 Meral Kayikcioglu
Meral Kayikcioglu Hasan Selcuk Ozkan
Hasan Selcuk Ozkan Burcu Yagmur
Burcu Yagmur Selen Bayraktaroglu
Selen Bayraktaroglu Asli Tetik Vardarli
Asli Tetik Vardarli- 1Department of Cardiology, Ege University School of Medicine, Izmir, Turkey
- 2Ege University School of Medicine, Izmir, Turkey
- 3Department of Radiology, Ege University School of Medicine, Izmir, Turkey
- 4Department of Medical Biology, Ege University School of Medicine, Izmir, Turkey
Background: Homozygous familial hypercholesterolemia (HoFH) is a rare and devastating genetic condition characterized by extremely elevated levels of low-density lipoprotein cholesterol (LDL-C) leading to an increased risk of premature atherosclerosis. Patients with Homozygous familial hypercholesterolemia mostly present with mutations in LDLR; however, herein, we present two cases with concomitant microsomal triglyceride transfer protein mutations, who showed different clinical courses and treatment adherence on long-term therapy with the new MTTP inhibitor lomitapide.
Objectives: We aimed to present the possibility of preventing the progression of atherosclerotic burden with effective and safe LDL-C reduction in patients with Homozygous familial hypercholesterolemia on low-dose lomitapide therapy and emphasize the role of treatment adherence in therapy success.
Methods: We present two patients with phenotypically Homozygous familial hypercholesterolemia, a compound heterozygous woman and a simple homozygous man, both with LDLR and additional MTTP mutations, who were treated with the MTTP-inhibiting agent lomitapide, with different treatment compliances. The role of impulsivity was investigated through Barratt Impulsivity Scale 11, and the extent of the atherosclerotic burden was followed up using coronary artery calcium scoring, echocardiographic and sonographic findings, and, eventually, through a strict follow-up of laboratory parameters. The patients were on lomitapide for 8 and 5 years, respectively, with no adverse effects.
Conclusion: When accompanied by good adherence to therapy, low-dose lomitapide on top of standard lipid-lowering therapy with decreased frequency of lipid apheresis prevented the progression of atherosclerotic burden. Non-compliance might occur due to patient impulsivity and non-adherence to a low-fat diet.
1 Introduction
Homozygous familial hypercholesterolemia (HoFH) is a rare metabolic disorder of mainly autosomal-dominant inheritance that causes extremely high low-density lipoprotein (LDL) cholesterol levels (Rosenson, 2021). Affected individuals show elevated LDL cholesterol levels from birth, which leads to premature atherosclerotic cardiovascular disease (ASCVD). Therefore, the early detection of this condition is critical to prevent early morbidity and mortality (Kayikcioglu et al., 2014; Zhang et al., 2020; Tokgozoglu and Kayikcioglu, 2021). At least four genes are associated with familial hypercholesterolemia (FH), including LDLR, APOB, PCSK9, and LDLRAP1, with phenotypic variations (Prince et al., 2013; Chemello et al., 2021; Tokgozoglu and Kayikcioglu, 2021). In most cases, the underlying mutation is in LDLR, which is associated with typical LDL cholesterol levels >500 mg/dl in patients with HoFH. Due to absent or defective LDL-receptor activity, individuals with HoFH are resistant to conventional lipid-lowering therapy (LLT) targeting LDL-cholesterol clearance by upregulating LDL receptors. Therefore, LDL apheresis is the most effective treatment for these patients (Kayikcioglu et al., 2018). However, the semi-invasive and time-consuming nature of apheresis is the major obstacle leading to decreased quality of life, increased risk of depression, and deterioration in mental status, leading to high treatment refusal and low adherence (Kayikcioglu et al., 2019; Tunçel et al., 2020). Therefore, effective pharmacotherapies with long-term safety are needed for patients with HoFH.
Lomitapide is a microsomal triglyceride transfer protein (MTTP) inhibitor that reduces LDL cholesterol levels through LDL receptor-independent pathways by directly decreasing apoprotein (Apo)-B levels (Cuchel et al., 2013). Herein, we present two patients with HoFH on long-term, low-dose lomitapide without drug adverse effects but different atheroma courses despite similar baseline LDL cholesterol levels.
2 Case descriptions
Both patients were undergoing long-term follow-up at the Ege University Medical School, Department of Cardiology, Lipid Clinic. They were selected by the means of need and after receiving consent for the compassionate use of lomitapide as an add-on therapy. The clinical characteristics of the patients before and after lomitapide therapy are shown in Table 1. Figure 1A,B depicts the timeline of the case symptoms, diagnosis, and management.
