
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 30 November 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1073880
This article is part of the Research Topic Genome-Wide Association Studies of COVID-19 Among Diverse Human Populations View all 12 articles
 Ping Li1†
Ping Li1† Yuehua Ke2†
Yuehua Ke2† Wenlong Shen1†
Wenlong Shen1† Shu Shi1
Shu Shi1 Yahao Wang1Kailin Lin3Xinjie Guo1
Yahao Wang1Kailin Lin3Xinjie Guo1 Changjun Wang2*
Changjun Wang2* Yan Zhang1*
Yan Zhang1* Zhihu Zhao1*
Zhihu Zhao1*The COVID-19 pandemic has resulted in great morbidity and mortality worldwide and human genetic factors have been implicated in the susceptibility and severity of COVID-19. However, few replicate researches have been performed, and studies on associated genes mainly focused on genic regions while regulatory regions were a lack of in-depth dissection. Here, based on previously reported associated variants and genes, we designed a capture panel covering 1,238 candidate variants and 25 regulatory regions of 19 candidate genes and targeted-sequenced 96 mild and 145 severe COVID-19 patients. Genetic association analysis was conducted between mild and severe COVID-19 patients, between all COVID-19 patients and general population, or between severe COVID-19 patients and general population. A total of 49 variants were confirmed to be associated with susceptibility or severity of COVID-19 (p < 0.05), corresponding to 18 independent loci. Specifically, rs1799964 in the promoter of inflammation-related gene TNF, rs9975538 in the intron of interferon receptor gene IFNAR2, rs429358 in the exon of APOE, rs1886814 in the intron of FOXP4-AS1 and a list of variants in the widely reported 3p21.31 and ABO gene were confirmed. It is worth noting that, for the confirmed variants, the phenotypes of the cases and controls were highly consistent between our study and previous reports, and the confirmed variants identified between mild and severe patients were quite different from those identified between patients and general population, suggesting the genetic basis of susceptibility and severity of SARS-CoV-2 infection might be quite different. Moreover, we newly identified 67 significant associated variants in the 12 regulatory regions of 11 candidate genes (p < 0.05). Further annotation by RegulomeDB database and GTEx eQTL data filtered out two variants (rs11246060 and rs28655829) in the enhancer of broad-spectrum antiviral gene IFITM3 that might affect disease severity by regulating the gene expression. Collectively, we confirmed a list of previously reported variants and identified novel regulatory variants associated with susceptibility and severity of COVID-19, which might provide biological and clinical insights into COVID-19 pathogenesis and treatment.
Coronavirus disease 2019 (COVID-19), an infectious disease caused by Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2) (Zhu et al., 2020), has spread worldwide, resulting more than 460 million infections and six million deaths up to 16 March 2022 (https://covid19.who.int/). The occurrence and clinical outcomes of COVID-19 have been revealed great heterogeneity, ranging from insensitive, asymptomatic, mild, moderate to severe, critical or even death (Wu and McGoogan, 2020). Host factors such as age, gender, comorbidities were reported to be associated with this heterogeneity (Zhou et al., 2020a; Richardson et al., 2020).
Host genetic variants might also affect susceptibility and severity of coronavirus infection, as indicated by previous studies of SARS, Middle East Respiratory Syndrome (MERS) and emerging studies of COVID-19 (Di Maria et al., 2020). The first genome-wide association study (GWAS) of COVID-19 reported two severity-associated loci in Italians and Spanish: the 3p21.31 locus containing several immune genes and ABO locus determining ABO blood groups (Ellinghaus et al., 2020). The COVID-19 Host Genetics Initiative (HGI) was established to bring together global effort to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic (Initiative, 2020). However, current reported genetic studies of COVID-19 are mainly based on European populations. Whether these findings could apply to other populations was unknown.
Besides GWAS studies, several candidate gene studies indicated that certain variants in the type I interferon (IFN) pathway genes and SARS-CoV-2 receptor/coreceptor genes were associated with susceptibility and severity of COVID-19 (Zhang et al., 2020a; Benetti et al., 2020; Zhang et al., 2020b; Kuo et al., 2020; Latini et al., 2020; Novelli et al., 2020). For example, rs12252, variant of IFITM3, was reported to be associated with severe COVID-19 (Zhang et al., 2020b). Many rare loss-of-function variants in IFN-pathway genes such as TLR3, IRF3, IRF7, IFNAR1, and IFNAR2 were identified to be associated with severe COVID-19 through impairing IFN immunity (Zhang et al., 2020a). Furthermore, in the ACE2 and TMPRSS2 genes, receptor and coreceptor gene for SARS-CoV-2 respectively, certain variants showed significantly different allele frequencies between COVID-19 patients and general population (Benetti et al., 2020; Latini et al., 2020; Novelli et al., 2020). However, these studies only focus on the genic region. The regulatory regions of these important genes are a lack of attention. Variants in regulatory regions, especially enhancers, could disrupt regulatory function, affect gene expression and thus contribute to susceptibility and severity of virus infection (Li et al., 2017; Downes et al., 2021). Therefore, it is necessary to pay more attention to variants in enhancers of important genes.
Here, we incorporated results of previous studies and designed a capture panel covering previously reported associated variants and regulatory regions of key genes. Using this panel, we targeted sequenced 96 mild and 145 severe COVID-19 patients. Genetic association analysis was conducted between mild and severe cases as well as ancestry-matched populations from 1000 Genomes Project. A list of previously reported associated variants was confirmed and two variants in the enhancers of IFITM3 genes that might affect disease severity by regulating the gene expression were newly identified, which will lead to a better understanding of the host genetic factors at play in COVID-19.
This study included 241 hospitalized COVID-19 patients recruited from Huoshenshan hospital at Wuhan city, Hubei province, China between 11 January 2020 and 11 March 2020. COVID-19 was diagnosed based on chest computed tomography (CT) manifestations and/or reverse transcription-polymerase chain reaction (RT-PCR) following the criteria of the New Coronavirus Pneumonia Prevention and Control Program (5th edition). In this study, the mild COVID-19 patients were those with no obvious clinical symptoms or with fever, respiratory symptoms, and radiological evidence of pneumonia. The severe COVID-19 patients were those having at least one of the following conditions: respiratory distress, respiratory rate ≤30 beats/minute; mean oxygen saturation ≤93% in a resting state; arterial blood oxygen partial pressure/oxygen concentration ≤300 mm·Hg; respiratory failure and requiring mechanical ventilation; shock; and admission to intensive care unit (ICU) with other organ function failure. Classifications of COVID-19 severity were taken as the worst classification during the patient’s hospital stay.
The clinical characteristics of the patients were extracted from the electronic medical records. We collected three broad classes of characteristics: 1) demographic variables (age, sex, and ethnicity); 2) symptoms (fever and diarrhea); and 3) comorbid conditions (hypertension, diabetes, cardiac disease, chronic bronchitis, chronic liver disease, chronic obstructive pulmonary disease, cerebrovascular disease, and cancer).
The Ethics Committee of Huoshenshan Hospital approved the study (HSSLL036). Given the urgency of the COVID-19 pandemic, the need for informed consent was waived by the ethnics boards of the hospital.
