
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 08 December 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1062715
This article is part of the Research Topic Genetics of Inborn Errors of Metabolism View all 30 articles
 Siyu Chang†
Siyu Chang† Yi Yang†
Yi Yang† Feng Xu
Feng Xu Wenjun JiXia ZhanXiaolan GaoTing Chen
Wenjun JiXia ZhanXiaolan GaoTing Chen Wenjuan Qiu
Wenjuan Qiu Huiwen Zhang
Huiwen Zhang Lili Liang
Lili Liang Deyun LuKaichuang Zhang
Deyun LuKaichuang Zhang Xuefan Gu
Xuefan Gu Lianshu Han*
Lianshu Han*Background: Primary carnitine deficiency (PCD) is an autosomal recessive disease caused by mutations in the SLC22A5 gene, which encodes the organic cation transporter 2 (OCTN2). Patients with PCD may be at risk of skeletal or cardiac myopathy, metabolic decompensation, and even sudden death. This study aimed to analyze the biochemical, clinical, and genetic characteristics of PCD patients identified by newborn screening (NBS) in Shanghai.
Methods: Dried blood spot (DBS) samples of newborns were analyzed through tandem mass spectrometry (MS/MS) from January 2003 to December 2021. Newborns with low free carnitine (C0) levels were recalled. Mutation in the SLC22A5 gene was analyzed on suspected positive newborns with low C0 levels after recall.
Results: 1,247,274 newborns were screened by MS/MS and 40 newborns were diagnosed with PCD, therefore the incidence of PCD in Shanghai was approximately 1:31,200. The mean C0 level in newborns with PCD was 5.37 ± 1.79 μmol/L before treatment and increased to 24.45 ± 10.87 μmol/L after treatment with L-carnitine. Twenty-three different variants were identified in the SLC22A5 gene, including 8 novel variants, of which c.51C>G (p.F17L) was the most frequent (27.27%, 18/66), followed by c.1400C>G (p.S467C) (25.76%, 17/66). Almost all the screened PCD patients were asymptomatic.
Conclusion: NBS via MS/MS was a quick and efficient method for the early diagnosis of PCD. The incidence of PCD in Shanghai was 1:31,200. Eight novel variants were identified, which greatly expanded the variant spectrum of SLC22A5. MS/MS combined with genetic testing could effectively improve the diagnostic accuracy of PCD.
Primary carnitine deficiency (PCD, OMIM 212140) refers to an autosomal recessive disorder of fatty acid oxidation caused by mutations in the SLC22A5 gene, which encodes the organic cation transporter 2 (OCTN2) (Tang et al., 1999; Cederbaum et al., 2002). The SLC22A5 gene is located on the chromosome 5q31.1 and contains over 150 reported disease-causing variants (Zhang et al., 2019). The incidence of PCD varies in different countries, which is estimated to be 1:142,000 in the United States, 1:40,000 in Japan, and even 1:300 in the Faroe Islands (Koizumi et al., 1999; Rasmussen et al., 2014; Therrell et al., 2014). Similarly, it varies in different regions throughout the China, ranging from 1:9,000 to 1:34,000 (Sun et al., 2017; Zhou et al., 2019; Lin et al., 2020; Lin et al., 2021). The clinical manifestations of PCD are diverse, mainly manifested as metabolic disorders, myocardial and skeletal muscle damage in infancy and early childhood, or muscle weakness and arrhythmia in adult (Saudubray and Garcia-Cazorla 2018). Patients with PCD may be at risk of sudden death due to cardiac failure (Longo et al., 2006; Kepka et al., 2011). Fortunately, early diagnosis by newborn screening (NBS) via tandem mass spectrometry (MS/MS) technology and timely supplementation of l-carnitine in the neonatal period can effectively prevent the poor outcome (Longo et al., 2016).
The main objective of this study was to analyze the clinical, biochemical, and molecular genetic characteristics of PCD patients identified by NBS in Shanghai. In this work, over 18-year experience of NBS for PCD was summarized and the genotype characteristics and clinical phenotype of the patients were compared, aiming to provide meaningful information for the large-scale screening and variant spectrum of SLC22A5 gene.
NBS via MS/MS was first introduced in China in 2003 at Xinhua Hospital, Shanghai Jiaotong University School of Medicine. From January 2003 to December 2021, 1,247,274 newborns were recruited for PCD screening in our hospital, accounting for approximately 70% of all infants born in Shanghai. This study was approved by the Ethical Committee of Xinhua Hospital, Shanghai Jiaotong University School of Medicine (Approval No. XHEC-D-2021-139). The written informed consent was obtained from the parents or guardians of newborns.
