthenica genome, including 160 typical genes that contained 80 coiled-coil (CC)-NBS-LRR (CNL) genes, 76 toll/interleukin-1 receptor (TIR)-NBS-LRR (TNL) genes, four resistance to powdery mildew 8 (RPW8)-NBS-LRR (RNL) subclass genes, and 178 atypical NLR genes encoding proteins without at least one important domain. Among its eight chromosomes, M. ruthenica chromosomes 3 and 8 contained most of the NLR genes. More than 40% of all NLR genes were located on these two chromosomes, mainly in multigene clusters. The NLR proteins of M. ruthenica had six highly conserved motifs: P-loop, GLPL, RNBS-D, kinase-2, RNBS-C, and MHDV. Phylogenetic analysis revealed that the NLR genes of M. ruthenica formed three deeply separated clades according to the N-terminal domain of the proteins encoded by these genes. Gene duplication and syntenic analysis suggested four gene duplication types in the NLR genes of M. ruthenica, namely, tandem, proximal, dispersed, and segmental duplicates, which involved 189, 49, 59, and 41 genes, respectively. A total of 41 segmental duplication genes formed 23 NLR gene pairs located on syntenic chromosomal blocks mainly between chromosomes 6 and 7. In addition, syntenic analysis between M. truncatula and M. ruthenica revealed 193 gene pairs located on syntenic chromosomal blocks of the two species. The expression analysis of M. ruthenica NLR genes showed that 303 (89.6%) of the NLR genes were expressed in different varieties. Overall, this study described the full NLR profile of the M. ruthenica genome to provide an important resource for mining disease-resistant genes and disease-resistant breeding.
Introduction
Plants are vulnerable to many pathogens during their natural growth, including fungi, bacteria, viruses, and nematodes (Kourelis and van der Hoorn, 2018). These pathogens affect the normal growth, reproduction, and physiological metabolism of plants, in addition to inducing the corresponding diseases. However, as sessile organisms, plants cannot actively avoid these microorganisms and lack a somatic adaptive immune system as is found in animals (Chisholm et al., 2006; Fujita et al., 2006). During their long-term interactions with pathogens, plants have evolved a set of effective self-protection mechanisms to resist pathogen invasion and harm. The innate defense system of plants consists of two main layers. The first layer is plant pattern recognition receptor (PRR) recognition of relatively conserved pathogen/microbe-associated molecular patterns (PAMPs/MAMPs) of the pathogens to stimulate downstream basic defense responses and hinder further pathogen growth. This reaction is termed PAMP/MAMP-triggered immunity (PTI) (Lacombe et al., 2010; Zipfel, 2014). At the same time, pathogens also interfere with the PTI response of host plants and enhance pathogen toxicity through excretive effectors. Effector-triggered immunity (ETI) is another plant immune response that recognizes some effectors directly or indirectly via resistance proteins (R proteins) that are encoded by resistance genes (R genes) in cells to inhibit further pathogen infection and reproduction (Thomma et al., 2011; Dou and Zhou, 2012). Therefore, R genes play an essential role in plant disease resistance processes.
To date, more than 300 R genes have been cloned from different plant species since the first R gene Hm1 of maize was cloned in 1992 (Johal and Briggs, 1992). Among them, more than 60% of genes belong to the nucleotide-binding site (NBS)-leucine-rich repeat receptor (LRR) (NBS-LRR, NLR) gene family (Kourelis and van der Hoorn, 2018). This gene family is the largest disease resistance gene family in plants, and some NLR genes are dominant with functions in plant immunity (Ellis and Jones, 1998; Shao et al., 2016; Kourelis and van der Hoorn, 2018). The proteins encoded by NLR genes contain three domains: the variable N-terminal domain, central NBS domain, and C-terminal domain LRR (Shi et al., 2018). The NBS domain consists of about 300 amino acid sequences and the C-terminal domain usually has 10–40 short LRR sequences (Traut, 1994; Jones and Jones, 1997). The NBS and LRR domains provide the energy required for signal transduction and recognition of specific pathogens, respectively (Goyal et al., 2020). According to the N-terminal domains, the proteins encoded by NLR genes can be further divided into coiled-coil (CC)-NBS-LRR (CNL), toll/interleukin-1 receptor (TIR)-NBS-LRR (TNL), and resistance to powdery mildew 8 (RPW8)-NBS-LRR (RNL) subclasses. Among them, the CNL and TNL subclasses are commonly used as sensors to detect pathogens, while the RNL subclass is used in immune signal transduction (Zhang et al., 2020).
With the publication of plant whole-genome data, NLR genes of many plant species [i.e., Arabidopsis thaliana (Meyers et al., 2003), rice (Zhou et al., 2004), maize (Cheng et al., 2012), barley (Li et al., 2021), tomato (Liu et al., 2014), potato (Jupe et al., 2012), pea (Djebbi et al., 2015), and alfalfa (Shao et al., 2014)] have been identified and analyzed, which has greatly promoted the understanding of their structures, mechanisms, and evolution. For example, Li et al. (2021) identified NLR genes in barely, which contains one RNL and 468 CNL genes. Qian et al. (2021) identified one RNL and 581 CNL genes in Secale cereale. Similarly, studies have reported no TNL subclass in monocotyledons, while both TNL and CNL subclasses have been found in dicotyledons, and RNL has been detected in angiosperms (Andersen et al., 2018; Zhang et al., 2020). In other words, NLR genes have differentiated during evolution in different plants. Moreover, NLR genes existed before the differentiation of green plants and greatly amplified after plants occupied land (Shao et al., 2019; Liu et al., 2021). The genetic diversity analysis of NLR genes based on whole-genome data has not only provided important resources for the exploration of functional disease-resistant genes but also has important value for molecular marker-assisted breeding in plants (Qian et al., 2021). Many genes or resistance gene analogs (RGAs), such as Rpi-amr3i from Solanum americanum (Witek et al., 2016), Sm1 from wheat (Walkowiak et al., 2020), and SRC7 from soybean (Yan et al., 2022), have been cloned by genome-wide NLR gene identification. Some of these have become the focus for efforts to improve disease resistance in plants and have been used in agricultural production. NLR genes are also widely involved in many other biological processes such as plant growth, development, environmental adaptation, and abiotic stress (Chini et al., 2004; MacQueen and Bergelson, 2016; Ren et al., 2020). Therefore, further studies on NLR genes are needed to expand the NLR gene database.
Medicago ruthenica (L.) (2n = 2x = 16) is a perennial legume forage grass species and homologous with M. sativa (Small and Jomphe, 1989). The latter is “the queen of forage crops” and one of the economically most important forage crops in the world (Brummer, 2004; Liu et al., 2013; Zhou et al., 2019). Because of its strong drought, cold, salt and alkali resistance and abundant leaves, good palatability, and high nutritional quality, M. ruthenica has been used not only in the genetic improvement of alfalfa but also as a high-quality pasture to provide nutrition for livestock (Wang et al., 2008; Li et al., 2013). M. ruthenica ‘Mengnong No. 1’ was approved for release by the Grass Variety Approval Committee of Inner Mongolia in 2019 (No. 2019003). Our previous study reported its average hay yield of 11,000–15,000 kg/hm2 after 3 years of cultivation, 113 first-order branches, and average crude protein content of 11–13% in Tumote, Hohhot, Inner Mongolia. However, like other crops, M. ruthenica is also susceptible to a variety of pathogens and diseases, such as powdery mildew, rust disease, and anthracnose. These diseases lead to serious yield and quality losses and further harm the health of livestock (Liu et al., 1989). Therefore, the study of the composition and distribution of R genes could help improve the recognition of disease resistance in M. ruthenica.
Fortunately, the whole genome of wild M. ruthenica has been sequenced and assembled (Wang et al., 2021). Specifically, PacBio, Illumina, 10× Genomics, and Hi-C technologies were used to assemble a 904.13 Mb genome with a scaffold N50 of 99.39 Mb. A total of 49,176 protein-encoding genes were annotated. This genome has provided information for the study of genome-wide NLR genes and a foundation for cloning the disease resistance genes of M. ruthenica. In this study, we made full use of the whole genome data of M. ruthenica to extract the coding sequences (CDSs) of NLR genes and analyze the NLR genes, including their classification, chromosome localization, conserved amino acid sequence, phylogenetics, duplication type, and synteny analysis. Furthermore, we analyzed the transcriptomes of powdery mildew resistance and susceptible varieties and characterized the expression of these NLR genes. The objective of this study was to provide comprehensive information on NLR genes in M. ruthenica. The results provide a reference for research on NLR gene functions and the genetic breeding of M. ruthenica.