We collected 30 unique variants associated with susceptibility or severity of SARS-CoV/SARS-CoV-2 infection from 21 papers (up to 29 November 2020), referred as “literature dataset”. In addition, we downloaded COVID19-hg GWAS meta-analysis results (release 4) produced by COVID-19 host genetics initiative (HGI) from https://www.covid19hg.org/results/r4/, and 1420 unique variants were selected that meet one of the following four requirements: 1) variants with p < 1E-5 in “A1_ALL” group, that is, phenotype of very severe respiratory confirmed covid vs. not hospitalized covid; 2) variants with p < 1E-5 in “B1_ALL” group, that is, phenotype of hospitalized covid vs. not hospitalized covid; 3) variants with p < 5E-7 in any one of group; 4) variants with p < 1E-5 in any three of groups except “A1_ALL” and “B1_ALL”. This was referred as “HGI dataset”. Two variants were overlapped in the two datasets, resulting in a total of 1448 unique variants collected.
IFNs play a central role in innate immunity against virus infection (Zhang et al., 2020a; Bastard et al., 2020) and cell receptors for virus are key determinant for viral entry (Hoffmann et al., 2020a; Shang et al., 2020). Therefore, We collected 19 IFN-pathway genes previously implicated in SARS-CoV/SARS-CoV-2 susceptibility and severity (Hamano et al., 2005; He et al., 2006; Ching et al., 2010; Zhang et al., 2020a; Zhang et al., 2020b) as well as 3 human cell receptors/co-receptors for SARS-CoV-2 (Hoffmann et al., 2020a; Hoffmann et al., 2020b; Zhou et al., 2020b; Shang et al., 2020). A total of 25 potential enhancers for these genes were obtained from GeneHancer database (GH score > 1, Gene Association > 100) (Fishilevich et al., 2017).
RNA probes were designed to cover these variants and gene enhancers. All the 25 enhancers and 1238 unique candidate variants were covered, including 29 variants in the literature dataset and 1211 variants in the HGI dataset (Supplementary Tables S1–S3).
Peripheral whole blood samples were collected from all participants. Genomic DNAs were extracted from 1 ml of peripheral whole blood, according to the manufacturer’s instructions (QIAamp DNA blood kits). The quality of the isolated genomic DNA was verified by the following two methods: 1) the DNA degradation and contamination were monitored in 1% agarose gels; and 2) the DNA concentration was measured using a Qubit DNA Assay Kit and a Qubit 3.0 Fluorometer (Life Technologies).
The targeted capture sequencing was conducted by iGeneTech Bioscience Corporation (Beijing, China). Briefly, Human genomic DNA was sheared to 150–200 bp by Bioruptor Pico (Diagenode). Then end repair, dA-tailing, and adapter ligation were performed. The ligation product was cleaned up and size-selected by using Beckman Ampure XP Beads (Beckman). The purified ligated product was amplified by using PCR. Then the library was in solution hybrid with biotinylated RNA probes, captured with Dynabeads MyOne Streptavidin T1 (Invitrogen), and amplified with PCR. The library was quantified by Qubit and fragment-size measured by Agilent 2100 Bioanalyzer system before high-throughput sequenced by NovaSeq.
Raw reads were firstly quality trimmed with Trimmomatic (Bolger et al., 2014). Clean reads were then aligned to the human reference genome (hg38) using BWA algorithm (Li and Durbin, 2009). PCR duplicates were removed using samtools (Li et al., 2009), and GATK software (McKenna et al., 2010) was used to call SNPs and indels. The detected variants were finally saved as VCF files. Data of autosomal biallelic variants for Han Chinese of 1000 Genomes Project (Auton et al., 2015) were downloaded from https://www.internationalgenome.org/. Genetic association analysis was conducted using PLINK 1.9 software (Chang et al., 2015).
p-values comparing demographics severe and mild disease groups were calculated by means of χ2 or Fisher exact test as appropriate, except for p-value for age which was calculated using student’s t test. Minor allele frequency (MAF) of variants was compared between case and control groups using Fisher exact test. Logistic regression analysis was also used to compute the contributing variants to severity with adjusting age, sex and comorbidities. Statistic power was calculated using G*Power 3 software (Faul et al., 2007). p < 0.05 was considered statistically significant.
A total of 241 COVID-19 patients were recruited, with 96 mild cases and 145 severe cases. Demographic and phenotypic data are shown in Table 1. Comparing mild patients with severe ones, the median age increased from 58 to 67 years (p < 0.0001). In addition, we observed a greater percentage of severe patients with comorbidities than mild patients (p = 0.03), specifically with diabetes (p = 0.04) and cerebrovascular disease (p = 0.03). This is in accordance with previous report that older COVID-19 patients and those with comorbidities were more likely to be severe in disease (Zhou et al., 2020a; Richardson et al., 2020). Previous report also indicated that severe patients had a greater percentage of male cases (Richardson et al., 2020). However, no obvious sex difference was found between mild and severe patients in our data (p = 0.17).
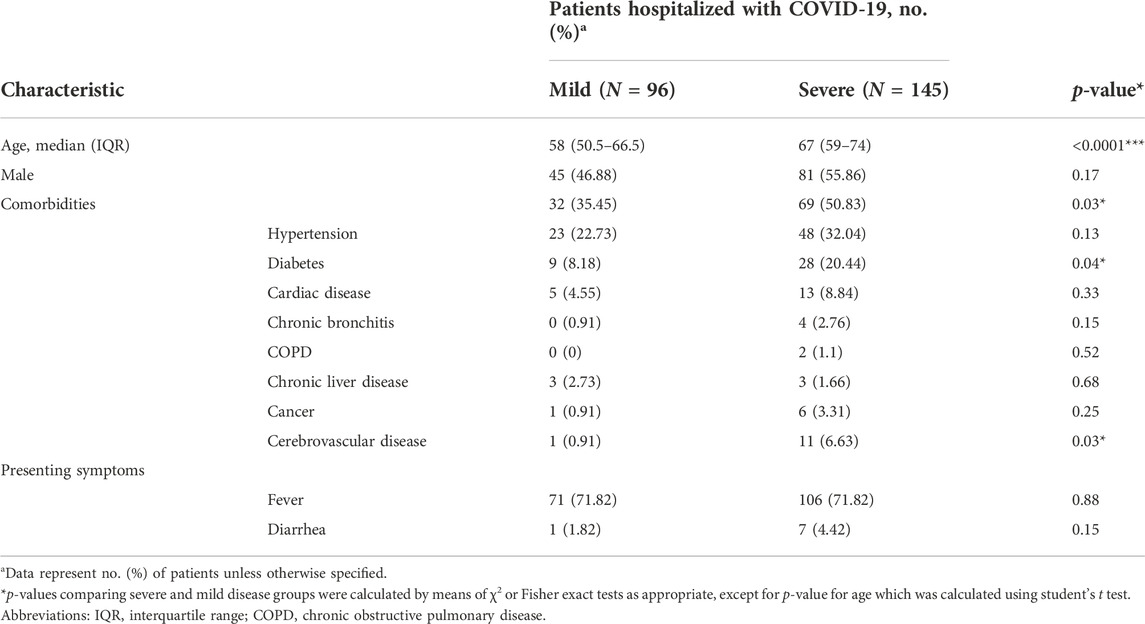
TABLE 1. Patient demographics by clinical phenotype.