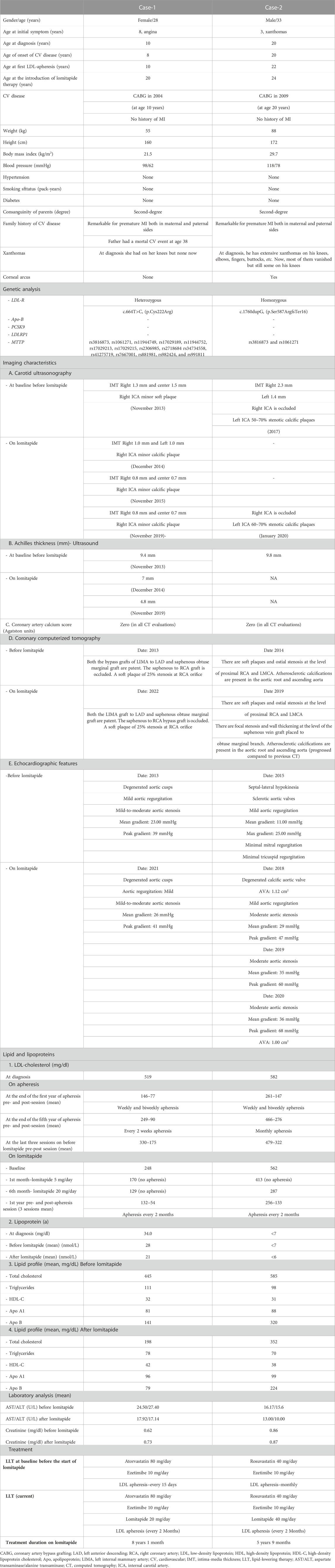
TABLE 1. Clinical characteristics of the cases.
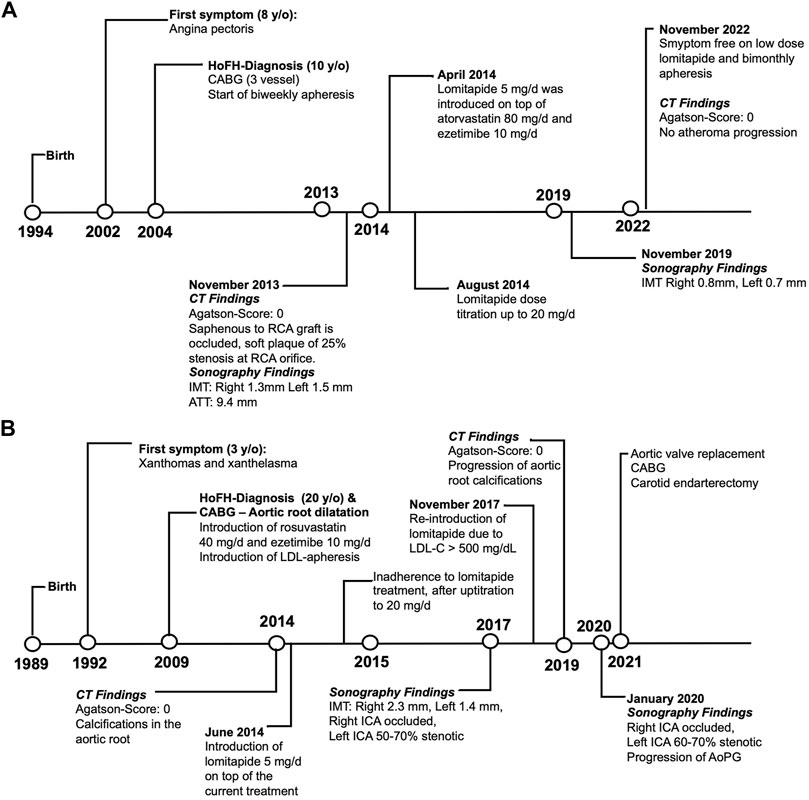
FIGURE 1. Timeline schema representing the clinical evolutions of Cases 1 (A) and 2 (B). Abbreviations: CABG, coronary artery bypass graft; CT, computed tomography; ICA, internal carotid artery; RCA, right coronary artery; IMT, intima-media thickness; ATT, Achilles tendon thickness; HoFH, homozygous familial hypercholesterolemia; AoPG, aortic peak gradient; LDL-C, low-density lipoprotein cholesterol.