Materials and methods
Data used in this study
The gff3 annotation files and genome sequences of M. ruthenica (https://doi.org/10.6084/m9. figshare.12726932) and amino acid sequences and gff3 annotation files of A. thaliana (https://www.ncbi.nlm.nih.gov/data-hub/genome/GCF_000001735.4/; https://www.arashare.cn/index/News/info/id/1699.html) and M. truncatula (http://plants.ensembl.org/Medicago_truncatula/Info/Index) were downloaded from public databases. Software “gffread” was used to extract the CDS sequences of M. ruthenica, and a Perl programming script was used to convert them into amino acid sequences.
Identification and classification of NLR genes in M. ruthenica
The NLR genes of M. ruthenica were identified from the whole genome using hidden Markov models search (HMMsearch) methods. The HMM profile (accession number PF00931) was downloaded from the Pfam database (http://pfam.xfam.org/) (Shao et al., 2014). NLR protein sequences were identified from whole-genome amino acid sequences of M. ruthenica using the hmmsearch command with an expectation value (E-value) of ≤0.0001. The obtained protein sequences were extracted using TBtools (Chen et al., 2020). These sequences were then compared to the NLR protein sequence from A. thaliana, and those that did not map onto the NLR protein family were eliminated. A. thaliana NLR genes were identified and classified based on whole-genome and gff3 files of A. thaliana, respectively. The extraction method for the A. thaliana NLR genes was consistent with that applied for M. ruthenica. Subsequently, the NLR genes identified in M. ruthenica were renamed according to the naming principles of Ameline-Torregrosa et al. (2008b).
The Pfam database (http://pfam.xfam.org/search) was used to scan for remaining proteins to confirm the presence of the NBS (also named NB-ARC) and TIR domains. Genes that did not encode a conserved NBS domain were removed from the subsequent analyses. The non-redundant candidate sequences were submitted to the NCBI Conserved Domains Database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) to identify the CC, RPW8, LRR, and other integrated domains (IDs). Meanwhile, proteins with no specific domains were compared to the CNL, RNL, and TNL subclasses in A. thaliana. Those with intact CC-NBS-LRR, TIR-NBS-LRR, or RPW8-NBS-LRR structures were divided into the corresponding gene subclass. The results from the two methods were combined, and the specific domains were selected for classification when the two results conflicted. Integrated domains with complete domains and E-value <10−5 were retained. Finally, the results were saved to Excel 2019 file format and viewed on an online website for data analysis (https://www.hiplot.com.cn/).
Chromosomal distribution of NLR genes in M. ruthenica genome
The location information of the NLR genes and the conserved regions were extracted using TBtools (Chen et al., 2020). The chromosomal distribution of M. ruthenica NLR genes was plotted using an online tool (https://hiplot-academic.com). A sliding window analysis was performed with 1 kb and 250 kb as the step and window sizes, respectively. The principle of clustering was that the distance between two NLR genes should be <250 kb on a chromosome (Ameline-Torregrosa et al., 2008b).
Motif analysis
MEME (https://meme-suite.org/meme/) was used to predict the motifs in the NBS domains of the identified NLR proteins. The maximum motif search value was set at 15, while the other parameters used the default settings (Bailey et al., 2015). The subsequent results of MEME were imported into TBtools for visualization (Chen et al., 2020). Finally, the highly conserved motifs were extracted and compared to reported amino acid sequences to confirm the type of conserved amino acid (Meyers et al., 2003; Ameline-Torregrosa et al., 2008b; Gong et al., 2021).
Phylogenetic analysis
M. ruthenica NLR proteins encoded by typical NLR genes with intact CNL, TNL, or RNL domains were selected for sequence alignment and phylogenetic analysis. MEGA 7.0 and ClustalW software were used to align and correct the amino acid sequences. The phylogenetic tree was constructed using Construct/Test Neighbor-Joining Tree in MEGA with a bootstrap test of 500 times. The image was further processed using the online software iTOL (https://itol.embl.de/).
Synteny and gene duplication analysis
NLR protein amino acid sequences from M. ruthenica were used as both a database and query to perform pair-wise all-against-all BLASTp. The results obtained with the gff3 annotation file, including chromosome number, gene identifier, and gene starting and ending positions, were input into MCScanX to classify the types of gene duplication using the duplicate gene classifier program (Wang et al., 2012). Similarly, the file obtained by BLASTp was also used for the analysis of intra-species collinearity using MCScanX software. Circos diagrams were drawn using Circos (v. 0.67; Krzywinski et al., 2009). For cross-species synteny analysis, the NLR proteins of M. truncatula (using an extraction method for genomic information consistent with that described for M. ruthenica) were used as queries and NLR proteins of M. ruthenica were used as a database for pair-wise all-against-all BLASTp. The remaining steps were consistent with the intra-species comparison.
Expression analysis
Two M. ruthenica varieties (‘Zhilixing’ and ‘Mengnong No. 1’) were used for the disease resistance experiment. Seedlings were transplanted in 2019 at Inner Mongolia Agricultural University (111.7°E, 40.8°N), with one plant per hole. The holes in each row were separated by 0.5 m, and the rows were separated by 0.5 m. Three replicates of each variety were set, and each replicate was grown in a 4.0 m × 5.0 m plot. The plants were cultivated using conventional field management. The materials were naturally infected with powdery mildew. The infection types of different M. ruthenica varieties were observed and photographed from July to September 2021.
To further characterize the expression patterns of NLR genes in different varieties of M. ruthenica, leaves of three plants were selected randomly for gRNA extraction and sequencing. The expression analysis was as follows. First, the genes related to disease resistance were selected from the whole-transcriptome data. Then, the NLR genes were screened out, and the fragments per kilobase of exon per million reads mapped (FPKM) values representing the expression of each gene in different samples were extracted (Li et al., 2009; Trapnell, C et al., 2012; Kim et al., 2019). Finally, genes with large differential expression in the two samples were selected and submitted to the online data analysis website (https://www.hiplot.com.cn/) to generate a heatmap.
Results
Identification and classification of M. ruthenica NLR genes
A total of 338 NLR genes with high confidence were identified from the sequenced and assembled genome of M. ruthenica (Wang et al., 2021). The resulting genes, their corresponding alias names, and original gene identifiers are listed in Supplementary Table S1. NLR genes accounted for 0.69% of all annotated protein-coding genes (49176). The obtained NLR genes were assigned into three subclasses (RNL, CNL, and TNL) based on the domain component of the protein analyses. These subclasses included 4, 198, and 136 genes, respectively. The specific information on each NLR gene, such as gene composition and gene annotation, provided the basic gene resources for the subsequent analyses.
Domain analysis
The protein sequences encoded by the different subclasses of NLR genes were further analyzed. The results were visualized according to the type of domain arrangement (Figure 1A; Supplementary Table S1). Among the 198 genes in the CNL subclass, 80 encoded intact CNL domains that simultaneously contained the typical N-terminal CC domain, the central NBS domain, and the C-terminal LRR domain, accounting for 40.4% of all CNL genes. A total of 22 genes did not encode the LRR domain (i.e., CN), 42 genes did not encode the CC domain (i.e., NL), and 54 genes did not encode either CC or LRR domains (i.e., N) in the CNL subclass. Similarly, a total of 76 proteins contained intact domains in the TNL subclass, with some atypical TNL genes also observed. A total of 28 and 16 TNL proteins did not contain TIR and LRR domains, respectively (i.e., TN or NL), and 16 proteins were missing both LRR and N terminal domains (i.e., N). All four RNL proteins contained intact domains. Among the 338 NLR proteins, four had an RPW8 domain, 104 had a TIR domain, 102 had a CC domain, 218 had an LRR domain, and all had an NBS domain. In general, 338 NLR proteins showed high diversity in their domain arrangements, with 160 (47.3%) typical proteins showing intact domains, and the rest (178) lacking at least one important domain. The ratio of typical CNL, TNL, and RNL proteins was 20:19:1. Some of the 338 NLR proteins also had additional integrated domains, such as PLN03210, PPP1R42, or CDC6.