The experimental design was illustrated in Figure 1. To validate previously reported variants associated with COVID-19 susceptibility and severity, we collected 30 unique variants from 21 papers (referred as “literature dataset”) and selected 1420 unique variants from COVID-19 host genetics initiative (HGI) release 4 (Initiative, 2020) (referred as “HGI dataset”). Additionally, to identify regulatory variants associated with COVID-19 susceptibility and severity in the enhancers of previously reported associated genes, we collected 19 IFN-pathway genes previously implicated in coronavirus susceptibility and severity (Hamano et al., 2005; He et al., 2006; Ching et al., 2010; Zhang et al., 2020a; Zhang et al., 2020b) as well as 3 human cell receptors/co-receptors for SARS-CoV-2 (Hoffmann et al., 2020a; Hoffmann et al., 2020b; Zhou et al., 2020b; Shang et al., 2020) and obtained 25 potential enhancers for these genes from GeneHancer database (Fishilevich et al., 2017) (referred as “enhancer dataset”).
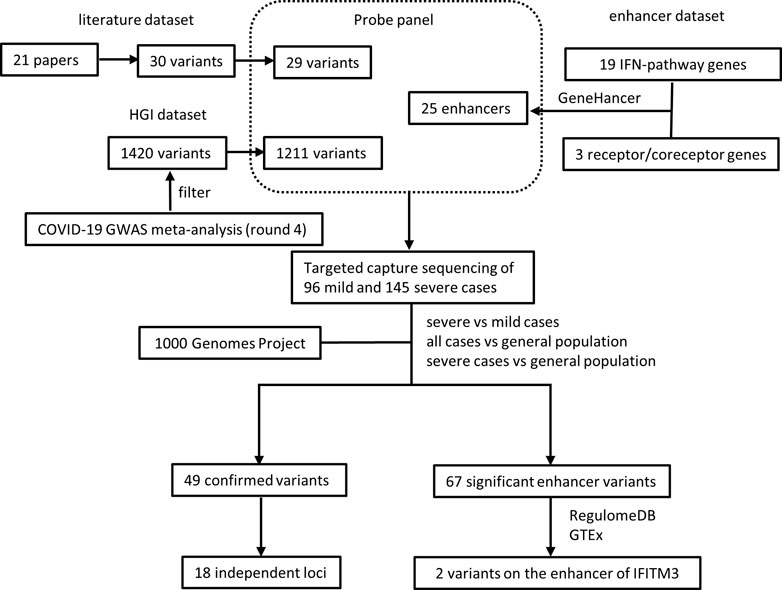
FIGURE 1. Illustration of experimental design and analysis.
A panel of RNA probes was designed to capture these variant and enhancers, resulting in 1238 unique variants and all the 25 potential enhancers covered (Supplementary Tables S1–S3). The 1238 unique variants included 29 variants in the literature dataset and 1211 variants in the HGI dataset, with two variants overlapped between the two datasets. Variants that fail to design probes might be due to GC content, repetitive sequence, dimer or secondary structure of probes.
The probe panel was used to targeted capture sequence the 241 COVID-19 patients. A median of 8.5 million raw reads were obtained for each sample. After filtering, a median of 8.3 million reads was kept as clean reads, with a median of average insert size of 173 bp. The reads were mapped to hg38 genome. The median mapping rate was 99.56%, with a median duplication rate of 27.47% (Supplementary Figure S1). Specifically, the median number of target mapped reads was 4.5 million, with a median target coverage rate of 99.57% and median target mean depth of 1472× (Supplementary Figure S2).
Out of the 1238 candidate variants in the panel, a total of 1006 variants in the panel were identified in the 241 COVID-19 patients, including 26 variants in the literature dataset and 982 variants in the HGI dataset.
Comparing 96 mild and 145 severe COVID-19 patients, seven variants were confirmed to be significantly different in minor allele frequency (MAF) (p < 0.05, Supplementary Table S2), corresponding to four independent loci. The four lead variants were shown in Table 2. Specifically, rs1799964 in the promoter of inflammation-related gene TNF was found to be associated with COVID-19 severity. CC genotype of this variant has been reported to be associated with femoral head necrosis after SARS-CoV infection (Wang et al., 2008). In addition, rs2224986 and rs13062942 were still associated with COVID-19 severity after adjusting age, sex and comorbidities on regression analysis. Both variants had a significant difference between hospitalized and non-hospitalized patients in the HGI dataset.
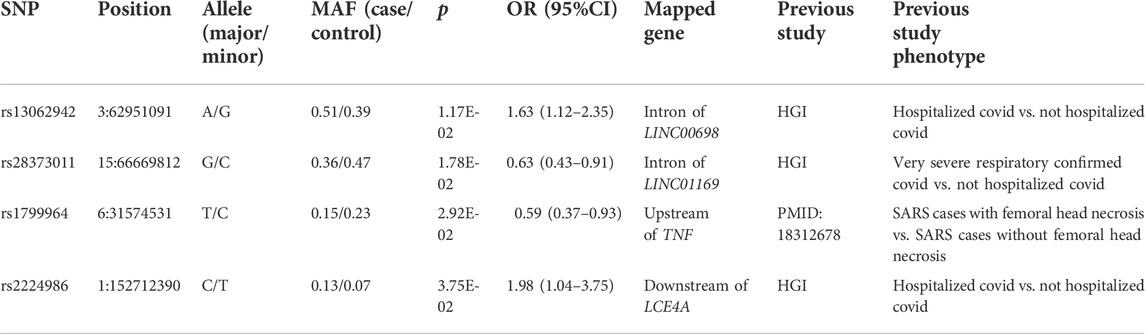
TABLE 2. Independent lead variants that were confirmed between mild and severe cases.
When comparing all COVID-19 patients with ancestry-matched general population from 1000 Genomes Project, we identified 39 significant variants (p < 0.05, Supplementary Table S5), corresponding 10 independent loci. The independent lead variants were shown in Table 3. Specifically, multiple genetic variants in the ABO gene locus and 3p21.31 region have been validated. In addition, the missense variant rs429358 in exon of APOE gene, which has been reported to be associated with COVID-19-positive in the UK Biobank data (Kuo et al., 2020), was confirmed in Chinese population. As for the confirmed variant rs1886814 in the intron of FOXP4-AS1 (forkhead box P4 antisense RNA 1), recent trans-ethnic genome-wide association study of severe COVID-19 that incorporated Chinese population and HGI results also revealed a significant variant nearby, rs1853837 (Wu et al., 2021), which is LD with rs1886814 (1000 Genomes Project CHB, r2 = 0.68).