2.1 Genetic analysis
Blood specimens were collected from the cases into tubes containing ethylenediaminetetraacetic acid. Genomic DNA was extracted from peripheral blood samples on a MagNA Pure LC instrument using the MagNA Pure LC DNA Isolation Kit I (Roche Applied Science, Germany). To amplify LDLR, APOB, PCSK9, LDLRAP1, and MTTP regions, the Ion AmpliSeq™ Designer tool was used to design specific assay primers to generate a custom FH-DNA panel based on the human (Hg19) reference genome (Ion AmpliSeq™ Target Technology, ThermoFisher Scientific, Waltham, MA). LDLR, APOB, PCSK9, LDLRAP1, and MTTP regions of the cases were sequenced on an Ion-Torrent PGM instrument (Life Technologies, Rockville, MD) (314-chip) according to the manufacturer’s recommended protocol. The Ion-Torrent suite 4.0 0.2 plugin was used to align the reads to the Hg19 reference genome. Integrative Genomics Viewer (IGV) v2.3 software http://www.broadinstitute.org/software/igv/download) was used to remove false-positive variations and possible PCR errors from the obtained variants. The Human Genetic Mutation Database (HGMD; http://www.hgmd.org/), the Variant Database (FHVD; http://www.ucl.ac.uk/ugi/fh), and https://varsome.com/databases were used for the annotation and prediction of the significance of the detected variants.
2.2 Case reports
Case 1 is a 28-year-old woman with HoFH receiving low-dose lomitapide combined with apheresis for 8 years. Her initial complaints started with chest pain at 8 years of age. She had undergone coronary artery bypass grafting (CABG) due to severe three-vessel coronary artery disease (CAD) at 10 years of age, when she was diagnosed with HoFH with an untreated LDL-cholesterol level ranging between 490 and 549 mg/dl. Her family history was remarkable for early-onset CAD and high cholesterol levels. She had received weekly or biweekly LDL-apheresis therapy since 10 years of age. As her LDL-cholesterol levels were far from the treatment goals, oral lomitapide 5 mg/dl was introduced on top of atorvastatin (80 mg/day) and ezetimibe (10 mg/day) at 20 years of age. The dose was titrated up to 20 mg/dl and has been maintained without adverse events. An additional 31% LDL-cholesterol reduction was observed in the first month of low-dose lomitapide (5 mg/day) therapy. At a dose of 20 mg/day, the LDL-cholesterol reduction reached 49% at the end of 6 months. As she declined to be weaned off apheresis, we reduced the apheresis frequency and she received low-dose lomitapide (20 mg/day) plus concomitant bimonthly apheresis. However, during the pandemic for 1 year, she could not access apheresis. Throughout the 8-year lomitapide treatment, she did not experience any increase in serum liver enzyme levels, weight loss, or liver steatosis. Moreover, no change was observed in hepatic fibro scans. She followed a fat-restricted diet compatible with lomitapide therapy to prevent steatorrhea, with concomitant supplementation of fat-soluble vitamins and essential fatty acids. Since the commencement of lomitapide therapy, the patient has remained stable at functional class (NYHA-I) without any adverse cardiovascular events.
Imaging work-up at diagnosis revealed mild aortic stenosis [aortic peak gradient (AoPG) of 30 mmHg-mean (AoMG) 23–18 mmHg] and mild aortic regurgitation with a normal ejection fraction (EF). During the 8-year follow-up, the AoMG was stable at around 25–30 mmHg. In 2022, cardiac catheterization revealed an AoPG of 30 mmHg with normal EF. The computed tomography (CT) angiography showed almost identical coronary and aortal involvement between the baseline (2013) and follow-up (2022) images (Figure 2A). These findings were confirmed by coronary angiography in 2022. Similarly, both the Achilles tendon thickness (ATT) on ultrasonography and carotid intima-media thickness (IMT) significantly decreased with lomitapide treatment. Moreover, the minor soft plaque at the orifice of the right carotid artery became calcified with no progression with lomitapide therapy.