Neonatal blood samples collected from heels were spotted on the filter papers from various maternity hospitals in Shanghai within 72 h to 7 days after birth. Dried blood spot (DBS) samples were sent to Xinhua Hospital Newborn Screening Center by cold-chain transportation after natural air drying. DBS samples were pre-processed using a derivatized method and analyzed by Micromass Quattro micro API Mass spectrometer (2000, 4000 or 4500). Sample preparation and MS/MS analysis strictly followed the laboratory standard operating procedures. The cut-off value of C0 in our laboratory was 10–60 μmol/L (Han et al., 2014). Newborns with low C0 levels and their mothers were recalled to collect the DBS samples for MS/MS detection. Subsequently, the mutations in SLC22A5 gene were analyzed on suspected positive newborns to further confirm the diagnosis.
Genomic DNA of suspected positive newborns and their parents was extracted from peripheral venous blood, primary screening or recalled DBS samples by using a blood DNA isolation kit (Tiangen Biotech Co. Ltd.). The 10 exons and their boundaries of the SLC22A5 gene were amplified by polymerase chain reaction and analyzed on an automated DNA sequencer (ABI3700, Applied Biosystems). The mutations in SLC22A5 gene were analyzed on suspected PCD patients using the Sanger sequencing. The mutations were identified using a normal human SLC22A5 sequence as a reference (NM_003060.4), and the gene sequencing results were compared with the data in the HGMD, LOVD, ClinVar, and dbSNP databases to obtain information about the pathogenic mutation sites. The DNA samples of their parents were used to verify if the mutation originated from the father or mother. The possible pathogenicity of novel variants was evaluated by using the PolyPhen-2, SIFT, LRT, Mutation Taster PROVEAN and GERP++. The effects of novel missense mutations on protein functions were predicted by HOPE (http://www.cmbi.ru.nl/hope/; Venselaar et al., 2010).
Patients were diagnosed with PCD based on C0 levels, genetic mutations, and clinical symptoms. Newborns with two variants in the SLC22A5 gene were diagnosed with PCD. The suspected positive newborns not performing genetic testing or carrying one variant after exclusion of maternal and secondary carnitine deficiencies but with consecutively lower C0 levels until treatment were diagnosed with PCD.
PCD patients were treated with l-carnitine supplementation as soon as it was diagnosed. Generally, the dosage of oral administration is 50–200 mg/kg/day, divided into three times a day. The dosage of l-carnitine supplementation was adjusted according to different C0 levels in individuals. PCD patients were followed up monthly during the initial treatment period and then once every 2–3 months after the C0 level was normal and stable. Other regular follow-up items included blood ammonia, blood sugar, liver function, creatine kinase, abdominal ultrasound, electrocardiogram, echocardiography, growth, and intelligence development assessment.
SPSS 16.0 (SPSS Inc., Chicago, IL, United States) was adopted for statistical analysis. The Student’s t-test was applied to compare the difference of C0 levels between two groups, and p < 0.05 meant the difference was statistically significant.
1,247,274 newborns were screened by MS/MS, and 984 infants primarily displayed low C0 levels, so the positive rate was 0.08%. Among them, 934 newborns were successfully recalled, and the positive recall rate was 95%. Eventually, 40 newborns including 18 males and 22 females were diagnosed with PCD, yielding a positive predictive value (PPV) of 4.28% (40/934). In addition, 15 newborns were diagnosed with maternal PCD. From these results, the incidence of PCD in Shanghai was approximately 1:31,200.
250,000 healthy newborns were randomly selected to describe the frequency distribution of C0 levels. C0 levels in the DBS samples from healthy newborns exhibited a normal distribution, with the 0.1%–99.9% and 0.5%–99.5% confidence intervals (CIs) of 9.84–62.52 μmol/L and 11.09–54.10 μmol/L, respectively (Figure 1). The C0 level in healthy newborns was 26.00 ± 8.01 μmol/L (mean ± standard deviation) (Figure 2). All newborns and mothers with PCD presented decreased C0 levels. C0 levels of PCD newborns at the primary screening and after the recall were 5.37 ± 1.79 μmol/L and 5.57 ± 2.13 μmol/L, respectively (Figure 2). The C0 level in mothers with PCD was 3.05 ± 1.28 μmol/L, and that in newborns with PCD was 20% of that in healthy newborns.
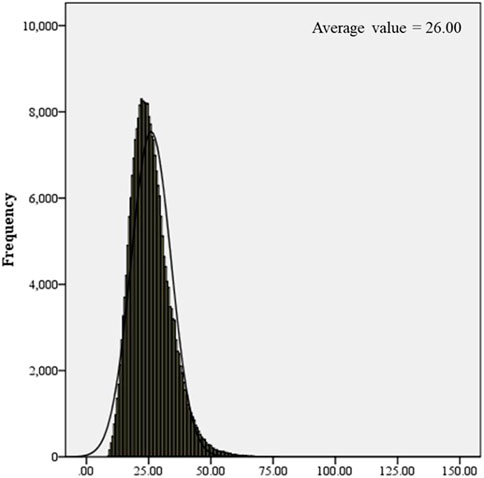
FIGURE 1. Frequency distribution histogram of C0.