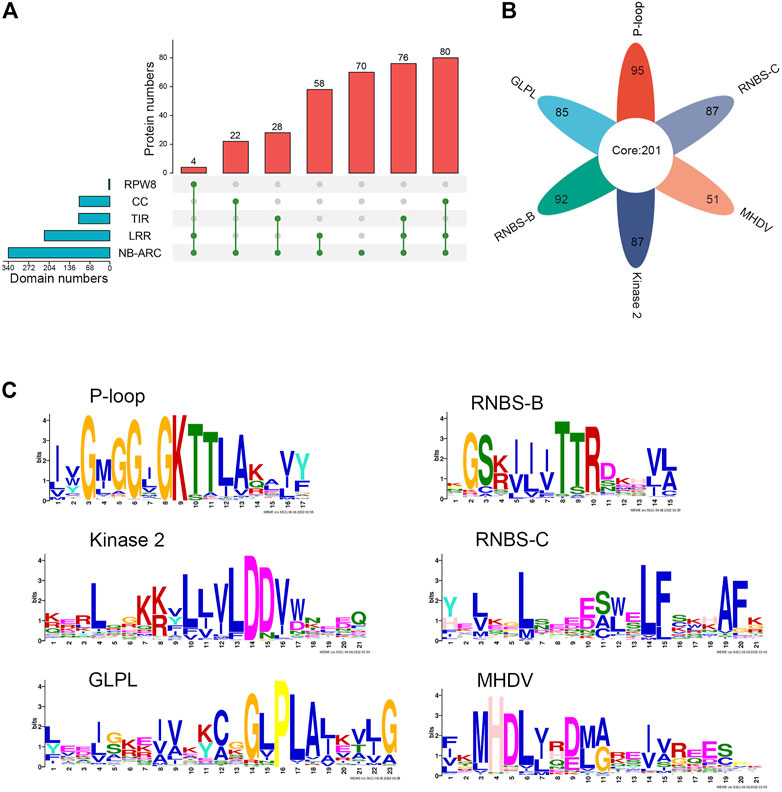
FIGURE 1. Identification and sequence analysis of NLR family genes in M. ruthenica. (A) Domain compositions of M. ruthenica NLR proteins. (B) Presence of six key motifs in the amino acid sequence of the NBS domain of M. ruthenica. (C) Amino acid features of the six key motifs in the amino acid sequence of the NBS domain of M. ruthenica.
Conserved motif analysis
MEME analysis was used to detect the presence of key motifs in the amino acid sequences of the NBS domains. The results revealed six motifs (P-loop, GLPL, RNBS-D, kinase-2, RNBS-C, and MHDV) that were readily detected and highly conserved in M. ruthenica NLR proteins, as has been reported for other angiosperms (Meyers et al., 2003; Ameline-Torregrosa, et al., 2008b) (Figures 1B, C). Specifically, 85% of the NLR proteins contained P-loop, GLPL, RNBS-D, kinase-2, and RNBS-C motifs, while 75% of the NLR proteins contained the MHDV motif (Supplementary Table S1). Meanwhile, 201 NLR proteins contained all six motifs simultaneously, accounting for 59.5% of all NLR proteins. Other proteins had lost at least one key motif in the NBS domain. Two NLR proteins did not contain any of the six key motifs, accounting for only 0.59% of the total (Supplementary Table S1). Except for RNBS-B, which was absent in RNL, the remaining motifs were present in most typical RNL, TNL, and CNL proteins, indicating that the RNL protein differed from TNL and CNL in terms of the length and amino acid composition of the conserved sequences. The NLR proteins of M. ruthenica also showed some exceptions, including those among the 80 typical CNL proteins, in which one lacked the GLPL motif, one lacked the RNBS-B motif, one lacked the Kinase 2 motif, and eight lacked the MHDV motif. These differences can be used to specifically identify the NLR genes of M. ruthenica and further distinguish these genes.
Distributions of NLR on the M. ruthenica chromosomes
Gene families dispersed in different positions generally show different patterns of expression regulation and perform important functions. Our analysis of their chromosomal distribution showed that all 338 NLR genes of M. ruthenica mapped to specific chromosomes (Figure 2A). Among them, the highest number of NLR genes mapped to chromosome 3 (75), followed by chromosome 8 (63). In contrast, the number was the lowest on chromosome 2 (23), which was only one less than on chromosome 1 (24). Chromosomes 4 and 7 possessed similar numbers of NLR genes (38 and 39). Chromosomes 5 and 6 contained 35 and 41 NLR genes, respectively. These results indicated that NLR genes were not evenly distributed on the eight chromosomes of M. ruthenica and that 138 (40.8%) of the 338 NLR genes were mainly distributed on chromosomes 3 and 8.
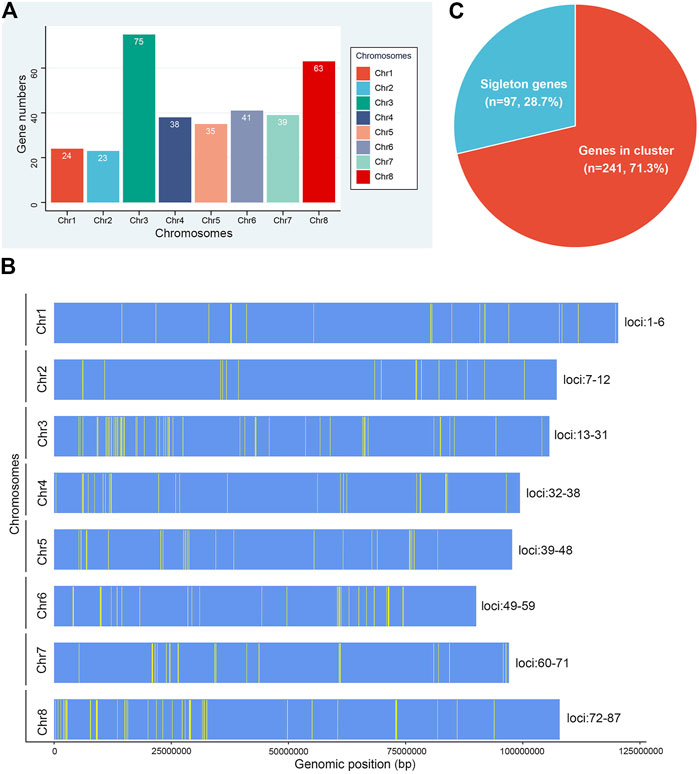
FIGURE 2. Chromosomal distribution of NLR genes in M. ruthenica. (A) Number of NLR genes on the eight chromosomes of M. ruthenica. (B) Physical locations of NLR genes on the eight chromosomes of M. ruthenica. (C) Proportions of NLR genes appearing as singletons (blue) and clusters (red) in the M. ruthenica genome.
The physical locations of the 338 NLR genes were extracted and further mapped on chromosomes (Figure 2B). The results showed the uneven distribution of these genes and that most were organized into clusters. A total of 87 clusters including 241 (71.3%) NLR genes were identified on eight chromosomes (Figure 2C). Chromosome 3 contained the most clusters (19), while chromosomes 1 and 2 had the fewest clusters (6). Three clusters, including seven NLR genes each, were considered the largest clusters. Overall, each cluster contained an average of three genes. Most clusters only contained two genes (47), accounting for 54.0% of all clusters. In addition, 97 NLR genes existed as singletons, accounting for 28.7% of all NLR genes. Overall, the NLR genes of M. ruthenica mainly existed in clusters, with few singletons. These results indicated that most NLR gene clusters of M. ruthenica mainly consisted of closely related genes, which shared a common ancestor, similar structure and function, and encoded similar protein products. In addition, the genes existing as singletons most likely evolved independently with new sources of mutation, or had close homologs elsewhere in the genome and might participate in more stable protein complexes or long-term protective functions (Mackey et al., 2003).
Phylogenetic analysis
To understand the separations and evolutionary relationships among the NLR genes of M. ruthenica, we performed phylogenetic analyses of amino acid sequences of 160 typical NLR genes. The results showed that M. ruthenica NLR genes formed three deeply separated clades, representing the three major subclasses RNL, TNL, and CNL, respectively (Figure 3A). The four RNL genes were further separated into two lineages, namely, RNL-1 and RNL-2. Mru1c19 and Mru1c20, and Mru8c330 and Mru8c334 formed highly supported lineages, respectively. The domain analysis showed that RNL-1 contained the integrated domain PLN03210, while RNL-2 did not (Supplementary Table S1).