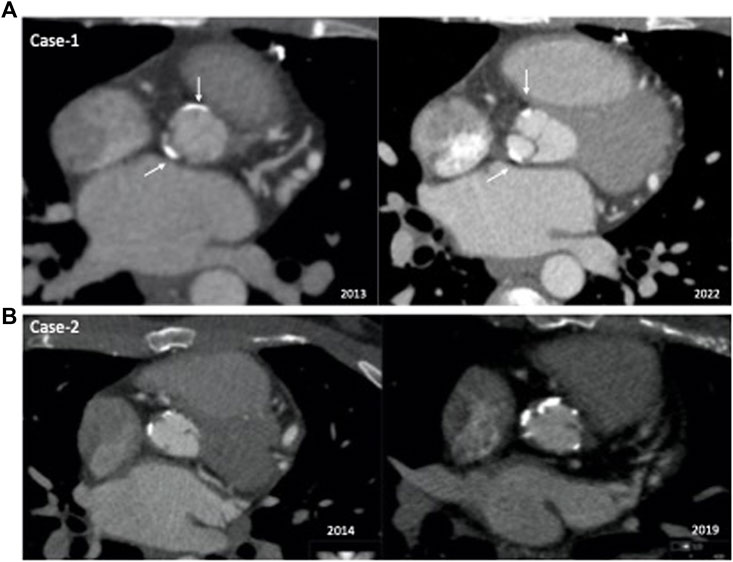
FIGURE 2. Comparison of baseline and follow-up computed tomography angiography images of the patients. (A) Case 1: Aortal involvement was almost identical between the baseline (2013) and follow-up (2022) images. (B) Case 2: Comparison of 2014 and 2019−ΔΔCT angiograms showed significant progression of calcifications in the aortic root.
Case 2 is a 33-year-old man with a remarkable history related to HoFH. His first symptoms were xanthomas on his ankles and xanthelasma which appeared at 3 years of age. However, he was not diagnosed with HoFH until 20 years of age. He has presented with exertional angina and angiographically extensive ostial coronary stenotic lesions and had undergone CABG accompanied by aortic root dilation at 20 years of age. One year later, he was referred to our lipid clinic for extremely high LDL-cholesterol levels ranging between 420 and 540 mg/dl. Xanthomas were apparent on his elbows, knees, and ankles. He was already prescribed rosuvastatin (40 mg/day) and ezetimibe treatment, and we introduced LDL-apheresis. He lived a 14-h drive from our center; therefore, he receive monthly apheresis. As his LDL-cholesterol levels were still >200 mg/dl post-apheresis (Mach et al., 2020), we introduced lomitapide in June 2014, when he was 25 years of age. At the initial dose of oral 5 mg/day, mild nausea was observed. With the up-titrated dose of 20 mg/day, his nausea did not increase; however, after 6 months, he declined to take his pills as he experienced an 8 kg weight loss. For the next 3 years, he continued monthly apheresis but no lomitapide. We re-introduced lomitapide in November 2017 due to persistent LDL-cholesterol levels >500 mg/dl. However, this time he was compliant with the low-fat diet and lomitapide. He did not experience any adverse symptoms or weight loss with an up-titrated dose to 40 mg/day. No alteration was observed in transaminase levels, and a recent hepatic fibroscan was normal. We observed an additional 27% LDL-cholesterol reduction for a 5 mg daily dose of lomitapide at the end of the first month, which increased to 49% at the end of 6 months at a dose of 20 mg daily. The patient has been taking the current dose of lomitapide (40 mg/day) for almost 5 years and concomitant apheresis (every 2–3 months). During the pandemic, his access to apheresis was disturbed for 1 year; however, he maintained his lomitapide therapy. He also received supplementation with omega 3-6-9 and vitamin E. However, he reported several times reasons other than adverse effects for not being adherent to lomitapide therapy. During the follow-up, his coronary lesions and aortic stenosis progressed, and he underwent aortic valve replacement (Benthal operation), carotid endarterectomy, and repeated CABG in 2021. Figure 2B depicts the comparison of 2014 and 2019−ΔΔCT angiograms showing significant progression of calcifications in the aortic root.
2.3 Genetic results
According to the sequencing results, Case 1 was heterozygous for LDLR [c.664T>C, (p.Cys222Arg)], while Case 2 was homozygous for LDLR [c.1760dupG, (p.Ser587ArgfsTer16)]. No clinically significant mutation was found in the APOB, PCSK9, and LDLRAP1 regions in both patients. Additionally, 16 different MTTP variants were detected in the two patients. Cases 1 and 2 shared 2 MTTP variants (rs3816873 and rs1061271). Case 2 also showed one mutation (c.*450A>G) with uncertain clinical significance in public databases, while Case 1 showed 13 mutations (rs11944749, rs17029189, rs11944752, rs17029213, rs17029215, rs2306985, rs2718684 rs34734558, rs41275719, rs7667001, rs881981, rs982424, and rs991811) in MTTP. Twelve of these 13 mutations were categorized as benign/likely benign in public databases; only MTTP c.3G>A (rs11944752) was associated with elevated plasma glucose, insulin (MAGIC Consortium HGVM 4589669), and cholesterol levels (Teslovich et al., 2010).