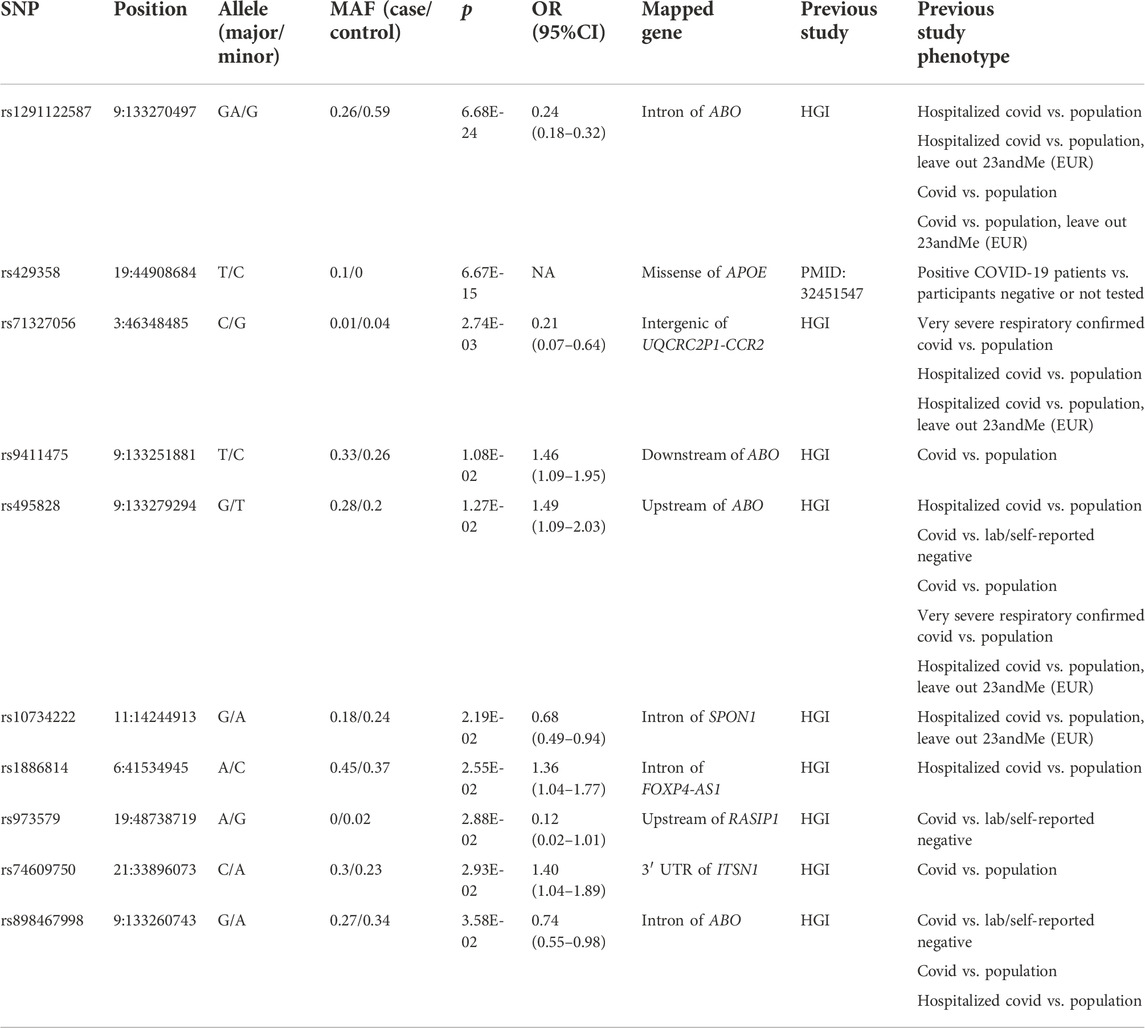
TABLE 3. Independent lead variants that were confirmed between all COVID-19 patients and general population.
When comparing severe COVID-19 patients with ancestry-matched general population from 1000 Genomes Project, we identified 23 significant variants (p < 0.05, Supplementary Table S6), corresponding 7 independent loci. The independent lead variants were shown in Table 4. Again, multiple genetic variants in the ABO gene locus and 3p21.31 region, rs429358 in exon of APOE gene and rs1886814 in the intron of FOXP4-AS1 have been validated. What’s more, rs9975538 in the intron of gene IFNAR2 was also confirmed. IFNAR2 gene, along with IFNAR1 encodes type I interferon receptor, thus the rs9975538 might affect type I interferon pathway.
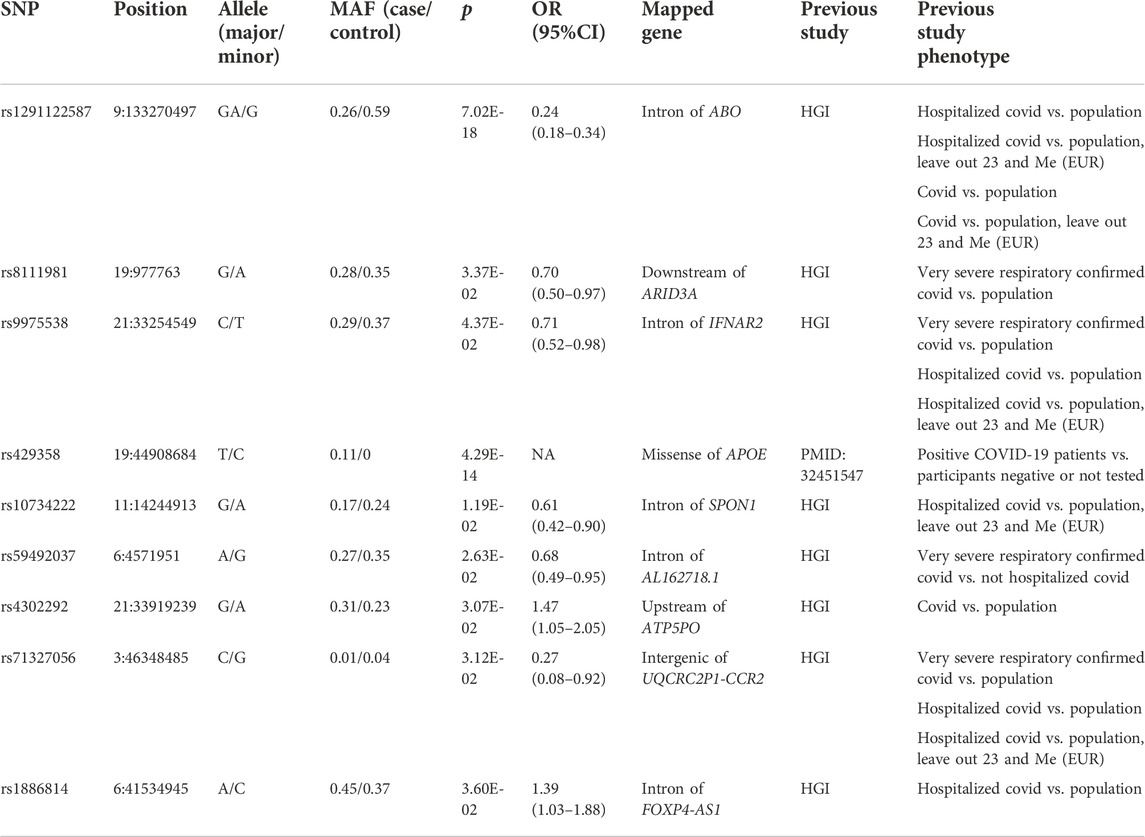
TABLE 4. Independent lead variants that were confirmed between severe COVID-19 patients and general population.