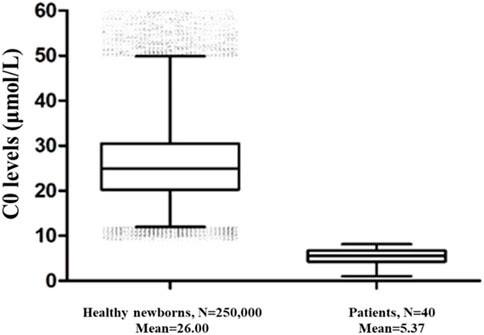
FIGURE 2. The C0 levels in healthy newborns and PCD patients. 250,000 healthy newborns were randomly selected for comparative analysis. The error bars are 1th and 99th percentiles.
PCD patients were treated with l-carnitine supplementation immediately after diagnosis, and the median age of patients at the initial treatment was 30 days. After treatment for about 1 month, the C0 level (24.45 ± 10.87 µmol) was significantly higher than that in primary screening (t = 9.668, p < 0.001). The follow-up period of PCD patients ranged from 1 month to 16 years, and the C0 level in most patients remained normal and stable. Case 3 claimed to be not good at sports. Case 15 was treated with l-carnitine supplementation since diagnosis to maintain the normal and stable C0 level, and suffered from easy fatigue and mild left ventricular enlargement. Case 17 was treated with l-carnitine supplementation for a short time after birth and then stopped treatment privately for 5 years; the C0 level in the most recent followed-up was 3.38 μmol/L, and muscle strength was weakness. The remaining PCD patients followed-up in our hospital were asymptomatic, with normal growth and development, no apparent abnormalities of heart and lung, and with good limb activity.
Thirty-six infants identified as having low C0 levels underwent SLC22A5 gene mutation analysis. Thirty newborns with PCD harbored two variants in SLC22A5, and six had one variant (Table 1). Twenty-three different variants were identified in SLC22A5 gene from 66 mutant alleles. Among them, 69.57% (16/23) were missense mutations, 17.39% (4/23) were non-sense mutations, 4.35% (1/23) were splicing mutations, and 8.70% (2/23) were frameshift mutations. Fifteen variants have been previously reported in the HGMD, LOVD, ClinVar, and dbSNP databases, and another eight were firstly found. Twelve variants were reported as pathogenic or likely pathogenic mutations, one variant is of uncertain clinical significance, and the remaining variants have no clinical classification (Table 2). The possible pathogenicity of 8 novel variants analyzed by PolyPhen-2, SIFT, LRT, Mutation Taster, PROVEAN, or GERP++ was listed in Table 3. The Sanger sequencing results of eight new mutations of SLC22A5 gene in PCD newborns and their parents was shown in Supplementary Figure S1. The mean primary screening and recalled C0 levels in four patients with c.51C>G (p.F17L) and a non-sense mutation or frameshift mutation (4.15 ± 0.67 μmol/L and 3.12 ± 0.58 μmol/L, respectively) were lower than those of 11 patients with c.51C>G (p.F17L) and missense mutation (5.92 ± 1.39 μmol/L and 6.55 ± 2.06 μmol/L, respectively), and the differences were statistically significant (t = 2.39, two-tailed p-value = 0.032, and t = 3.23, two-tailed p-value = 0.006, respectively).