3 Discussion
We present two real-life cases of HoFH with completely different disease courses after the commencement of lomitapide therapy. Case 1 demonstrated that long-term effective LLT with low-dose lomitapide combined with apheresis safely prevented the progression of atheroma both in coronary and vascular territories and retarded the progression of aortic stenosis. However, Case 2 showed a completely different course, with significant progression of his coronary lesions and aortic involvement. The patient had to undergo re-operation despite similar baseline LDL-cholesterol levels to those in Case 1. The difference between these patients’ disease courses suggests the importance of adherence to therapy for the clinical success of therapy.
Lomitapide is a new-generation potent lipid-lowering agent that acts independently of LDL receptors. Lomitapide inhibits MTTP, a cellular protein that transports neutral lipids between membrane vesicles and acts as a chaperone for the synthesis of apoB-containing triglyceride-rich lipoproteins. Lomitapide has a critical role in the assembly and secretion of apoB-containing lipoproteins in the liver and intestines (Hussain et al., 2012). Thus, lomitapide, besides triglycerides, effectively reduces LDL-cholesterol levels in patients who are lacking or have mutant LDLR. Lomitapide has been approved by the Food and Drug Administration as a lipid-lowering agent for patients with HoFH as an adjunct to standard LLT and by the European Medicines Agency for patients treated with standard LLT with or without apheresis.
Clinical evidence indicates lomitapide as a potent LDL-receptor-independent agent that has made it possible for patients with HoFH to reach their LDL-cholesterol goals. The phase 3 open-label dose-escalation trial demonstrated that lomitapide at maximal-tolerated doses (5–60 mg/day) reduced LDL-cholesterol by 50% at 26 weeks when added to the standard of care including lipid apheresis in 29 patients with HoFH (Cuchel et al., 2013). Moreover, long-term lomitapide treatment (range 2.1–5.7 years) with a median daily dose of 40 mg reduced LDL-cholesterol levels by an average of 45.5% in 19 patients with HoFH (Blom et al., 2017). During the 246-week therapy, 14 (74%) patients achieved the LDL-cholesterol target of 100 mg/dl and 11 (58%) patients reached the target of 70 mg/dl on at least 1 occasion. We observed a 31% additional reduction in LDL-cholesterol after 1 month of very low-dose (5 mg/day) lomitapide and 49% at the end of 6 months of therapy for a dose of only 20 mg/day in Case 1. In Case 2, the LDL-cholesterol levels were reduced by an additional 27% and 49% for 5 and 20 mg daily doses of lomitapide at the end of 1 and 6 months, respectively. Due to the effective reductions in LDL-cholesterol levels, we decreased the frequency of the apheresis sessions to every 2 months. The LDL-cholesterol reductions achieved in our cases are consistent with those in an HoFH case series reporting >50% reductions for doses of 10–40 mg/day in real-life settings (Stefanutti et al., 2016), which likely led to clinically significant reductions in ASCVD. Consequently, Case 1, who showed a consistent reduction of LDL-cholesterol levels, showed no ASCVD events and no atheroma progression. However, Case 2 showed a different course, with atherosclerosis progression during the 5-year therapy. The difference in therapy adherence was likely the major explanation for the diverse disease courses after lomitapide therapy in our patients. Adherence to LLT favors the survival of patients with ASCVD (Dopheide et al., 2021). Case 1 was adherent since the first dose of therapy. However, in Case 2, the first attempt to use lomitapide was not successful as the patient declined its use due to weight loss; another major obstacle was non-compliance to the low-fat diet, which is a requirement for lomitapide use. After 3 years, we re-introduced the drug. While he was adherent to the low-fat diet, the patient had many excuses for his lack of therapy adherence, likely due to his impulsive behaviors. Barratt impulsivity Scale 11 (Tamam et al., 2013) demonstrated the high impulsivity of Case 2 and the low impulsivity of Case 1. Treatment adherence is inversely related to impulsive and compulsive behaviors (López-Torrecillas et al., 2021).