In total, 49 unique variants were confirmed to be associated with susceptibility or severity of COVID-19 (p < 0.05), corresponding 18 independent loci. It is worth noting that, for the confirmed variants, the phenotypes of the cases and controls were highly consistent between our study and previous reports, and the confirmed variants identified between mild and severe patients were quite different from those identified between patients and general population, suggesting the genetic basis of susceptibility and severity of SARS-CoV-2 infection might be quite different.
Previous candidate gene study of genetic association with COVID-19 mainly focused on the exon region of important genes (Zhang et al., 2020a; Novelli et al., 2020). However, regulatory regions, particularly enhancers, play a vital role in gene expression and may affect disease susceptibility and severity when misfunction (Claringbould and Zaugg, 2021). To investigate the role of enhancer variants of key genes in COVID-19, we also included 25 enhancers of 16 IFN-pathway genes and 3 SARS-CoV-2 receptor/co-receptor genes in the probe panel. These enhancers were predicted by GeneHancer database. Genetic association analysis was conducted between mild and severe COVID-19 patients, between all COVID-19 patients and ancestry-matched general population from 1000 Genomes Project, or between severe COVID-19 patients and ancestry-matched general population from 1000 Genomes Project.
In total, we identified 67 variants in the enhancer region of the panel that were associated with susceptibility or severity of COVID-19, relating to 12 enhancers of 11 genes (p < 0.05, Supplementary Tables S7–S9). Further annotation by RegulomeDB database filtered out five potential regulatory variants for broad-spectrum antiviral gene IFITM3 and one for SARS-CoV-2 co-receptor gene TMPRSS2 (probability score > 0.8, Table 5). Specifically, among the six variants, GTEx revealed two variants affecting the expression of IFITM3, that is, T allele of rs11246060 in the enhancer of IFITM3, which protected COVID-19 patients from severe outcomes when comparing mild and severe patients (p = 2.38E-2, OR = 0.39, 95% CI = 0.17–0.89), was associated with increased expression of IFITM3 in whole blood (p = 1.72E-10), while T allele of rs28655829 in the enhancer of IFITM3, which increased the risk of severity when comparing mild and severe patients (p = 2.38E-2, OR = 0.39, 95% CI = 0.17–0.89), was associated with decreased expression of IFITM3 in cultured fibroblasts (p = 7.55E-8). This indicated that these variants might confer genetic risk or protection by affecting the gene expression and highlighted the importance of IFITM3 gene in the defense of SARS-CoV-2 infection.
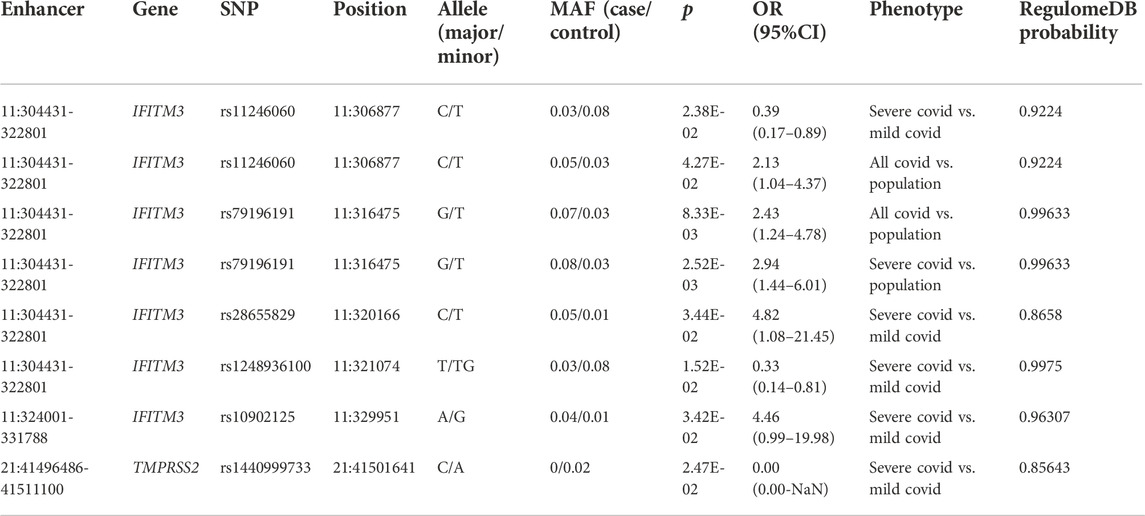
TABLE 5. Significant enhancer variants that were associated with susceptibility and severity of COVID-19 and predicted regulatory by RegulomeDB.
In this study, out of 1238 variant previously reported to be association with susceptibility and severity of COVID-19, we confirmed 49 variants, corresponding to 18 independent loci, including 3p21.31 locus, ABO, IFNAR2, TNF, APOE, and FOXP4-AS1 gene.
3p21.31 locus has been identified as a risk factor by GWAS studies of Italian and Spanish (Ellinghaus et al., 2020), British (Wang et al., 2008), Americans (Shelton et al., 2021), and meta-GWAS study of HGI (Initiative, 2021). However, most participants of the studies are Europeans and the major genetic risk factor in 3p21.31 for severe COVID-19 is proposed to be inherited from Neanderthals (Zeberg and Pääbo, 2020), which is almost absent in East Asians. In accordance, previous COVID-19 GWAS study of Chinese population was unable to replicate the locus (Wang et al., 2020; Wu et al., 2021). In our study, consistent with the low frequency of the previously reported lead variant rs11385942 in 3p21.31, only one individual was identified to harbor this variant. This individual had severe COVID-19, which might be due to the risk variant rs11385942. On the other hand, we confirmed another variant in 3p21.31, rs71327056, which is an intergenic variant between UQCRC2P1 and CCR2 genes and is not linkage disequilibrium (LD) with rs11385942 (Ellinghaus et al., 2020) (1000 Genomes Project CEU, r2 = 0.19), suggesting there might be more than one independent variant in 3p21.31 contribution to COVID-19 susceptibility and severity. Notably, the minor allele G of rs71317056 was found to increase severity in HGI release4 where most of individuals were Europeans while our study revealed that the minor allele of rs71317056 effected oppositely in Chinese population (Tables 3, 4). We speculated this might due to the interaction with another risk haplotype which Europeans inherited from Neanderthals but was absent in Asians. In addition, rs71317056 is in LD with rs35943069 (1000 Genomes Project CEU, r2 = 1; CHB, r2 = 1), which resides in a potentially enhancer region that is annotated by GeneHancer (Fishilevich et al., 2017). The minor allele is associated with increased CCR1 gene expression in cultured fibroblasts (GTEx V8, Supplementary Table S10) (Consortium, 2020), suggesting that it might function through expression regulation of CCR1, receptor for a C-C type chemokine which play an important role in immune system against viral infection (Zlotnik and Yoshie, 2012).
The ABO locus has also been revealed to be associated with COVID-19 severity (Ellinghaus et al., 2020). However, the previously reported lead variant rs657152 had no significant difference in allele frequency between cases and controls of our study. Instead, we confirmed several other variants in the ABO locus when comparing all COVID-19 cases with general Chinese population. Specifically, one of the lead variants, rs34357864 was also been confirmed when comparing severe COVID-19 cases with general Chinese population. Similar to our study, HGI data revealed that this variant was significant when either all COVID-19 patients or hospitalized COVID-19 patients compared with general population, suggesting this is a variant associated with susceptibility.