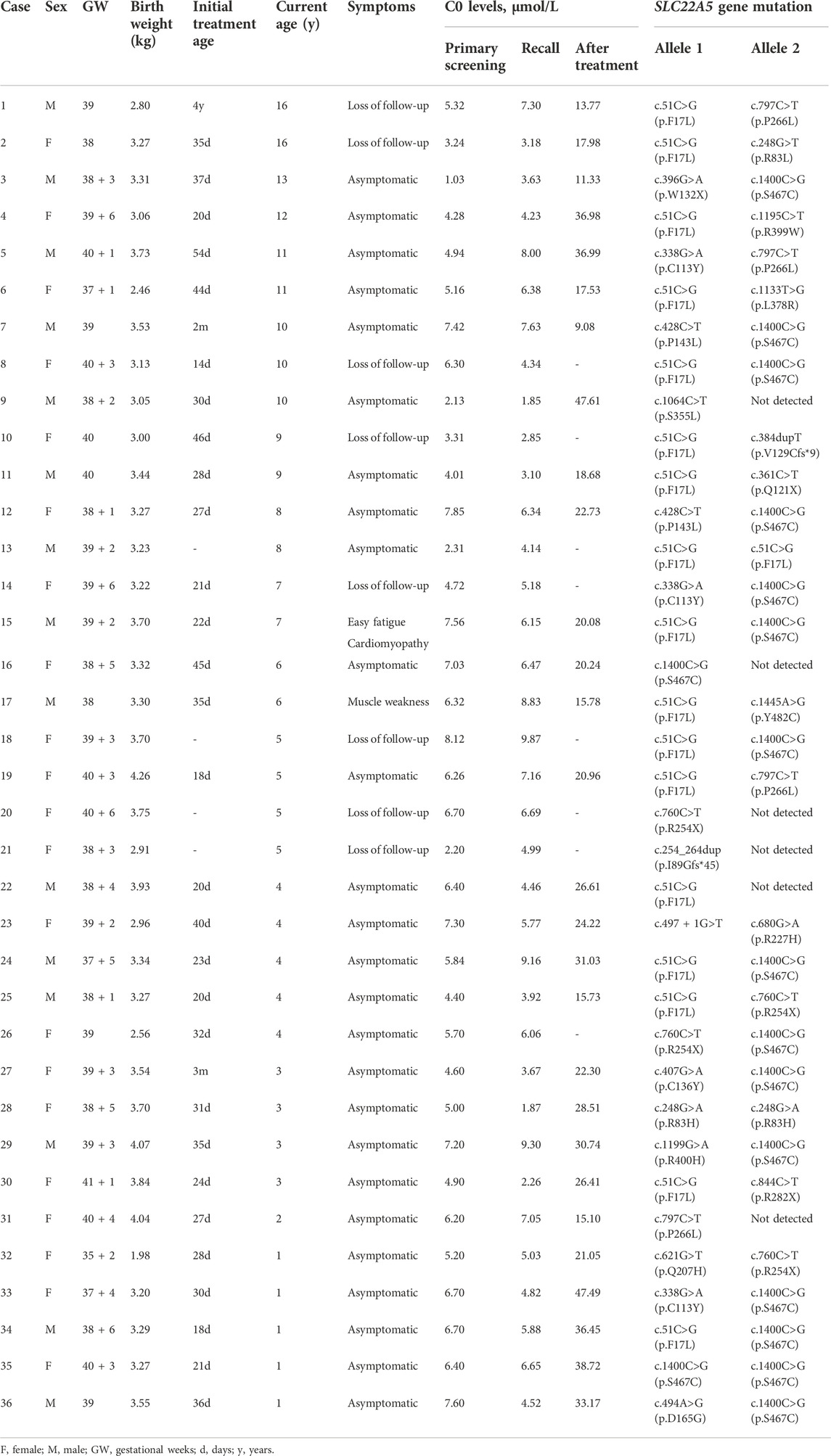
TABLE 1. Clinical, biochemical, and molecular characteristics of PCD patients identified by NBS.
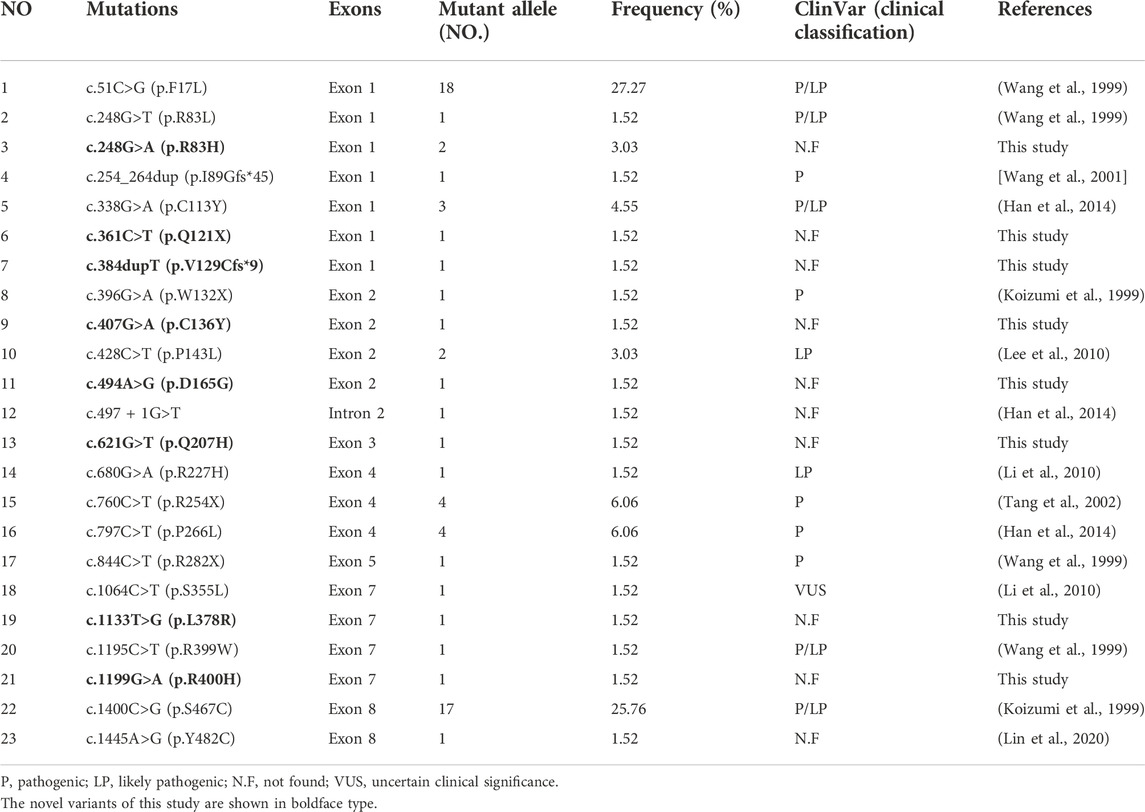
TABLE 2. Frequencies, locations, and clinical classification of detected mutations.