Hepato-steatosis and liver-enzyme elevation are well-known phenomena associated with lomitapide treatment. However, none of these were observed in either of our patients with the long-term use of lomitapide at doses lower than those used in clinical trials. The only adverse effect was the weight loss reported by Case 2. These findings suggest that most of the adverse events can be avoided by lowering the dosing of lomitapide, as assessed by the LOWER study, in which the long-term (5-year) follow-up of patients on lomitapide (10–20 mg/dl) showed significantly lower adverse event rates compared to the results of the Phase-3 study conducted with doses of 40–60 mg/dl (Underberg et al., 2020). Furthermore, the 20 mg daily dose of lomitapide effectively reduced LDL-cholesterol levels almost to the targets in Case 1 in the present study. Increasing the dose to 40 mg likely would have allowed us to stop apheresis; however, as the patient insisted on apheresis therapy, we continued with 20 mg lomitapide combined with bi-monthly apheresis for >7 years without adverse effects. Low-dose lomitapide plus conventional therapy (statin and ezetimibe) with a decreased apheresis frequency probably increased the adherence to long-term therapy besides efficient LDL-lowering. Similarly, the use of combination therapy was associated with a higher proportion and greater odds of achieving the therapeutic LDL goals of <70 mg/dl and <55 mg/dl,, particularly with the combination of five drugs in the HICC registry (Vallejo-Vaz et al., 2021).
An important aspect of Case 1 in this study is the stabilized course of the aortic atheroma. In patients with late apheresis initiation after 8–10 years of age, the progression of aortic atheroma to stenosis reportedly cannot be prevented even if the LDL goals are attained (Cuchel et al., 2014; Kayikcioglu et al., 2018; Kayikcioglu, 2021). However, in Case 1, after commencing lomitapide use, the aortic involvement of the valvular stenosis and aortic plaques remained stable. This is also contrary to our clinical experience that despite meeting the LDL goals, children with late-onset apheresis therapy (after 8 years of age) show a gradual progression of aortic valve sclerosis or mild stenosis to moderate and severe stenosis.
In addition, our experiences with both of these patients call attention to lipid apheresis as a treatment difficult to access during the pandemic. Thus, novel effective LDL-lowering agents such as oral lomitapide may be useful for patients with HoFH during lockdowns due to pandemics or any other disaster that could disrupt access to apheresis as a hospital-based therapy (Kayikcioglu et al., 2020). Moreover, apheresis cessation or decreased frequency with an effective LDL-lowering agent will improve the psychosocial wellbeing of patients. We previously documented that apheresis as a semi-invasive, time-consuming therapy leads to a decreased quality of life, increased risk of depression, and deterioration in mental status, which lead to high refusal and low adherence rates (Kayikcioglu et al., 2019; Tunçel et al., 2020) noticeably leading to undertreatment of patients with HoFH, consequently affecting ASCVD outcomes.
Although both patients were phenotypically HoFH with untreated LDL-cholesterol levels >500 mg/dl, xanthomas appearing at early ages, and parental clinical features of heterozygous FH, Case 1 was heterozygous and Case 2 was homozygous for LDLR mutations. In addition to standard genetic analysis of the four associated genes, this study is the first to study MTTP in patients with HoFH. Loss-of-function mutations within MTTP result in severe autosomal recessive diseases, causing ataxia, failure to thrive, steatorrhea, and muscle weakness known as abetalipoproteinemia (Hussain et al., 2012). However, gain-of-function mutations in MTTP are not yet well understood. We identified several variants in both patients that may alter lipid levels or affect the clinical efficacy and adverse reactions to lomitapide.
The cases presented have some limitations and strengths. The evaluation of only five genes affecting LDL-cholesterol levels might be a limitation as other genes may explain the severe FH phenotype of the patients, especially in Case 1. However, the evaluation of MTTP variants is an important strength as these are the first case presentations of HoFH with MTTP variants. Detailed long-term follow-up of the cases and evaluation of impulsivity are additional strengths that allow the evaluation of adherence, clinical course, and the associated factors. Moreover, Case 1 is the first reported case to show the cessation of aortic atheroma progression despite the late onset of apheresis therapy, suggesting the importance of effective LDL-cholesterol lowering in combination with lomitapide and apheresis. Moreover, the comparison of two HoFH patients with similar lipid profiles at baseline and who received the same therapy allowed the assessment of the impact of adherence to therapy.