The T allele of rs9975538 in the intron of gene IFNAR2 had a lower frequency in severe COVID-19 patients compared with general Chinese population, consistent with results of HGI release 4 comparing either very severe respiratory confirmed COVID-19 patients or hospitalized COVID-19 patients with general population. rs9975538 was also in LD with rs2236757 (1000 Genomes Project CEU, r2 = 0.75; CHB, r2 = 1), the previously reported variant that was associated with critical illness of COVID-19 (Pairo-Castineira et al., 2021). Notably, the T allele of rs9975538 increased the expression of IFNAR2 and IL10RB in lung (GTEx V8, p = 1.09E-5, p = 1.99E-5 respectively). As IFNAR2 and IL10RB are type I and III IFN receptor respectively, this variant might confer protection by increasing IFN receptor expression and thus upregulating the antiviral activity of IFN pathway. In addition, IFNAR2 play a vital role in multiple sclerosis, a chronic autoimmune disorder characterized by inflammation of the central nervous system, demyelination and axonal damage (Gilli et al., 2008; Órpez-Zafra et al., 2017). This leads to a hypothesis that COVID-19 patients harboring IFNAR2 rs9975538 variant might be more likely to develop neurological disorders (Douaud et al., 2022; Lee et al., 2022), possibly though neuroinflammatory pathways.
Rs1799964, which is located upstream of TNF gene, was found to be associated with disease severity, with C allele being associated with mild phenotype. TNF is multifunctional proinflammatory cytokine and plays a key role in regulating the immunological response to infections (Waters et al., 2013). The C allele of rs1799964 was associated with increased expression of TNF than T allele (Nourian et al., 2017) and increased lymphocyte counts (Chen et al., 2020), which might protect the body from severe disease.
APOE gene polymorphisms have been reported to be associated with susceptibility or severity of COVID-19 in British, Czech, Spanish, Finnish and Kurdish population (Kuo et al., 2020; Al-Jaf et al., 2021; Del Ser et al., 2021; Hubacek et al., 2021; Kurki et al., 2021). Consistent with above findings, we revealed an association of APOE variant rs429358 with susceptibility to COVID-19 in Chinese population. Given that APOE is associated with Alzheimer’s and cardiovascular diseases and type 2 diabetes (Liu et al., 2013; Mahley, 2016; Liu et al., 2019), comorbidities that are related to COVID-19 susceptibility and severity, the effect of the APOE variant on COVID-19 could be indirect. Meanwhile, recent researches indicated that APOE might also affect SARS-CoV-2 infection directly by interacting with ACE2 inhibiting SARS-CoV-2 cellular entry (Zhang et al., 2022), regulating cellular cholesterol homeostasis (Gao et al., 2022) and modulating antiviral immunity (Ostendorf et al., 2022). Notably, another variant that determines APOE isoforms, rs7412, did not pass the significant threshold, probably because this variant mainly contributes to APOE ε2 isoform while COVID-19 is more associated with APOE ε4 isoform (Kuo et al., 2020; Al-Jaf et al., 2021; Del Ser et al., 2021; Hubacek et al., 2021; Kurki et al., 2021).
Rs1886814 in the intron of the lncRNA FOXP4-AS1 was found to be associated with disease susceptibility when comparing all COVID-19 patients or severe COVID-19 patients with general Chinese population. In HGI release 4 datasets, it is associated with COVID-19 hospitalization when comparing with general population. Recent trans-ethnic genome-wide association study of severe COVID-19 that incorporated Chinese population and HGI results revealed another significant variant in the intron of FOXP4-AS1, rs1853837, which is LD with rs1886814 in Chinese population (1000 Genomes Project CHB, r2 = 0.68) (Wu et al., 2021). The risk allele C of rs1886814 is an eQTL in positive association with the expression of FOXP4 in lung (GETx V8, p = 3.28E-6) (Consortium, 2020). FOXP4 is a transcription factor expressed in both thymocytes and peripheral CD4+ and CD8+ T cells, and is necessary for normal T cell cytokine recall responses to antigen following pathogenic infection (Wiehagen et al., 2012).
We noted that the overall confirmation rate was not high, possibly due to different population structure and limited sample size of our study. The statistic power was provided in Supplementary Figure S3. On the other hand, it is worth noting that for the validated variants, the phenotypes of cases and controls were highly coordinated in our study and original study. All variants validated in our mild and severe group were specific to be identified in previous association study of severity, that is, when comparing hospitalized or very severe respiratory confirmed COVID-19 patients with not hospitalized ones. Nearly all variants validated in the COVID-19 patients and general Chinese population group were specific to be identified in previous association study of susceptibility, that is, comparing COVID-19 patients with general population. The remarkable specificity suggested that susceptibility and severity might have different genetic basis and also indicated the accuracy of our study.
In addition, we also identified 67 variants in the 12 regulatory regions of 11 candidate genes associated with susceptibility or severity of COVID-19, which have not been reported before. Further annotation by RegulomeDB and GTEx database revealed two variants affected the expression of IFITM3 and conferred genetic risk and protection respectively. IFITM3 gene encodes a transmembrane protein that could be induced by interferons and function as a broad-spectrum antiviral effector molecule by directly limiting cellular entry of a number of pathogenic viruses, including influenza A virus, West Nile virus, dengue virus, SARS-CoV and SARS-CoV-2 (Diamond and Farzan, 2013; Shi et al., 2021). Moreover, rs12252 variant in the gene has been found to be associated with COVID-19 severity (Zhang et al., 2020b; Alghamdi et al., 2021; Gómez et al., 2021). Our results indicated that, in addition to genetic variants, enhancer variants of IFITM3 might confer genetic risk or protection by affecting gene expression as well. Though larger cohort studies are needed to confirm these genetic associations, our data presented here also highlighted the important role of IFITM3 in host defense against SARS-CoV-2.
In conclusion, we confirmed a list of previously reported variants associated with susceptibility and severity of COVID-19, and identified several enhancer variants potentially regulating expression of genes associated with COVID-19. Though larger cohort studies and further experiments are needed to confirm these genetic associations and explore the molecular mechanism, elucidation of host genetic factors contributing to susceptibility to severe infection will provide the opportunity for clinical risk profiling of patients with COVID-19, mechanistic understanding of the underlying pathophysiology and further identification of potential therapeutic targets.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Ethics Committee of Huoshenshan Hospital (HSSLL036). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Conceptualization: ZZ, YZ, and CW; Methodology: YZ and PL; Formal analysis: PL and WS; Resources: CW, YK; Data Curation: SS, YW, KL, and XG; Writing—original draft: PL; Writing—review and editing: PL and all; Supervision: ZZ, YZ, and CW; Project administration: YZ and YK; Funding acquisition: PL and YZ.
This work was supported by grants from the National Key Research and Development Program of China (2018YFC1200704, 2018YFA0900801) and the National Natural Science Foundation of China (31801087).