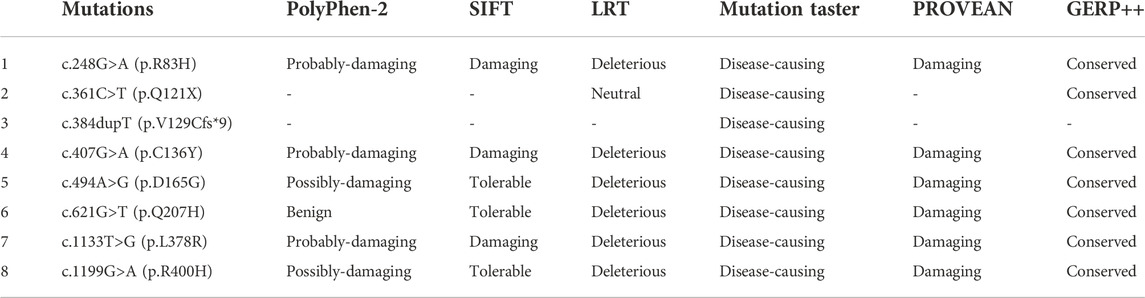
TABLE 3. List of the 8 novel variants and their possible pathogenicity analysis.
The most common mutation in PCD newborns was c.51C>G (p.F17L) with a frequency of 27.27% (18/66), followed by c.1400C>G (p.S467C) (25.76% (17/66)). In addition, c.760C>T (p.R254X) (6.06%), c.797C>T (p.P266L) (6.06%), and c.338G>A (p.C113Y) (4.55%) were relatively common. Almost all the identified variants were scattered on exons except one on intron.
The effects of protein structure and function on the 6 novel missense mutations were predicted by HOPE, and the diagram of 3D-protein structure was shown in Supplementary Figure S2. 1) p. R83H: the mutant residue was smaller than the wild-type Arg 83 residue. This difference in size might lead to loss of interactions. 2) p. C136Y: the mutant residue was bigger than the wild-type Cys 136 residue, which might lead to bumps. The mutant residue was located near a highly conserved position and probably damaged the protein. 3) p. D165G: the mutation introduced a very flexible glycine at position 165, which can disturb the required rigidity of the protein. The domain of mutant residue located was important for binding with other molecules. The charge of the wild-type Asp 165 residue was lost due to this mutation, which can cause loss of interactions with other molecules. 4) p. Q207H: the wild-type Glu 207 residue and mutant residue differ in size. The mutant residue was bigger than the wild-type residue, which can affect the contact with the lipid-membrane. 5) p. L378R: the mutant residue was bigger than the wild-type Leu 378 residue and could affect the contact with the lipid-membrane. The wild-type residue was more hydrophobic than the mutation, which might result in the loss of hydrophobic interactions with other molecules on the protein surface. 6) p. R400H: the mutant residue was smaller than the wild-type residue, and the charge of the wild-type residue was lost by this mutation, which might cause a possible loss of external interactions.
This NBS program for PCD comprised a large cohort of 1,247,274 newborns, most of them were from eastern China. A total of 40 newborns were diagnosed with PCD, yielding a prevalence of PCD in Shanghai of 1:31,200. We further analyzed the clinical, biochemical, and genetic characteristics of patients with PCD, and investigated the correlation between biochemistry and different mutation types. Moreover, we identified 8 novel variants from the SLC22A5 gene and predicted the protein structure effects on the new missense mutations. These findings expanded the PCD disease spectrum in Chinese patients and increased concerns about early diagnosis and therapy.