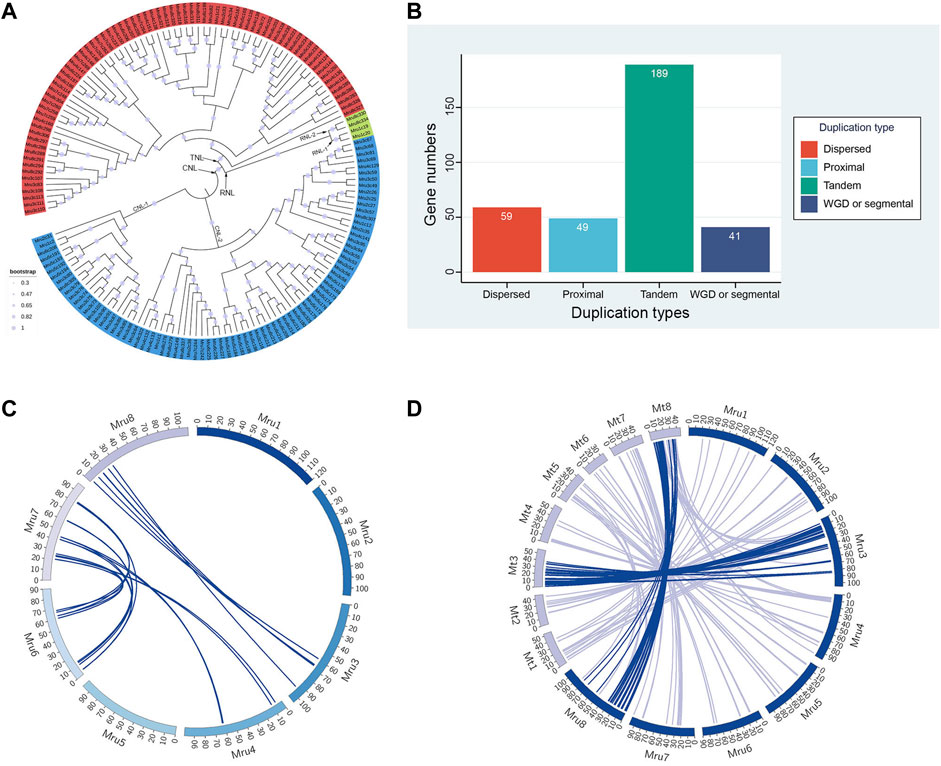
FIGURE 3. Phylogenetic, gene duplication, and synteny analyses of NLR genes in M. ruthenica. (A) Phylogenetic analysis of typical NLR genes in M. ruthenica. RNL, CNL, and TNL genes are shown in green, blue, and red, respectively. (B) Numbers of each duplication type of NLR genes. (C) Synteny analysis of NLR genes in M. ruthenica. (D) Synteny analysis of NLR genes between M. ruthenica and M. truncatula.
A total of 80 CNL genes in M. ruthenica formed two clades, including 21 genes in CNL-1 and 59 genes in CNL-2. CNL-2 was further divided into two or three lineages, which were more closely related. This phenomenon was also observed in the TNL clade. Specifically, a total of 76 TNL genes in M. ruthenica formed two clades, in which the gene Mru8c327 became a single clade closer to CNL, while the other genes belonged to another clade. The possible reasons for this may be related to the PLN03150 domain, which was not detected in other genes (Supplementary Table S1). In general, the evolution of NLR genes in M. ruthenica was mainly affected by the different types of N-terminal domains. In addition, other integration domains such as PLN03210 and PLN03150 also influenced the genetic relationship between different genes.
Gene duplication analysis
The duplication types of the 338 NLR genes were analyzed using MCScanX (Wang et al., 2012). The results revealed four different duplication types (e.g., tandem array, proximal, dispersed, and segmental duplicate) in M. ruthenica (Figure 3B; Supplementary Table S1). The largest group contained 189 (55.9%) tandem array genes. Proximal, dispersed, and segmental duplicate types included 49 (14.5%), 59 (17.5%), and 41 (12.1%) genes, respectively.
Synteny analysis of NLR genes in M. ruthenica
Pair-wise all-against-all BLASTp of the 338 NLR genes in M. ruthenica was used to determine and visualize as circle plots the intra-species genomic synteny (Figure 3C; Supplementary Table S2). Synteny analysis identified 23 NLR gene pairs located on syntenic chromosomal blocks. Among the eight chromosomes, the highest number of syntenic NLR gene pairs was observed between chromosomes 6 and 7 (12 pairs). In contrast, six and five pairs of syntenic genes were identified between chromosomes 3 and 8 and between chromosomes 4 and 7, respectively. Some genes were associated with two different genes on different chromosomes to form two different syntenic gene pairs. For example, Mru7c261 and Mru7c262 located on chromosome 7 were collinear with Mru4c147 and Mru4c148 located on chromosome 4, and Mru6c196 and Mru6c197 located on chromosome 6, respectively (Supplementary Table S2). Some genes were associated with two different genes on the same chromosome to constitute different syntenic gene pairs. For example, Mru6c196 and Mru6c221 were, respectively, collinear with Mru7c261 and Mru7c269, as well as Mru7c268 and Mru7c253 located on chromosome 7. In general, the multiple copies of NLR genes in M. ruthenica were mainly distributed on chromosomes 6 and 7.
To confirm the homology of NLR genes between M. ruthenica and its related species, the NLR genes from M. truncatula were extracted for inter-species analysis with pair-wise all-against-all BLASTp (Figure 3D; Supplementary Table S2). Synteny analysis revealed 193 NLR gene pairs located on syntenic chromosomal blocks of the two species. Among the eight chromosomes, the highest number (63) of syntenic NLR gene pairs was located on chromosome 3. However, chromosome 1 of M. ruthenica and chromosome 3 of M. truncatula had seven pairs of syntenic NLR genes. These results indicated the significant synteny relationship between these chromosomes in M. ruthenica and M. truncatula.
Expression analysis of NLR genes in M. ruthenica
The transcriptome data of M. ruthenica ‘Zhilixing’ and M. ruthenica ‘Mengnong No. 1’ (Unpublished) were used to characterize the different expression levels of the 338 NLR genes. ‘Zhilixing’ is more susceptible to powdery mildew than ‘Mengnong No. 1’ in the initial flowering stage (Figures 4A, B). Overall, 303 of 338 NLR genes showed differences in expression levels (Supplementary Table S3). Specifically, 72 NLR genes of Mengnong No. 1 showed at least 1.5-fold higher expression than ‘Zhilixing’ (Figure 4C). In addition, 22 genes, including Mru3c66, Mru3c69, and Mru8c295, were not expressed in ‘Zhilixing’ but were expressed or slightly expressed in Mengnong No. 1 (Supplementary Table S3). The expression analysis revealed 303 NLR genes with different expression between the two varieties, which may play an important role in M. ruthenica powdery mildew resistance responses in the field.
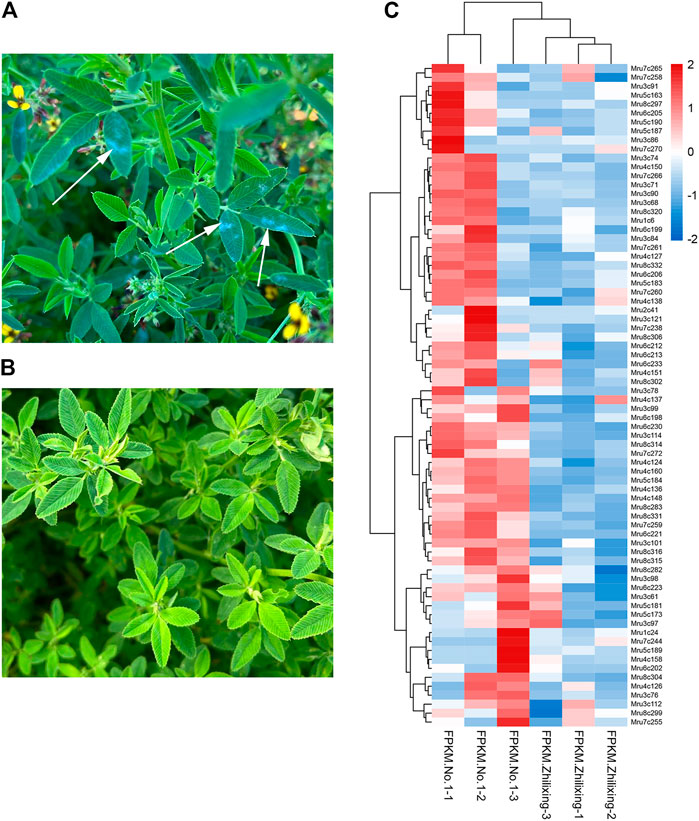
FIGURE 4. Powdery mildew resistance and expression analysis of different M. ruthenica varieties at the initial flowering stage in Inner Mongolia. (A) M. ruthenica ‘Zhilixing’. Arrows: powdery mildew. (B) M. ruthenica ‘Mengnong No. 1’. (C) Expression heatmap of 72 NLR genes differentially expressed between M. ruthenica ‘Zhilixing’ and ‘Mengnong No. 1’.
Discussion
M. ruthenica, a relative species of M. sativa, is often used as a high-quality genetic resource to improve the abiotic stress resistance of alfalfa and other pasture species because of its high tolerance to various extreme environmental conditions (Campbell et al., 1999; Guan et al., 2009). Therefore, increasing numbers of studies have focused on the response mechanisms, resistance breeding, and the screening and cloning of abiotic stress resistance genes in M. ruthenica (Shu et al., 2018; Shi et al., 2021; Yin et al., 2021). However, research on the resistance of M. ruthenica to biological stress is lacking, especially in disease resistance breeding and cloning disease resistance genes. The genome-wide identification of the NLR gene family would accelerate the process of cloning resistance genes and understanding the corresponding resistance mechanisms (Xue et al., 2020). The genome-wide identification of NLR genes in angiosperms has recently been reported. These studies have mainly focused on NLR gene numbers, classification, distribution, and conserved domains (Huang et al., 2008; Lozano et al., 2015). The present study is the first to systematically study the NLR genes of M. ruthenica through bioinformatics analysis based on whole-genome data. The results showed that M. ruthenica contains 338 NLR genes, 160 of which encode NLR proteins with intact domains, representing approximately 47.3% of the total NLR genes. The remaining genes encode only partial domains of NLR proteins with high similarity to domains with full-sized NLR in A. thaliana. The number of NLR genes in M. ruthenica was comparable to that of M. truncatula A17 and common bean. Specifically, M. truncatula A17 has 333 NLR genes containing 177 CNL and 156 TNL, while the common bean has 337 NLR genes containing 103 TNL, 224 CNL, and 10 RNL (Ameline-Torregrosa et al., 2008b; Shao et al., 2014). The total number of NLR genes in M. ruthenica represented 0.69% of the total annotated genes, which is consistent with the proportions (0.6%–1.76%) of NLR genes in other plants (Porter et al., 2009). Thus, although the NLR genes evolved rapidly, they have largely not been lost in M. ruthenica.