In conclusion, long-term, low-dose lomitapide was an effective and well-tolerated adjunct to conventional LLT and lipid apheresis in real life. Adherence to lomitapide and a low-fat diet in addition to standard LLT with decreased apheresis frequency appeared to be associated with more effective and safer management of HoFH and prevented the progression of atheroma burden in both the coronaries and aorta and regression in CIMT and ATT. Further studies are needed to illuminate the role of gain-of-function mutations in MTTP on lipid levels and response to lomitapide in patients with HoFH.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the patients for the publication of any potentially identifiable images or data included in this article.
Author contributions
MK and HO designed this study. MK, HO, BY, and SB collected data. MK and HO drafted the manuscript. AV performed the genetic analysis. AV searched for genetic literature. MK, HO, and BY conducted the literature search. All authors contributed to the manuscript and approved the final version for submission.
Funding
Article processing charges were funded by Amryt Pharmaceuticals DAC. Amryt Pharmaceutical DAC was not involved in data analysis, the preparation of the manuscript, nor the decision to submit it for publication.
Conflict of interest
MK has received honoraria (for lectures and consultancy) from Abbott, Jansen, AstraZeneca, Abdi Ibrahim, Nova Nordisk, Novartis, and Sanovel; has received research funding from AstraZeneca, Amryt Pharma; and has participated in clinical trials with Amgen, Bayer Schering, LIB Therapeutics, Lilly, Novartis, and Medpace for the last 3 years.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Blom, D. J., Averna, M. R., Meagher, E. A., du Toit Theron, H., Sirtori, C. R., Hegele, R. A., et al. (2017). Long-term efficacy and safety of the microsomal triglyceride transfer protein inhibitor lomitapide in patients with homozygous familial hypercholesterolemia. Circulation 136 (3), 332–335. doi:10.1161/CIRCULATIONAHA.117.028208
Chemello, K., Garcia-Nafria, J., Gallo, A., Martin, C., Lambert, G., and Blom, D. (2021). Lipoprotein metabolism in familial hypercholesterolemia. J. Lipid Res. 62, 100062. doi:10.1016/j.jlr.2021.100062
Cuchel, M., Bruckert, E., Ginsberg, H. N., Raal, F. J., Santos, R. D., Hegele, R. A., et al. (2014). Homozygous familial hypercholesterolaemia: New insights and guidance for clinicians to improve detection and clinical management. A position paper from the consensus panel on familial hypercholesterolaemia of the European atherosclerosis society. Eur. Heart J. 35 (32), 2146–2157. doi:10.1093/EURHEARTJ/EHU274
Cuchel, M., Meagher, E. A., du Toit Theron, H., Blom, D. J., Marais, A. D., Hegele, R. A., et al. (2013). Effi cacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 381 (9860), 40–46. doi:10.1016/S0140-6736(12)61731-0
Dopheide, J. F., Veit, J., Ramadani, H., Adam, L., Papac, L., Vonbank, A., et al. (2021). Adherence to statin therapy favours survival of patients with symptomatic peripheral artery disease. Eur. Heart J. Cardiovasc. Pharmacother. 7 (4), 263–270. doi:10.1093/EHJCVP/PVZ081
Hussain, M. M., Rava, P., Walsh, M., Rana, M., and Iqbal, J. (2012). Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. 9, 14–16. doi:10.1186/1743-7075-9-14
Kayikcioglu, M., Kismali, E., Can, L., and Payzin, S. (2014). Long-term follow-up in patients with homozygous familial hypercholesterolemia; 13-year experience of a University hospital lipid clinic. Turk Kardiyol. Dern. Ars. 42 (7), 599–611. doi:10.5543/tkda.2014.09633
Kayikcioglu, M., Kuman-Tuncel, O., Pirildar, S., Yilmaz, M., Kaynar, L., Aktan, M., et al. (2019). Clinical management, psychosocial characteristics, and quality of life in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis in Turkey: Results of a nationwide survey (A-HIT1 registry). J. Clin. Lipidol. 13 (3), 455–467. doi:10.1016/j.jacl.2019.02.001
Kayikcioglu, M. (2021). LDL apheresis and lp (a) apheresis: A clinician’s perspective. Curr. Atheroscler. Rep. 23 (4), 15. doi:10.1007/s11883-021-00911-w