MS/MS is currently recognized as the most reliable and quick method for the early diagnosis of PCD by detecting the C0 level. However, C0 levels in infants shortly after birth reflect the carnitine levels of their mothers, because carnitine is transferred from the placenta to the fetus during pregnancy, which may lead to false-positive and false-negative diagnoses (El-Hattab et al., 2010; Magoulas and El-Hattab, 2012). Moreover, premature birth, mitochondrial diseases, insufficient intake, several organic acidemias and fatty acid oxidation defects, can lead to secondary carnitine deficiency (Lin et al., 2020). Therefore, a further differential diagnosis is required to rule out carnitine deficiency caused by these factors, so as to determine whether it is PCD. Notably, case 18 showed slightly decreased C0 levels at initial screening and recall (8.12 and 9.87 μmol/L, respectively) and harbored two pathogenic variants in the SLC22A5 gene, so that he may be readily missed if the recall procedures are not strictly followed. In addition, some PCD patients with one mutation were identified in this study, which might be related to sequence technology since it cannot detect variants in the regulatory regions or deep introns, and large deletions or duplications. This is also a limitation of this study, especially in patients with severely low C0 levels, such as case 9, which might present an important undetected variant. Collectively, MS/MS combined with genetic testing become an effective strategy to avoid missed diagnoses, especially in newborns with C0 levels closing to the lower reference limit.
The SLC22A5 gene is located on chromosome 5q31.1, containing 10 exons and three introns. So far, more than 150 disease-causing variants have been reported, most of which are missense mutations. The variant spectrum of SLC22A5 differs in different races and regions. For instance, the mutation c.95A>G (p.N32S) is common in Faroe Islands (Rasmussen et al., 2014), c.136C>T (p.P46S) is common in the Texas state of United States (Li et al., 2010), c.454G>C (p.G152R) and c.760C>T (p.R254X) are common in Turkey, and c.1400C>G (p.S467C) is common in Japan (Koizumi et al., 1999; Kilic et al., 2012). In China, the variant spectrum of SLC22A5 is also varied in different regions and provinces. A previous study found that c.1400C>G (p.S467C), c.51C>G (p.F17L), and c.760C > T (p.R254X) are common in Chinese patients (Lin et al., 2020). Among them, c.1400C>G (p.S467C) is the most common variant in Zhejiang, Jiangsu, and Henan provinces, and c.760C > T (p.R254X) is common in Taiwan (Tang et al., 2002; Lee et al., 2010; Sun et al., 2017; Zhou et al., 2019; Lin et al., 2020). In contrast to these reports, this study found that c.51C>G (p.F17L) and c.1400C>G (p.S467C) were the most common variants in PCD newborns, and the frequencies of these two mutations among the identified variants showed no significant difference, which indicated that these two variants might be hotspot mutations in eastern China. Furthermore, 8 novel variants were identified in this work: c.248G>A (p.R83H), c.361C>T (p.Q121X), c.384dupT (p.V129Cfs*9), c.407G>A (p.C136Y), c.494A>G (p.D165G), c.621G>T (p.Q207H), c.1133T>G (p.L378R), and c.1199G>A (p.R400H), expanding the variant spectrum of the SLC22A5 gene. The possible pathogenicity and protein structure effect on the novel mutations predicted by the bioinformatic analysis indicate that these variants may be potential etiologies of PCD, and further functional research is required to evaluate the effects of these new mutations.
Consistent with previous report, early diagnosis and treatment have good outcomes for patients with PCD (Tang et al., 2002). Within our cohort, 70% of patients were asymptomatic, and 5% suffered from easy fatigue, mild left ventricular enlargement, and muscle weakness. Unfortunately, 25% of the patients were lost to follow-up, and the possible reason was poor treatment compliance because they might have no symptoms without treatment. In this study, c.760C > T (p.R254X) was the most common non-sense mutation, which can cause a premature termination codon at amino acid 254 and prevent OCTN2, leading to obvious clinical manifestations (Tang et al., 2002; Han et al., 2014; Frigeni et al., 2017). Four PCD patients with R254X mutation identified by NBS showed no obvious clinical manifestation after carnitine supplementation and dietary guidance. However, PCD patients with two R254X mutations diagnosed in the clinic presented muscle weakness and cardiomyopathy, those with R254X and other mutations suffered from heart failure, cardiomyopathy, hepatomegaly, diarrhea, feeding difficulties, fever, and even death (Han et al., 2014). Therefore, standardized medication instruction and targeted treatment adjustment are essential for Chinese PCD patients, while asymptomatic patients also need long-term follow-up and treatment to monitor their health conditions, highlighting the importance of NBS.