The NLR proteins contained typical domains with highly similar sequences, which are an important component of NLR proteins and have important functions in disease resistance (Bai et al., 2002). Among the three major domains (TIR/CC/RPW8, NBS, and LRR), NBS is the core domain of NLR proteins and is present in G-protein and GTP/ATP-binding protein families, which bind and hydrolyze ATP and GTP and participate in the nucleoside triphosphate metabolic pathway (Downward, 1990; Bourne et al., 1991; Kaziro et al., 1991; Traut, 1994; Tameling et al., 2002; Rojas et al., 2012). Several studies have reported that the LRR domain specifically recognizes pathogens through protein–protein interactions (Kobe and Deisenhofer, 1995; Ellis and Jones, 1998; Leister and Katagiri, 2000; Hulbert et al., 2001; Belkhadir et al., 2004). Moreover, differences in LRR number are the main factor of variation in NLR gene length (Jones and Jones, 1997). Mutations in the LRR region confer disease resistance in A. thaliana (Bent et al., 1994; Grant et al., 1995; Warren et al., 1998; Gassmann et al., 1999) and rice (Bryan et al., 2000). TIR and CC structures stimulate downstream signaling systems in EDS1 and NPR1 types after R gene and pathogen recognition and participate in plant immune response (Medzhitov et al., 1997; Ellis and Jones, 1998; Bai et al., 2002; Monosi et al., 2004). Finally, RPW8 has demonstrated broad-spectrum resistance to mildew in A. thaliana (Xiao et al., 2001).
Each domain or variety of domain arrangements of NLR proteins performs different functions and plays an important role in defending against pathogen invasion (Ameline-Torregrosa et al., 2008b). Therefore, the structural domains of NLR proteins were analyzed. Among the proteins encoded by 338 NLR genes of M. ruthenica, four had the RPW8 domain, 102 had the CC structure, 104 had the TIR domain, 218 contained the LRR domain, and all contained the NBS domain. Overall, 108 of the 338 NLR proteins contained two domains simultaneously, including 58 that contained NL, 22 that contained CN, and 28 that contained TN. Similar various atypical domains have been observed in M. truncatula (Ameline-Torregrosa et al., 2008b). In addition, the 338 NLR genes of M. ruthenica included 160 typical NLR genes with intact CNL (80), TNL (76), or RNL (4) structures. This result differs from that of many other plants. For example, the ratios of CNL and TNL genes were nearly 1:2 in A. thaliana and potato and 1:3 in grape (Meyers et al., 2003; Lozano et al., 2012; The Tomato Genome Consortium, 2012). Moreover, no TNL genes have been detected in many Gramineous, and only a minority of RNL genes have been found (Cheng et al., 2012; The Tomato Genome Consortium, 2012; Li et al., 2021; Qian et al., 2021). However, in our study, the ratio of typical CNL and TNL was close to 1:1, with slight differences in the numbers of genes in the CNL (198) and TNL (136) subclasses. The result was similar for M. truncatula whose CNL, TNL, and RNL genes accounted for 48.9%, 48.1%, and 3% of these genes, respectively (Shao et al., 2016). These results suggested that CNL and TNL played equally important roles in disease resistance in Medicago (Leister, 2004; Jupe et al., 2012).
Phylogenetic analysis has been used to show the relationship between different individuals (Kumar et al., 2016). Phylogenetic analyses of NLR genes have shown that non-TNL (lacking the TIR domain, usually known as CNL and RNL subclasses) were more diverse than TNL genes. Many studies have also suggested that non-TNL subclasses are older than TNL subclasses (Cannon et al., 2002; Meyers et al., 2002; 2003; Mun et al., 2009). The TNL and CNL subclasses were first identified among land plants in Physcomitrella patens and Selaginella moellendorffii, respectively (Yue et al., 2012). Subsequently, the R genes of these two subclasses were extended to higher plants and the RPW8 domain has recently been identified in angiosperms (Zhang et al., 2020). However, genes with RPW8 structure were assigned to the CNL lineage in some phylogenetic analyses, indicating a closer evolutionary relationship among the genes in these two subclasses (Ameline-Torregrosa et al., 2008b; Lozano et al., 2015). In contrast, in our study, instead of being assigned to the CNL subclasses, genes encoding the RPW8 domain in their N-terminal region formed a strong branch distinguished from CNL in M. ruthenica. This showed that the RNL clade was not classified into the CNL clade but rather formed an independent monophyletic clade with TNL and CNL. This finding has also been observed in other legumes, including M. truncatula, pigeon pea, soybean, and common bean (Shao et al., 2014). Meyers et al. (2003) also reported that RNL genes formed a separate lineage group (CNL-A group) in A. thaliana and that its principle of action also differed from that of most CNL genes. In addition, CNL, TNL, and RNL genes continued to be divided into smaller clades due to different integration domains. In short, the evolutionary relationships of the NLR gene family in M. ruthenica are mainly affected by the CC, TIR, and RPW8 domains and other accessory domains. Moreover, the function and phylogeny of RNL genes differed significantly between the CNL and TNL clades in M. ruthenica.
Polyploidy and multiple forms of gene doubling increase the copy numbers of homologous genes. Gene duplication is one of the most important evolutionary processes, which generates genetic diversity and new functions, and plays a crucial role in adaptation and speciation (Magadum et al., 2013). In this study, we identified 189, 49, 59, and 41 tandem, proximal, dispersed, and segmental duplicate genes, accounting for 55.9%, 14.5%, 17.5%, and 12.1% of all NLR genes, respectively. These results were consistent with the findings of Duan et al. (2019) who performed phylogenetic analysis and reported a large number of tandem duplications of NLR genes in legumes, including Glycine max, Arachis duranensis, and M. truncatula. Thus, tandem duplications may have occurred before the doubling of legumes and may have been conducive to the expansion of family gene copy numbers (Ratnaparkhe et al., 2011).
Syntenic analysis could explain the conservation of gene types and the relative order among different species diverging from the same ancestry. A large amount of homology information is contained in syntenic chromosome blocks at the DNA level between related species (Wang et al., 2012). Intra-species syntenic analysis could be used to confirm the homology of different chromosomes and analyze the duplication of regions or genes with multiple copies, while inter-species analysis could show genome homology (Qian et al., 2021). The syntenic analysis of NLR genes in M. ruthenica revealed 23 NLR gene pairs located on syntenic chromosomal blocks associated with chromosomes 6 and 7, 3 and 8, and 4 and 7. The same genes may have different homologs on different chromosomes, indicating that the NLR genes of M. ruthenica have multiple copies in different directions.
M. truncatula (2n = 2x = 16), a member of the Medicago genus, has been used as a model species of legume (Barker et al., 1990; Cook, 1999). A17-Jemalong is one genotype of M. truncatula with high resistance to many diseases such as anthracnose and powdery mildew (Ameline-Torregrosa et al., 2008a). Therefore, it was selected as the target species for syntenic analysis with M. ruthenica. The results revealed 193 NLR gene pairs located on syntenic chromosomal blocks of chromosome 3 between M. ruthenica and M. truncatula, indicating that NLR genes expanded similarly in these two species (Qian et al., 2021). However, earlier studies demonstrated significant loss and rearrangement in the M. truncatula genome, especially the NLR genes, which suggested rapid evolution and may have eliminated any evidence of syntropy (Ameline-Torregrosa et al., 2008b). Overall, the NLR gene family is a large multi-gene family with a complex gene structure and evolutionary process. More comprehensive and detailed analyses are required to better support breeding for disease resistance.