We thank all the patients participating in this study. We thank the COVID-19 Host Genetic Initiative for sharing the HGI release4 summary statistics, and the 1000 Genomes Project for sharing the human genetic variation data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1073880/full#supplementary-material
Al-Jaf, S. M. A., Niranji, S. S., Ali, H. N., and Mohammed, O. A. (2021). Association of Apolipoprotein e polymorphism with SARS-CoV-2 infection. Infect. Genet. Evol. 95, 105043. doi:10.1016/j.meegid.2021.105043
Alghamdi, J., Alaamery, M., Barhoumi, T., Rashid, M., Alajmi, H., Aljasser, N., et al. (2021). Interferon-induced transmembrane protein-3 genetic variant rs12252 is associated with COVID-19 mortality. Genomics 113 (4), 1733–1741. doi:10.1016/j.ygeno.2021.04.002
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526 (7571), 68–74. doi:10.1038/nature15393
Bastard, P., Rosen, L. B., Zhang, Q., Michailidis, E., Hoffmann, H-H., Zhang, Y., et al. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370 (6515), eabd4585. doi:10.1126/science.abd4585
Benetti, E., Tita, R., Spiga, O., Ciolfi, A., Birolo, G., Bruselles, A., et al. (2020). ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. 28 (11), 1602–1614. doi:10.1038/s41431-020-0691-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Chen, M-H., Raffield, L. M., Mousas, A., Sakaue, S., Huffman, J. E., Moscati, A., et al. (2020). Trans-ethnic and ancestry-specific blood-cell genetics in 746, 667 individuals from 5 global populations. Cell 182 (5), 1198–1213. doi:10.1016/j.cell.2020.06.045
Ching, J. C-Y., Chan, K. Y. K., Lee, E. H. L., Xu, M-S., Ting, C. K. P., So, T. M. K., et al. (2010). Significance of the myxovirus resistance A (MxA) gene -123C>a single-nucleotide polymorphism in suppressed interferon beta induction of severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 201 (12), 1899–1908. doi:10.1086/652799
Claringbould, A., and Zaugg, J. B. (2021). Enhancers in disease: Molecular basis and emerging treatment strategies. Trends Mol. Med. 27 (11), 1060–1073. doi:10.1016/j.molmed.2021.07.012
Consortium, G. T. (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369 (6509), 1318–1330. doi:10.1126/science.aaz1776
Del Ser, T., Fernández-Blázquez, M. A., Valentí, M., Zea-Sevilla, M. A., Frades, B., Alfayate, E., et al. (2021). Residence, clinical features, and genetic risk factors associated with symptoms of COVID-19 in a cohort of older people in madrid. Gerontology 67 (3), 281–289. doi:10.1159/000513182
Di Maria, E., Latini, A., Borgiani, P., and Novelli, G. (2020). Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): Rapid systematic review and field synopsis. Hum. Genomics 14 (1), 30. doi:10.1186/s40246-020-00280-6
Diamond, M. S., and Farzan, M. (2013). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13 (1), 46–57. doi:10.1038/nri3344
Douaud, G., Lee, S., Alfaro-Almagro, F., Arthofer, C., Wang, C., McCarthy, P., et al. (2022). SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 604 (7907), 697–707. doi:10.1038/s41586-022-04569-5
Downes, D. J., Cross, A. R., Hua, P., Roberts, N., Schwessinger, R., Cutler, A. J., et al. (2021). Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat. Genet. 53 (11), 1606–1615. doi:10.1038/s41588-021-00955-3
Ellinghaus, D., Degenhardt, F., Bujanda, L., Buti, M., Albillos, A., Invernizzi, P., et al. (2020). Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. 383 (16), 1522–1534. doi:10.1056/NEJMoa2020283
Faul, F., Erdfelder, E., Lang, A-G., and Buchner, A. G. (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39 (2), 175–191. doi:10.3758/bf03193146
Fishilevich, S., Nudel, R., Rappaport, N., Hadar, R., Plaschkes, I., Iny Stein, T., et al. (2017). GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Oxford): Database.
Gao, P., Ji, M., Liu, X., Chen, X., Liu, H., Li, S., et al. (2022). Apolipoprotein E mediates cell resistance to influenza virus infection. Sci. Adv. 8 (38), eabm6668. doi:10.1126/sciadv.abm6668
Gilli, F., Valentino, P., Caldano, M., Granieri, L., Capobianco, M., Malucchi, S., et al. (2008). Expression and regulation of IFNalpha/beta receptor in IFNbeta-treated patients with multiple sclerosis. Neurology 71 (24), 1940–1947. doi:10.1212/01.wnl.0000327340.50284.8d
Gómez, J., Albaiceta, G. M., Cuesta-Llavona, E., García-Clemente, M., López-Larrea, C., Amado-Rodríguez, L., et al. (2021). The Interferon-induced transmembrane protein 3 gene (IFITM3) rs12252 C variant is associated with COVID-19. Cytokine 137, 155354. doi:10.1016/j.cyto.2020.155354
Hamano, E., Hijikata, M., Itoyama, S., Quy, T., Phi, N. C., Long, H. T., et al. (2005). Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem. Biophys. Res. Commun. 329 (4), 1234–1239. doi:10.1016/j.bbrc.2005.02.101
He, J., Feng, D., de Vlas, S. J., Wang, H., Fontanet, A., Zhang, P., et al. (2006). Association of SARS susceptibility with single nucleic acid polymorphisms of OAS1 and MxA genes: A case-control study. BMC Infect. Dis. 6, 106. doi:10.1186/1471-2334-6-106
Hoffmann, M., Kleine-Weber, H., and Pöhlmann, S. (2020). A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78 (4), 779–784. doi:10.1016/j.molcel.2020.04.022
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181 (2), 271–280. doi:10.1016/j.cell.2020.02.052
Hubacek, J. A., Dlouha, L., Dusek, L., Majek, O., and Adamkova, V. (2021). Apolipoprotein E4 allele in subjects with COVID-19. Gerontology 67 (3), 320–322. doi:10.1159/000516200
Initiative, C-H. G. (2021). Mapping the human genetic architecture of COVID-19. Nature 600 (7889), 472–477.