In order to gain further insight into the biochemistry and genotype correlation in PCD, we analyzed the C0 levels of different mutation types in the patients. Based on a previous report, PCD patients with non-sense mutations or frameshift mutations show extremely feeble OCTN2 activity, while those with missense mutations have partial residual OCTN2 activity (Kilic et al., 2012). Nowadays, the relationship between the C0 level and SLC22A5 genotype in PCD patients identified by NBS is unclear (Zhang et al., 2019). However, it was found in this work that the primary screening and recalled C0 levels in patients with c.51C>G (p.F17L) and a non-sense mutation or frameshift mutation were lower than those with c.51C>G (p.F17L) and other missense mutations. All compound heterozygous PCD patients with c.51C>G (p.F17L) and a non-sense mutation or frameshift mutation have a C0 value of <5 μmol/L. The C0 level (1.03 μmol/L) of case 3 with c.396G>A (p.W132X) and c.1400C>G (p.S467C) was the lowest among all screened PCD patients, and was 15% of those with c.1400C>G (p.S467C) and other missense mutations (6.72 ± 1.16 μmol/L). In addition, 2 novel variants were identified from the non-sense or frameshift mutations, c.361C>T (p.Q121X) can result in a premature termination of translation and presented between transmembrane domains 1 and 2, c.384dupT can cause a frameshift of p. V129Cfs*9 at the first extracellular topological domain of OCTN2. Both patients with these two mutations had a C0 value of <5 μmol/L, which further suggested that non-sense or frameshift mutations might affect the C0 levels of PCD newborns more greatly.
In conclusion, NBS via MS/MS was an efficient and reliable method for early diagnosis of PCD. The incidence of PCD in Shanghai was 1:31,200. Eight novel variants were identified in this study, greatly expanding the variant spectrum of SLC22A5 gene. c.51C>G (p.F17L) and c.1400C>G (p.S467C) were the most common variants of newborns in Shanghai. The combination of biochemical and molecular analysis could effectively increase the diagnostic accuracy of PCD.
The data presented in the study are deposited in the SRA database, accession numbers: SRR22420523-SRR22420530.
The studies involving human participants were reviewed and approved by The Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine (Approval No. XHEC-D-2021-139). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
SC contributed to detecting neonatal samples, reorganizing and analyzing the clinical data of the patients, and drafting the manuscript. YY contributed to reorganizing and analyzing the clinical data of the patients and revising the manuscript. FX, WJ, XZ, XG, and TC contributed to detecting neonatal dried blood samples by tandem mass spectrometry. WQ, HZ, LL, DL, KZ, and XG contributed to collecting and treating the patients and providing the clinical data. LH, as the doctor of most patients, contributed to designing the research, treating the patients, providing the clinical data, and revising the manuscript. All authors contributed to the article.
This work was supported by the National Key Research and Development Program of China (No. 2016YFC0901505).
We are very grateful to the patients and their families for their active participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1062715/full#supplementary-material
Cederbaum, S. D., Koo-McCoy, S., Tein, I., Hsu, B. Y., Ganguly, A., Vilain, E., et al. (2002). Carnitine membrane transporter deficiency: A long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol. Genet. Metab. 77 (3), 195–201. doi:10.1016/s1096-7192(02)00169-5
El-Hattab, A. W., Li, F. Y., Shen, J., Powell, B. R., Bawle, E. V., Adams, D. J., et al. (2010). Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects. Genet. Med. 12 (1), 19–24. doi:10.1097/GIM.0b013e3181c5e6f7
Frigeni, M., Balakrishnan, B., Yin, X., Calderon, F. R. O., Mao, R., Pasquali, M., et al. (2017). Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 38 (12), 1684–1699. doi:10.1002/humu.23315
Han, L., Wang, F., Wang, Y., Ye, J., Qiu, W., Zhang, H., et al. (2014). Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency. Eur. J. Med. Genet. 57 (10), 571–575. doi:10.1016/j.ejmg.2014.08.001
Kepka, A., Szajda, S. D., Waszkiewicz, N., Pludowski, P., Chojnowska, S., Rudy, M., et al. (2011). Carnitine function metabolism and value in hepatic failure during chronic alcohol intoxication. Postepy Hig. Med. Dosw. 65, 645–653. doi:10.5604/17322693.962226