Kayikcioglu, M., Tokgozoglu, L., Tuncel, O. K., Pirildar, S., and Can, L. (2020). Negative impact of COVID-19 pandemic on the lifestyle and management of patients with homozygous familial hypercholesterolemia. J. Clin. Lipidol. 14 (6), 751–755. doi:10.1016/J.JACL.2020.09.002
Kayikcioglu, M., Tokgozoglu, L., Yilmaz, M., Kaynar, L., Aktan, M., Durmus, R. B., et al. (2018). A nation-wide survey of patients with homozygous familial hypercholesterolemia phenotype undergoing LDL-apheresis in Turkey (A-HIT 1 registry). Atherosclerosis 270, 42–48. doi:10.1016/j.atherosclerosis.2018.01.034
López-Torrecillas, F., Castillo-Fernandez, E., Ramirez-Ucles, I., and Martin, I. (2021). Impulsivity and compulsivity and their relationship with non-adherence to treatment in the prison population. Int. J. Environ. Res. Public Health 18 (16), 8300. doi:10.3390/ijerph18168300
Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., et al. (2020). 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 41, 111–188. doi:10.1093/eurheartj/ehz455
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 9 (1), 63–75. doi:10.1016/j.jalz.2012.11.007
Rosenson, R. S. (2021). Existing and emerging therapies for the treatment of familial hypercholesterolemia. J. Lipid Res. 62, 100060. doi:10.1016/j.jlr.2021.100060
Stefanutti, C., Morozzi, C., Di Giacomo, S., Sovrano, B., Mesce, D., and Grossi, A. (2016). Management of homozygous familial hypercholesterolemia in real-world clinical practice: A report of 7 Italian patients treated in rome with lomitapide and lipoprotein apheresis. J. Clin. Lipidol. 10 (4), 782–789. doi:10.1016/j.jacl.2016.02.009
Tamam, L., Karataş, G., and Güleç, H. (2013). Barratt dürtüsellik ölçeği kısa formu (BIS-11-KF) türkçe uyarlama çalışması. npa. 50 (2), 130–134. doi:10.4274/npa.y6296
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466 (7307), 707–713. doi:10.1038/nature09270
Tokgozoglu, L., and Kayikcioglu, M. (2021). Familial hypercholesterolemia: Global burden and approaches. Curr. Cardiol. Rep. 23 (10), 151. doi:10.1007/s11886-021-01565-5
Tuncel, K., Kayikcioglu, M., Pirildar, S., Yilmaz, M., Kaynar, L., and Aktan, M. (2020). Mental status and physical activity in patients with homozygous familial hypercholesterolemia: A subgroup analysis of a nationwide survey (A-HIT1 registry). J. Clin. Lipidol. 14 (3), 361–370. doi:10.1016/j.jacl.2020.04.006
Underberg, J. A., Cannon, C. P., Larrey, D., Makris, L., Blom, D., and Phillips, H. (2020). Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: Five-year data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER). J. Clin. Lipidol. 14 (6), 807–817. doi:10.1016/j.jacl.2020.08.006
Vallejo-Vaz, A. J., Stevens, C. A., Lyons, A. R., Dharmayat, K. I., Freiberger, T., Hovingh, G. K., et al. (2021). Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS familial hypercholesterolaemia studies collaboration (FHSC). Lancet 398 (10312), 1713–1725. doi:10.1016/S0140-6736(21)01122-3
Zhang, Y., Jin, J. L., Cao, Y. X., Zhang, H. W., Guo, Y. L., Wu, N. Q., et al. (2020). Lipoprotein (a) predicts recurrent worse outcomes in type 2 diabetes mellitus patients with prior cardiovascular events: A prospective, observational cohort study. Cardiovasc. Diabetol. 19 (1), 111. doi:10.1186/s12933-020-01083-8
Keywords: MTTP, lomitapide, homozygous familial hypercholesterolemia, lipoprotein apheresis, low-density lipoprotein receptor, genetics
Citation: Kayikcioglu M, Ozkan HS, Yagmur B, Bayraktaroglu S and Vardarli AT (2023) Case report: Therapy adherence, MTTP variants, and course of atheroma in two patients with HoFH on low-dose, long-term lomitapide therapy. Front. Genet. 13:1087089. doi: 10.3389/fgene.2022.1087089
Received: 01 November 2022; Accepted: 21 November 2022;
Published: 04 January 2023.
Edited by:
Alberico Luigi Catapano, University of Milan, ItalyReviewed by:
Marat V. Ezhov, Ministry of Health of the Russian Federation, RussiaLin Zhu, Vanderbilt University Medical Center, United States
Copyright © 2023 Kayikcioglu, Ozkan, Yagmur, Bayraktaroglu and Vardarli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meral Kayikcioglu, bWVyYWwua2F5aWtjaW9nbHVAZ21haWwuY29t