NLR gene expression patterns have been reported for many plants, which provide more evidence to understand NLR gene structures, functions, and applications (Lowe et al., 2017). Yu et al. (2021) reported that NLR gene expression levels in Akebia trifoliata were higher in rind tissue that was vulnerable to pathogens. Similarly, Ren et al. (2020) also observed significantly higher NLR gene expression in orchardgrass with high resistance to rust fungus compared to the expression in highly sensitive materials. The same results were reported in other plants, including Cucumis sativus (Zhang et al., 2022), Helianthus annuus (Neupane et al., 2018), Hordeum vulgare (Wang et al., 2013), and Populus (Bresson et al., 2011). In the present study, we found that 303 (89.6%) NLR genes were expressed in different M. ruthenica leaf tissues. Most showed no significant differences in expression, possibly due to the low sequencing coverage or pseudogenization (Frazier et al., 2016; Neupane et al., 2018). However, 72 of 303 NLR genes showed higher expression in resistant varieties compared to susceptible varieties, which indicated that these NLR genes might play important roles in the resistance of M. ruthenica to powdery mildew and may provide new resistance sources for the improvement of M. ruthenica disease resistance, although their specific functions and regulatory mechanisms are still unclear. Additional study on the functions of NLR genes of M. ruthenica under abiotic stress conditions is needed. The results of the present study have laid the foundation for further understanding. In general, NLR genes in M. ruthenica play important roles in pathogen defense to guarantee the normal growth and development of M. ruthenica in different ecological environments.
Conclusion
Analysis of whole-genome data of M. ruthenica uncovered a draft NLR gene family, which included 338 NLR genes with high confidence and diversity. These genes can be divided into three subclasses (TNL, CNL, and RNL) according to their domain arrangements. Six motifs (P-loop, GLPL, RNBS-D, kinase-2, RNBS-C, and MHDV) exist in the amino acid sequences of the NBS domains. The NLR genes of M. ruthenica mainly mapped to chromosomes 3 and 8, and most were organized in gene clusters. Phylogeny analysis revealed that genes in the TNL, CNL, and RNL subclasses formed three deeply separated clades. Collinearity analysis showed that the NLR genes of M. ruthenica had four duplication types, with tandem array genes the most numerous. Moreover, segmental duplication genes formed 23 NLR gene pairs located on syntenic chromosomal blocks of M. ruthenica. Meanwhile, 193 syntenic NLR gene pairs were identified between M. ruthenica and M. truncatula. Differential expression analysis showed that 303 (89.6%) NLR genes could play essential roles in powdery mildew resistance. These results further refine and expand information about M. ruthenica disease resistance gene families, which could lay a foundation for screening and cloning disease resistance genes and molecular breeding of disease resistance in M. ruthenica and its related species.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author contributions
CT and FS conceived and designed the study. CT obtained and analyzed the data. CT drafted the manuscript. YZ provided transcriptome data. All authors read and approved the final manuscript.
Funding
This research was supported financially by the Inner Mongolia Technology Planning Project (2020GG0063) and the Key Project in Science and Technology of Inner Mongolia (2021ZD0031).
Acknowledgments
The authors thank Guotang Yang of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, for assistance in data analysis and manuscript revision.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1088763/full#supplementary-material
References
Ameline-Torregrosa, C., Cazaux, M., Danesh, D., Chardon, F., Cannon, S. B., Esquerré-Tugayé, M. T., et al. (2008a). Genetic dissection of resistance to anthracnose and powdery mildew in Medicago truncatula. Mol. Plant Microbe. Interact. 21, 61–69. doi:10.1094/MPMI-21-1-0061
Ameline-Torregrosa, C., Wang, B. B., O'Bleness, M. S., Deshpande, S., Zhu, H., Roe, B., et al. (2008b). Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21. doi:10.1104/pp.107.104588
Andersen, E. J., Ali, S., Byamukama, E., Yen, Y., and Nepal, M. P. (2018). Disease resistance mechanisms in plants. Genes. (Basel) 9, 339. doi:10.3390/genes9070339
Bai, J., Pennill, L. A., Ning, J., Lee, S. W., Ramalingam, J., Webb, C. A., et al. (2002). Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 12, 1871–1884. doi:10.1101/gr.454902
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi:10.1093/nar/gkv416
Barker, D. G., Bianchi, S., Blondon, F., Datte´e, F., Duc, Y., Essad, S., et al. (1990). Medicago truncatula, a model plant for studying the molecular genetics of the Rhizobium-legume symbiosis. Plant Mol. Biol. Rep. 8, 40–49. doi:10.1007/bf02668879
Belkhadir, Y., Subramaniam, R., and Dangl, J. L. (2004). Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Curr. Opin. Plant Biol. 7, 391–399. doi:10.1016/j.pbi.2004.05.009
Bent, A. F., Kunkel, B. N., Dahlbeck, D., Brown, K. L., Schmidt, R., Giraudat, J., et al. (1994). RPS2 of Arabidopsis thaliana: A leucine-rich repeat class of plant disease resistance genes. Science 265, 1856–1860. doi:10.1126/science.8091210
Bourne, H. R., Sanders, D. A., and McCormick, F. (1991). The GTPase superfamily: Conserved structure and molecular mechanism. Nature 349, 117–127. doi:10.1038/349117a0
Bresson, A., Jorge, V., Dowkiw, A., Guerin, V., Bourgait, I., Tuskan, G. A., et al. (2011). Qualitative and quantitative resistances to leaf rust finely mapped within two nucleotide-binding site leucine-rich repeat (NBS-LRR)-rich genomic regions of chromosome 19 in poplar. New Phytol. 192, 151–163. doi:10.1111/j.1469-8137.2011.03786.x
Brummer, E. C. (2004). Applying genomics to alfalfa breeding programs. Crop Sci. 44, 1904–1907. doi:10.2135/cropsci2004.1904
Bryan, G. T., Wu, K. S., Farrall, L., Jia, Y., Hershey, H. P., McAdams, S. A., et al. (2000). tA single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell. 12, 2033–2046. doi:10.1105/tpc.12.11.2033
Campbell, T. A., Bao, G., and Xia, Z. L. (1999). Completion of the agronomic evaluations of Medicago ruthenica [(L.) Ledebour] germplasm collected in Inner Mongolia. Genet. Resour. Crop Ev. 46, 477–484. doi:10.1023/A:1008730322306
Cannon, S. B., Zhu, H., Baumgarten, A. M., Spangler, R., May, G., Cook, D. R., et al. (2002). Diversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS-LRR resistance gene subfamilies. J. Mol. Evol. 54, 548–562. doi:10.1007/s0023901-0057-2
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi:10.1016/j.molp.2020.06.009
Cheng, Y., Li, X., Jiang, H., Ma, W., Miao, W., Yamada, T., et al. (2012). Systematic analysis and comparison of nucleotide-binding site disease resistance genes in maize. FEBS J. 279, 2431–2443. doi:10.1111/j.1742-4658.2012.08621.x
Chini, A., Grant, J. J., Seki, M., Shinozaki, K., and Loake, G. J. (2004). Drought tolerance established by enhanced expression of the CC-NBS-LRR gene, ADR1, requires salicylic acid, EDS1 and ABI1. Plant J. 38, 810–822. doi:10.1111/j.1365-313X.2004.02086.x
Chisholm, S. T., Coaker, G., Day, B., and Staskawicz, B. J. (2006). Host-microbe interactions: Shaping the evolution of the plant immune response. Cell. 124, 803–814. doi:10.1016/j.cell.2006.02.008
Cook, D. R. (1999). Medicago truncatula—A model in the making. Curr. Opin. Plant Biol. 2, 301–304. doi:10.1016/s1369-5266(99)80053-3
Djebbi, S., Bouktila, D., Makni, H., Makni, M., and Mezghani-Khemakhem, M. (2015). Identification and characterization of novel NBS-LRR resistance gene analogues from the pea. Genet. Mol. Res. 14, 6419–6428. doi:10.4238/2015.June.11.18
Dou, D., and Zhou, J. M. (2012). Phytopathogen effectors subverting host immunity: Different foes, similar battleground. Cell. Host Microbe 12, 484–495. doi:10.1016/j.chom.2012.09.003