Kilic, M., Ozgul, R. K., Coskun, T., Yucel, D., Karaca, M., Sivri, H. S., et al. (2012). Identification of mutations and evaluation of cardiomyopathy in Turkish patients with primary carnitine deficiency. JIMD Rep. 3, 17–23. doi:10.1007/8904_2011_36
Koizumi, A., Nozaki, J., Ohura, T., Kayo, T., Wada, Y., Nezu, J., et al. (1999). Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 8 (12), 2247–2254. doi:10.1093/hmg/8.12.2247
Lee, N. C., Tang, N. L., Chien, Y. H., Chen, C. A., Lin, S. J., Chiu, P. C., et al. (2010). Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol. Genet. Metab. 100 (1), 46–50. doi:10.1016/j.ymgme.2009.12.015
Li, F. Y., El-Hattab, A. W., Bawle, E. V., Boles, R. G., Schmitt, E. S., Scaglia, F., et al. (2010). Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum. Mutat. 31 (8), E1632–E1651. doi:10.1002/humu.21311
Lin, Y., Xu, H., Zhou, D., Hu, Z., Zhang, C., Hu, L., et al. (2020). Screening 3.4 million newborns for primary carnitine deficiency in Zhejiang Province, China. Clin. Chim. Acta. 507, 199–204. doi:10.1016/j.cca.2020.04.039
Lin, Y., Zhang, W., Huang, C., Lin, C., Lin, W., Peng, W., et al. (2021). Increased detection of primary carnitine deficiency through second-tier newborn genetic screening. Orphanet J. Rare Dis. 16 (1), 149. doi:10.1186/s13023-021-01785-6
Longo, N., Amat di San Filippo, C., and Pasquali, M. (2006). Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 142C (2), 77–85. doi:10.1002/ajmg.c.30087
Longo, N., Frigeni, M., and Pasquali, M. (2016). Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 1863 (10), 2422–2435. doi:10.1016/j.bbamcr.2016.01.023
Magoulas, P. L., and El-Hattab, A. W. (2012). Systemic primary carnitine deficiency: An overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 7, 68. doi:10.1186/1750-1172-7-68
Rasmussen, J., Nielsen, O. W., Janzen, N., Duno, M., Gislason, H., Kober, L., et al. (2014). Carnitine levels in 26, 462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J. Inherit. Metab. Dis. 37 (2), 215–222. doi:10.1007/s10545-013-9606-2
Saudubray, J. M., and Garcia-Cazorla, A. (2018). Inborn errors of metabolism overview: Pathophysiology, manifestations, evaluation, and management. Pediatr. Clin. North Am. 65 (2), 179–208. doi:10.1016/j.pcl.2017.11.002
Sun, Y., Wang, Y. Y., and Jiang, T. (2017). Clinical features and genotyping of patients with primary carnitine deficiency identified by newborn screening. J. Pediatr. Endocrinol. Metab. 30 (8), 879–883. doi:10.1515/jpem-2017-0002
Tang, N. L., Ganapathy, V., Wu, X., Hui, J., Seth, P., Yuen, P. M., et al. (1999). Mutations of OCTN2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum. Mol. Genet. 8 (4), 655–660. doi:10.1093/hmg/8.4.655
Tang, N. L., Hwu, W. L., Chan, R. T., Law, L. K., Fung, L. M., and Zhang, W. M. (2002). A founder mutation (R254X) of SLC22A5 (OCTN2) in Chinese primary carnitine deficiency patients. Hum. Mutat. 20 (3), 232. doi:10.1002/humu.9053
Therrell, B. L., Lloyd-Puryear, M. A., Camp, K. M., and Mann, M. Y. (2014). Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol. Genet. Metab. 113 (1-2), 14–26. doi:10.1016/j.ymgme.2014.07.009
Venselaar, H., Te Beek, T. A., Kuipers, R. K., Hekkelman, M. L., and Vriend, G. (2010). Protein structure analysis of mutations causing inheritable diseases. An e-science approach with life scientist friendly interfaces. BMC Bioinform. 11, 548. doi:10.1186/1471-2105-11-548
Wang, Y., Ye, J., Ganapathy, V., and Longo, N. (1999). Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc. Natl. Acad. Sci. USA 96 (5), 2356–2360. doi:10.1073/pnas.96.5.2356
Zhang, Y., Li, H., Liu, J., Yan, H., Liu, Q., Wei, X., et al. (2019). Molecular investigation in Chinese patients with primary carnitine deficiency. Mol. Genet. Genomic Med. 7 (9), e901. doi:10.1002/mgg3.901
Keywords: primary carnitine deficiency, SLC22A5, newborn screening, tandem mass spectrometry, free carnitine
Citation: Chang S, Yang Y, Xu F, Ji W, Zhan X, Gao X, Chen T, Qiu W, Zhang H, Liang L, Lu D, Zhang K, Gu X and Han L (2022) Clinical, biochemical, and molecular genetic characteristics of patients with primary carnitine deficiency identified by newborn screening in Shanghai, China. Front. Genet. 13:1062715. doi: 10.3389/fgene.2022.1062715
Received: 06 October 2022; Accepted: 22 November 2022;
Published: 08 December 2022.
Edited by:
Elsayed Abdelkreem, Sohag University, EgyptReviewed by:
Kevin E. Glinton, Baylor College of Medicine, United StatesCopyright © 2022 Chang, Yang, Xu, Ji, Zhan, Gao, Chen, Qiu, Zhang, Liang, Lu, Zhang, Gu and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lianshu Han, aGFubGlhbnNodUB4aW5odWFtZWQuY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.