Initiative, C-H. G. (2020). The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28 (6), 715–718. doi:10.1038/s41431-020-0636-6
Kuo, C-L., Pilling, L. C., Atkins, J. L., Masoli, J. A. H., Delgado, J., Kuchel, G. A., et al. (2020). APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 75 (11), 2231–2232. doi:10.1093/gerona/glaa131
Kurki, S. N., Kantonen, J., Kaivola, K., Hokkanen, L., Mäyränpää, M. I., Puttonen, H., et al. (2021). APOE ε4 associates with increased risk of severe COVID-19, cerebral microhaemorrhages and post-COVID mental fatigue: A Finnish biobank, autopsy and clinical study. Acta Neuropathol. Commun. 9 (1), 199. doi:10.1186/s40478-021-01302-7
Latini, A., Agolini, E., Novelli, A., Borgiani, P., Giannini, R., Gravina, P., et al. (2020). COVID-19 and genetic variants of protein involved in the SARS-CoV-2 entry into the host cells. Genes 11 (9), E1010. doi:10.3390/genes11091010
Lee, M. H., Perl, D. P., Steiner, J., Pasternack, N., Li, W., Maric, D., et al. (2022). Neurovascular injury with complement activation and inflammation in COVID-19. Brain. 145 (7), 2555–2568. doi:10.1093/brain/awac151
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, P., Shi, M-L., Shen, W-L., Zhang, Z., Xie, D-J., Zhang, X-Y., et al. (2017). Coordinated regulation of IFITM1, 2 and 3 genes by an IFN-responsive enhancer through long-range chromatin interactions. Biochim. Biophys. Acta. Gene Regul. Mech. 1860 (8), 885–893. doi:10.1016/j.bbagrm.2017.05.003
Liu, C-C., Liu, C-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9 (2), 106–118. doi:10.1038/nrneurol.2012.263
Liu, S., Liu, J., Weng, R., Gu, X., and Zhong, Z. (2019). Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 19 (1), 213. doi:10.1186/s12872-019-1194-0
Mahley, R. W. (2016). Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 94 (7), 739–746. doi:10.1007/s00109-016-1427-y
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Nourian, M., Chaleshi, V., Pishkar, L., Azimzadeh, P., Baradaran Ghavami, S., Balaii, H., et al. (2017). Evaluation of tumor necrosis factor (TNF)-α mRNA expression level and the rs1799964 polymorphism of the TNF-α gene in peripheral mononuclear cells of patients with inflammatory bowel diseases. Biomed. Rep. 6 (6), 698–702. doi:10.3892/br.2017.908
Novelli, A., Biancolella, M., Borgiani, P., Cocciadiferro, D., Colona, V. L., D'Apice, M. R., et al. (2020). Analysis of ACE2 genetic variants in 131 Italian SARS-CoV-2-positive patients. Hum. Genomics 14 (1), 29. doi:10.1186/s40246-020-00279-z
Órpez-Zafra, T., Pavía, J., Hurtado-Guerrero, I., Pinto-Medel, M. J., Rodriguez Bada, J. L., Urbaneja, P., et al. (2017). Decreased soluble IFN-β receptor (sIFNAR2) in multiple sclerosis patients: A potential serum diagnostic biomarker. Mult. Scler. 23 (7), 937–945. doi:10.1177/1352458516667564
Ostendorf, B. N., Patel, M. A., Bilanovic, J., Hoffmann, H. H., Carrasco, S. E., Rice, C. M., et al. (2022). Common human genetic variants of APOE impact murine COVID-19 mortality. Nature 611, 346–351. doi:10.1038/s41586-022-05344-2
Pairo-Castineira, E., Clohisey, S., Klaric, L., Bretherick, A. D., Rawlik, K., Pasko, D., et al. (2021). Genetic mechanisms of critical illness in COVID-19. Nature 591 (7848), 92–98. doi:10.1038/s41586-020-03065-y
Richardson, S., Hirsch, J. S., Narasimhan, M., Crawford, J. M., McGinn, T., Davidson, K. W., et al. (2020). Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York city area. JAMA 323 (20), 2052–2059. doi:10.1001/jama.2020.6775
Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., Auerbach, A., et al. (2020). Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 117 (21), 11727–11734. doi:10.1073/pnas.2003138117
Shelton, J. F., Shastri, A. J., Ye, C., Weldon, C. H., Filshtein-Sonmez, T., Coker, D., et al. (2021). Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 53 (6), 801–808. doi:10.1038/s41588-021-00854-7
Shi, G., Kenney, A. D., Kudryashova, E., Zani, A., Zhang, L., Lai, K. K., et al. (2021). Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 40 (3), e106501. doi:10.15252/embj.2020106501
Wang, F., Huang, S., Gao, R., Zhou, Y., Lai, C., Li, Z., et al. (2020). Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 6 (1), 83. doi:10.1038/s41421-020-00231-4
Wang, S., Wei, M., Han, Y., Zhang, K., He, L., Yang, Z., et al. (2008). Roles of TNF-alpha gene polymorphisms in the occurrence and progress of SARS-cov infection: A case-control study. BMC Infect. Dis. 8, 27. doi:10.1186/1471-2334-8-27
Waters, J. P., Pober, J. S., and Bradley, J. R. (2013). Tumour necrosis factor in infectious disease. J. Pathol. 230 (2), 132–147. doi:10.1002/path.4187
Wiehagen, K. R., Corbo-Rodgers, E., Li, S., Staub, E. S., Hunter, C. A., Morrisey, E. E., et al. (2012). Foxp4 is dispensable for T cell development, but required for robust recall responses. PloS one 7 (8), e42273. doi:10.1371/journal.pone.0042273
Wu, P., Ding, L., Li, X., Liu, S., Cheng, F., He, Q., et al. (2021). Trans-ethnic genome-wide association study of severe COVID-19. Commun. Biol. 4 (1), 1034. doi:10.1038/s42003-021-02549-5
Wu, Z., and McGoogan, J. M. (2020). Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA 323 (13), 1239–1242. doi:10.1001/jama.2020.2648
Zeberg, H., and Pääbo, S. (2020). The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587 (7835), 610–612. doi:10.1038/s41586-020-2818-3
Zhang, H., Shao, L., Lin, Z., Long, Q-X., Yuan, H., Cai, L., et al. (2022). APOE interacts with ACE2 inhibiting SARS-CoV-2 cellular entry and inflammation in COVID-19 patients. Signal Transduct. Target. Ther. 7 (1), 261. doi:10.1038/s41392-022-01118-4
Zhang, Q., Bastard, P., Liu, Z., Le Pen, J., Moncada-Velez, M., Chen, J., et al. (2020)., 370. New York, NY), eabd4570. doi:10.1126/science.abd4570Inborn errors of type I IFN immunity in patients with life-threatening COVID-19Science6515
Zhang, Y., Qin, L., Zhao, Y., Zhang, P., Xu, B., Li, K., et al. (2020). Interferon-induced transmembrane protein 3 genetic variant rs12252-C associated with disease severity in coronavirus disease 2019. J. Infect. Dis. 222 (1), 34–37. doi:10.1093/infdis/jiaa224
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in wuhan, China: A retrospective cohort study. Lancet 395 (10229), 1054–1062. doi:10.1016/S0140-6736(20)30566-3
Zhou, P., Yang, X-L., Wang, X-G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579 (7798), 270–273. doi:10.1038/s41586-020-2012-7
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., et al. (2020). A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382 (8), 727–733. doi:10.1056/NEJMoa2001017
Keywords: COVID-19, SARS-CoV-2, targeted capture sequencing, SNP, genetic variant, enhancer, suscepibility
Citation: Li P, Ke Y, Shen W, Shi S, Wang Y, Lin K, Guo X, Wang C, Zhang Y and Zhao Z (2022) Targeted screening of genetic associations with COVID-19 susceptibility and severity. Front. Genet. 13:1073880. doi: 10.3389/fgene.2022.1073880
Received: 19 October 2022; Accepted: 18 November 2022;
Published: 30 November 2022.
Edited by:
Zhongshan Cheng, St. Jude Children’s Research Hospital, United StatesReviewed by:
Valerio Caputo, University of Rome Tor Vergata, ItalyCopyright © 2022 Li, Ke, Shen, Shi, Wang, Lin, Guo, Wang, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changjun Wang, c2NpZW5jZTIwMDhAaG90bWFpbC5jb20=; Yan Zhang, emFueTE5ODNAMTYzLmNvbQ==; Zhihu Zhao, emhhb3poQGJtaS5hYy5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.