Downward, J. (1990). The ras superfamily of small GTP-binding proteins. Trends biochem. Sci. 15, 469–472. doi:10.1016/0968-0004(90)90300-z
Duan, X. Q., Shen, Y. S., Shen, S. Q., Meng, F. B., Liu, Y., Zhang, L., et al. (2019). Multiple genome alignment analysis of the NBS-LRR resistant genes evolution in Legume. Mol. plant Breed. 17, 3145–3156. doi:10.13271/j.mpb.017.003145
Ellis, J., and Jones, D. (1998). Structure and function of proteins controlling strain-specific pathogen resistance in plants. Curr. Opin. Plant Biol. 1, 288–293. doi:10.1016/1369-5266(88)80048-7
Frazier, T. P., Palmer, N. A., Xie, F., Tobias, C. M., Donze-Reiner, T. J., Bombarely, A., et al. (2016). Identification, characterization, and gene expression analysis of nucleotide binding site (NB)-type resistance gene homologues in switchgrass. BMC Genom 17, 892. doi:10.1186/s12864-016-3201-5
Fujita, M., Fujita, Y., Noutoshi, Y., Takahashi, F., Narusaka, Y., Yamaguchi-Shinozaki, K., et al. (2006). Crosstalk between abiotic and biotic stress responses: A current view from the points of convergence in the stress signaling networks. Curr. Opin. Plant Biol. 9, 436–442. doi:10.1016/j.pbi.2006.05.014
Gassmann, W., Hinsch, M. E., and Staskawicz, B. J. (1999). The Arabidopsis RPS4 bacterial-resistance gene is a member of the TIR-NBS-LRR family of disease-resistance genes. Plant J. 20, 265–277. doi:10.1046/j.1365-313x.1999.t01-1-00600.x
Gong, Y. H., Zhao, C. L., Wang, X., Li, Y. L., Ding, G. Z., and Chen, L. (2021). Identification and analysis of NBS-LRR gene family in sugar beet. J. plant Prot. 59, 1–18. doi:10.13802/j.cnki.zwbhxb.2021.2021023
Goyal, N., Bhatia, G., Sharma, S., Garewal, N., Upadhyay, A., Upadhyay, S. K., et al. (2020). Genome-wide characterization revealed role of NBS-LRR genes during powdery mildew infection in Vitis vinifera. Genomics 112, 312–322. doi:10.1016/j.ygeno.2019.02.011
Grant, M. R., Godiard, L., Straube, E., Ashfield, T., Lewald, J., Sattler, A., et al. (1995). Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 269, 843–846. doi:10.1126/science.7638602
Guan, B., Zhou, D., Zhang, H., Tian, Y., Japhet, W., and Wang, P. (2009). Germination responses of Medicago ruthenica seeds to salinity, alkalinity, and temperature. J. Arid. Environ. 73, 135–138. doi:10.1016/j.jaridenv.2008.08.009
Huang, C. L., Hwang, S. Y., Chiang, Y. C., and Lin, T. P. (2008). Molecular evolution of the Pi-ta gene resistant to rice blast in wild rice (Oryza rufipogon). Genetics 179, 1527–1538. doi:10.1534/genetics.108.089805
Hulbert, S. H., Webb, C. A., Smith, S. M., and Sun, Q. (2001). Resistance gene complexes: Evolution and utilization. Annu. Rev. Phytopathol. 39, 285–312. doi:10.1146/annurev.phyto.39.1.285
Johal, G. S., and Briggs, S. P. (1992). Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258, 985–987. doi:10.1126/science.1359642
Jones, D. A., and Jones, J. D. G. (1997). The role of leucine-rich repeat proteins in plant defences. Adv. Bot. Res. 24, 89–167. doi:10.1016/S0065-2296(08)60072-5
Jupe, F., Pritchard, L., Etherington, G. J., Mackenzie, K., Cock, P. J., Wright, F., et al. (2012). Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genom 15 (13), 75. doi:10.1186/1471-2164-13-75
Kaziro, Y., Itoh, H., Kozasa, T., Nakafuku, M., and Satoh, T. (1991). Structure and function of signal-transducing GTP-binding proteins. Annu. Rev. Biochem. 60, 349–400. doi:10.1146/annurev.bi.60.070191.002025
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
Kobe, B., and Deisenhofer, J. (1995). A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature 374, 183–186. doi:10.1038/374183a0
Kourelis, J., and van der Hoorn, R. A. L. (2018). Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell. 30, 285–299. doi:10.1105/tpc.17.00579
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi:10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Lacombe, S., Rougon-Cardoso, A., Sherwood, E., Peeters, N., Dahlbeck, D., van Esse, H. P., et al. (2010). Interfamily transfer of a plant pattern-recognition receptor confers broad-spectrum bacterial resistance. Nat. Biotechnol. 28, 365–369. doi:10.1038/nbt.1613
Leister, D. (2004). Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 20, 116–122. doi:10.1016/j.tig.2004.01.007
Leister, R. T., and Katagiri, F. (2000). A resistance gene product of the nucleotide binding site —Leucine rich repeats class can form a complex with bacterial avirulence proteins in vivo. Plant J. 22, 345–354. doi:10.1046/j.1365-313x.2000.00744.x
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Li, H. Y., Li, Z. Y., Cai, L. Y., Shi, W. G., Mi, F. G., and Shi, F. L. (2013). Analysis of genetic diversity of ruthenia medic (Medicago ruthenica (l.) trautv.) in inner Mongolia using ISSR and SSR markers. Genet. Resour. Crop Ev. 60, 1687–1694. doi:10.1007/s10722-012-9950-3
Li, Q., Jiang, X. M., and Shao, Z. Q. (2021). Genome-wide analysis of NLR disease resistance genes in an updated reference genome of barley. Front. Genet. 24, 694682. doi:10.3389/fgene.2021.694682
Liu, R., Liang, Y. F., Xue, F. X., Song, D. H., Yang, Z. X., Cai, L., et al. (1989). The first report on Medicago ruthenica var. inschanicus disease. Pratacult. Sci. 6, 4.
Liu, Y. F., Wan, H. J., Wei, Y. P., Li, Z. M., Ye, Q. J., Wang, R. Q., et al. (2014). Genome-wide analysis of NBS-LRR resistance genes in tomato. J. Nucl. Agric. Sci. 28, 790. doi:10.11869/j.issn.100-8551.2014.05.0790
Liu, Y., Zeng, Z., Zhang, Y. M., Li, Q., Jiang, X. M., Jiang, Z., et al. (2021). An angiosperm NLR Atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion. Mol. Plant 14, 2015–2031. doi:10.1016/j.molp.2021.08.001
Liu, Z., Chen, T., Ma, L., Zhao, Z., Zhao, P. X., Nan, Z., et al. (2013). Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS One 8, e83549. doi:10.1371/journal.pone.0083549
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput. Biol. 13, e1005457. doi:10.1371/journal.pcbi.1005457
Lozano, R., Hamblin, M. T., Prochnik, S., and Jannink, J. L. (2015). Identification and distribution of the NBS-LRR gene family in the Cassava genome. BMC Genom 16, 360. doi:10.1186/s12864-015-1554-9
Lozano, R., Ponce, O., Ramirez, M., Mostajo, N., and Orjeda, G. (2012). Genome-wide identification and mapping of NBS-encoding resistance genes in Solanum tuberosum group phureja. PLoS One 7, e34775. doi:10.1371/journal.pone.0034775
Mackey, D., Belkhadir, Y., Alonso, J. M., Ecker, J. R., and Dangl, J. L. (2003). Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell. 112, 379–389. doi:10.1016/s0092-8674(03)00040-0
MacQueen, A., and Bergelson, J. (2016). Modulation of R-gene expression across environments. J. Exp. Bot. 67, 2093–2105. doi:10.1093/jxb/erv530
Magadum, S., Banerjee, U., Murugan, P., Gangapur, D., and Ravikesavan, R. (2013). Gene duplication as a major force in evolution. J. Genet. 92, 155–161. doi:10.1007/s12041-013-0212-8
Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397. doi:10.1038/41131
Meyers, B. C., Kozik, A., Griego, A., Kuang, H., and Michelmore, R. W. (2003). Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell. 15, 809–834. doi:10.1105/tpc.009308
Meyers, B. C., Morgante, M., and Michelmore, R. W. (2002). TIR-X and TIR-NBS proteins: Two new families related to disease resistance TIR-NBS-LRR proteins encoded in Arabidopsis and other plant genomes. Plant J. 32, 77–92. doi:10.1046/j.1365-313x.2002.01404.x
Monosi, B., Wisser, R. J., Pennill, L., and Hulbert, S. H. (2004). Full-genome analysis of resistance gene homologues in rice. Theor. Appl. Genet. 109, 1434–1447. doi:10.1007/s00122-004-1758-x
Mun, J. H., Yu, H. J., Park, S., and Park, B. S. (2009). Genome-wide identification of NBS-encoding resistance genes in Brassica rapa. Mol. Genet. Genomics 282, 617–631. doi:10.1007/s00438-009-0492-0
Neupane, S., Andersen, E. J., Neupane, A., and Nepal, M. P. (2018). Genome-wide identification of NBS-encoding resistance genes in sunflower (Helianthus annuus L.). Genes. (Basel) 9, 384. doi:10.3390/genes9080384
Porter, B. W., Paidi, M., Ming, R., Alam, M., Nishijima, W. T., and Zhu, Y. J. (2009). Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol. Genet. Genomics 281, 609–626. doi:10.1007/s00438-009-0434-x
Qian, L. H., Wang, Y., Chen, M., Liu, J., Lu, R. S., Zou, X., et al. (2021). Genome-wide identification and evolutionary analysis of NBS-LRR genes from Secale cereale. Front. Genet. 12, 771814. doi:10.3389/fgene.2021.771814
Ratnaparkhe, M. B., Wang, X., Li, J., Compton, R. O., Rainville, L. K., Lemke, C., et al. (2011). Comparative analysis of peanut NBS-LRR gene clusters suggests evolutionary innovation among duplicated domains and erosion of gene microsynteny. New Phytol. 192, 164–178. doi:10.1111/j.1469-8137.2011.03800.x
Ren, S., Sun, M., Yan, H., Wu, B., Jing, T., Huang, L., et al. (2020). Identification and distribution of NBS-encoding resistance genes of Dactylis glomerata L. and its expression under abiotic and biotic stress. Biochem. Genet. 58, 824–847. doi:10.1007/s10528-020-09977-8
Rojas, A. M., Fuentes, G., Rausell, A., and Valencia, A. (2012). The ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell. Biol. 196, 189–201. doi:10.1083/jcb.201103008
Shao, Z. Q., Xue, J. Y., Wang, Q., Wang, B., and Chen, J. Q. (2019). Revisiting the origin of plant NBS-LRR genes. Trends Plant Sci. 24, 9–12. doi:10.1016/j.tplants.2018.10.015
Shao, Z. Q., Xue, J. Y., Wu, P., Zhang, Y. M., Wu, Y., Hang, Y. Y., et al. (2016). Large-scale analyses of angiosperm nucleotide-binding site-leucine-rich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns. Plant Physiol. 170, 2095–2109. doi:10.1104/pp.15.01487
Shao, Z. Q., Zhang, Y. M., Hang, Y. Y., Xue, J. Y., Zhou, G. C., Wu, P., et al. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: Understanding gained from and beyond the legume family. Plant Physiol. 166, 217–234. doi:10.1104/pp.114.243626
Shi, J., Zhang, M., Zhai, W., Gao, H., Zhang, W., et al. (2018). Genome-wide analysis of nucleotide binding site-leucine-rich repeats (NBS-LRR) disease resistance genes in Gossypium hirsutum. Physiol. Mol. Plant P. 104, 1–8. S0885576518301607-. doi:10.1016/j.pmpp.2018.07.007
Shi, R., Jiao, W., Yun, L., Zhang, Z., Zhang, X., Wang, Q., et al. (2021). Utilization of transcriptome, small RNA, and degradome sequencing to provide insights into drought stress and rewatering treatment in Medicago ruthenica. Front. Plant Sci. 12, 675903. doi:10.3389/fpls.2021.675903
Shu, Y., Li, W., Zhao, J., Liu, Y., and Guo, C. (2018). Transcriptome sequencing and expression profiling of genes involved in the response to abiotic stress in Medicago ruthenica. Genet. Mol. Biol. 41, 638–648. doi:10.1590/1678-4685-GMB-2017-0284
Small, E., and Jomphe, M. (1989). A synopsis of the genus Medicago (Leguminosae). Can. J. Bot. 67, 3260–3294. doi:10.1139/b89-405
Tameling, W. I., Elzinga, S. D., Darmin, P. S., Vossen, J. H., Takken, F. L., Haring, M. A., et al. (2002). The tomato R gene products I-2 and MI-1 are functional ATP binding proteins with ATPase activity. Plant Cell. 14, 2929–2939. doi:10.1105/tpc.005793
Thomma, B. P., Nürnberger, T., and Joosten, M. H. (2011). Of PAMPs and effectors: The blurred PTI-ETI dichotomy. Plant Cell. 23, 4–15. doi:10.1105/tpc.110.082602
Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi:10.1038/nature11119
Trapnell, C., Roberts, A., Goff, L. A., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 7, 562–578. doi:10.1038/nprot.2012.016
Traut, T. W. (1994). The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem. 222, 9–19. doi:10.1111/j.1432-1033.1994.tb18835.x
Walkowiak, S., Gao, L., Monat, C., Haberer, G., Kassa, M. T., Brinton, J., et al. (2020). Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283. doi:10.1038/s41586-020-2961-x
Wang, D. K., Hong, L. I., and Luo, X. Y. (2008). Crossbreeding of Melilotoides ruthenicus and Medicago sativa. Acta Agrestia Sin. 16, 458–465. doi:10.3901/CJME.2008.04.040
Wang, T., Ren, L., Li, C., Zhang, D., Zhang, X., Zhou, G., et al. (2021). The genome of a wild Medicago species provides insights into the tolerant mechanisms of legume forage to environmental stress. BMC Biol. 19, 96. doi:10.1186/s12915-021-01033-0
Wang, X., Richards, J., Gross, T., Druka, A., Kleinhofs, A., Steffenson, B., et al. (2013). The rpg4-mediated resistance to wheat stem rust (Puccinia graminis) in barley (Hordeum vulgare) requires Rpg5, a second NBS-LRR gene, and an actin depo-lymerization factor. Mol. Plant Microbe. Interact. 26, 407–418. doi:10.1094/MPMI-06-12-0146-R
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Warren, R. F., Henk, A., Mowery, P., Holub, E., and Innes, R. W. (1998). A mutation within the leucine-rich repeat domain of the Arabidopsis disease resistance gene RPS5 partially suppresses multiple bacterial and downy mildew resistance genes. Plant Cell. 10, 1439–1452. doi:10.1105/tpc.10.9.1439
Witek, K., Jupe, F., Witek, A. I., Baker, D., Clark, M. D., and Jones, J. D. (2016). Accelerated cloning of a potato late blight-resistance gene using RenSeq and SMRT sequencing. Nat. Biotechnol. 34, 656–660. doi:10.1038/nbt.3540
Xiao, S., Ellwood, S., Calis, O., Patrick, E., Li, T., Coleman, M., et al. (2001). Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science 291, 118–120. doi:10.1126/science.291.5501.118
Xue, J. Y., Takken, F. L. W., Nepal, M. P., Maekawa, T., and Shao, Z. Q. (2020). Editorial: Evolution and functional mechanisms of plant disease resistance. Front. Genet. 11, 593240. doi:10.3389/fgene.2020.593240
Yan, T., Zhou, Z., Wang, R., Bao, D., Li, S., Li, A., et al. (2022). A cluster of atypical resistance genes in soybean confers broad-spectrum antiviral activity. Plant Physiol. 188, 1277–1293. doi:10.1093/plphys/kiab507
Yin, M., Zhang, S., Du, X., Mateo, R. G., Guo, W., Li, A., et al. (2021). Genomic analysis of Medicago ruthenica provides insights into its tolerance to abiotic stress and demographic history. Mol. Ecol. Resour. 21, 1641–1657. doi:10.1111/1755-0998.13363
Yu, X., Zhong, S., Yang, H., Chen, C., Chen, W., Yang, H., et al. (2021). Identification and characterization of NBS resistance genes in Akebia trifoliata. Front. Plant Sci. 12, 758559. doi:10.3389/fpls.2021.758559
Yue, J. X., Meyers, B. C., Chen, J. Q., Tian, D., and Yang, S. (2012). Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol. 193, 1049–1063. doi:10.1111/j.1469-8137.2011.04006.x
Zhang, W., Yuan, Q., Wu, Y., Zhang, J., and Nie, J. (2022). Genome-wide identification and characterization of the CC-NBS-LRR gene family in cucumber (Cucumis sativus L.). Int. J. Mol. Sci. 23, 5048. doi:10.3390/ijms23095048
Zhang, Y. M., Chen, M., Sun, L., Wang, Y., Yin, J., Liu, J., et al. (2020). Genome-wide identification and evolutionary analysis of NBS-LRR genes from Dioscorea rotundata. Front. Genet. 11, 484. doi:10.3389/fgene.2020.00484
Zhou, Q., Jia, C., Ma, W., Cui, Y., Jin, X., Luo, D., et al. (2019). MYB transcription factors in alfalfa (Medicago sativa): Genome-wide identification and expression analysis under abiotic stresses. PEERJ 7, e7714. doi:10.7717/peerj.7714
Zhou, T., Wang, Y., Chen, J. Q., Araki, H., Jing, Z., Jiang, K., et al. (2004). Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS-LRR genes. Mol. Genet. Genomics 271, 402–415. doi:10.1007/s00438-004-0990-z
Keywords: Medicago ruthenica, NLR gene family, chromosome distribution, phylogenetic analysis, syntenic analysis, expression analysis
Citation: Tong C, Zhang Y and Shi F (2023) Genome-wide identification and analysis of the NLR gene family in Medicago ruthenica. Front. Genet. 13:1088763. doi: 10.3389/fgene.2022.1088763
Received: 03 November 2022; Accepted: 22 December 2022;
Published: 10 January 2023.
Edited by:
Xianzhong Huang, Anhui Science and Technology University, ChinaReviewed by:
Zhu-Qing Shao, Nanjing University, ChinaKaijing Zhang, Anhui Science and Technology University, China
Copyright © 2023 Tong, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengling Shi, bm1jenlzZmxAMTI2LmNvbQ==